Mechanistic Insights into the Role of Molecular Chaperones in Protein Misfolding Diseases: From Molecular Recognition to Amyloid Disassembly


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
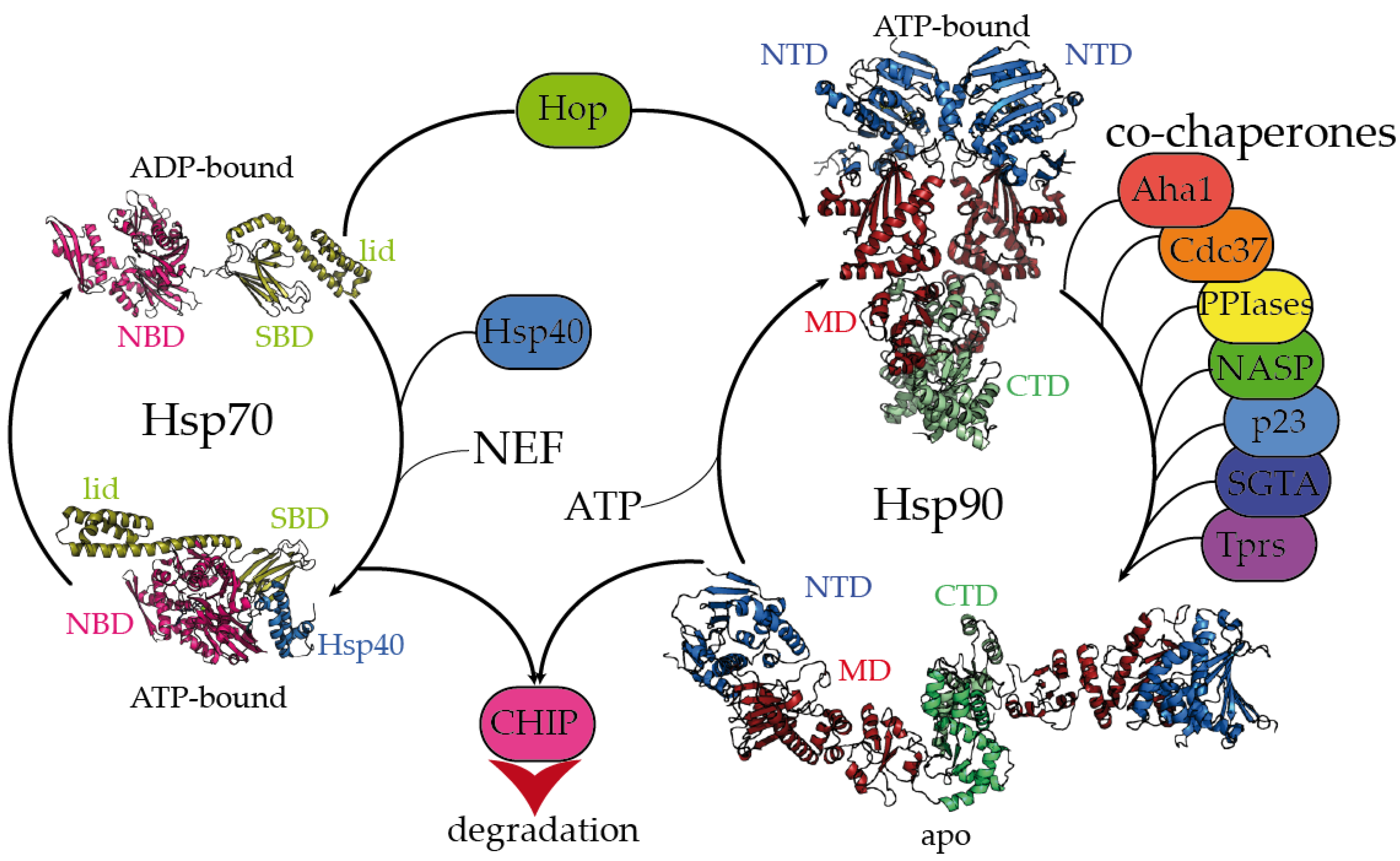
2. Dynamic Hsp70 and Hsp90 Are Closely Monitored by Co-Chaperones
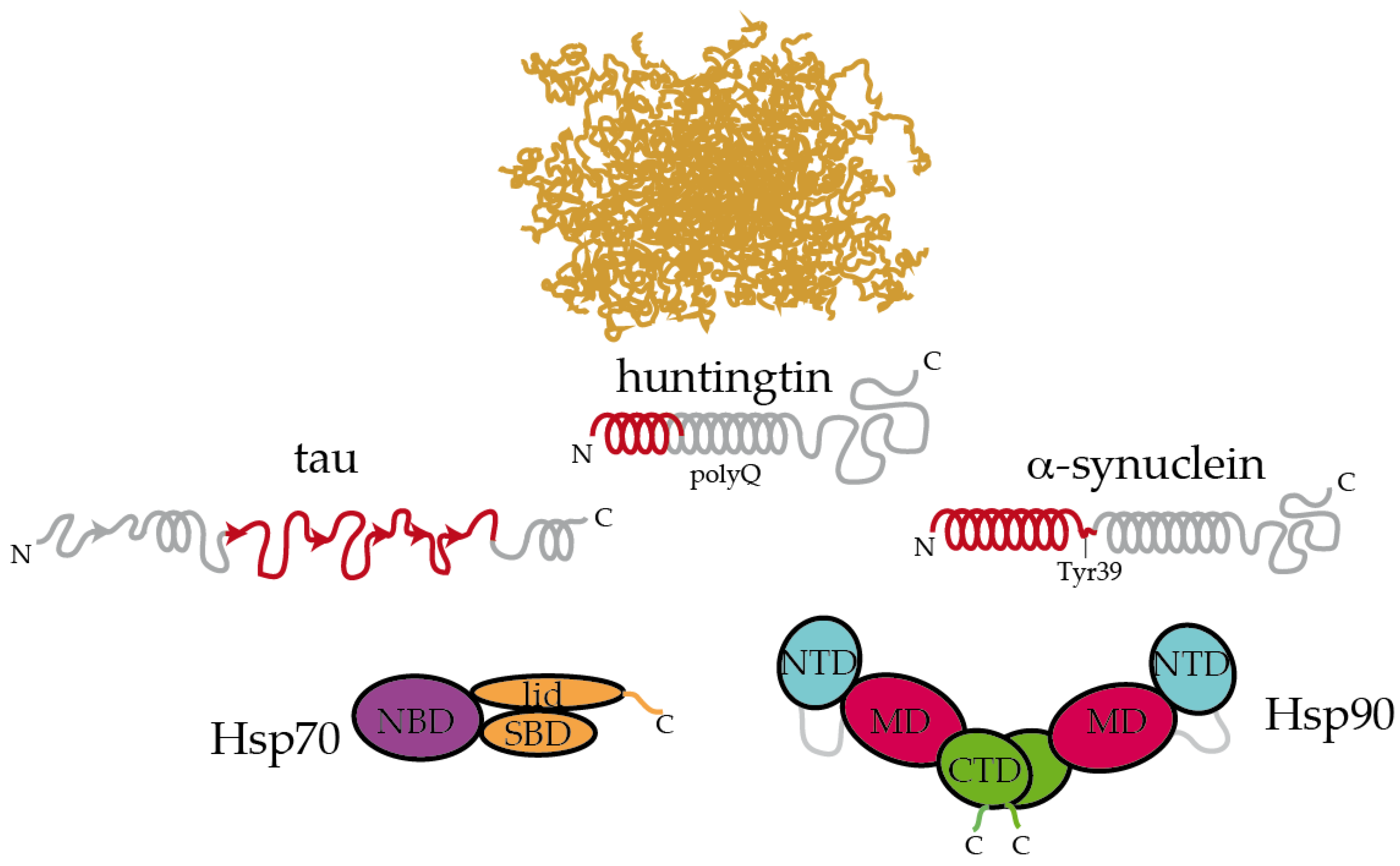
3. Selective Recognition of Misfolded Proteins by Hsp70 and Hsp90
4. Hsp70 and Hsp90 Play Crucial Roles in Disease-Related LLPS
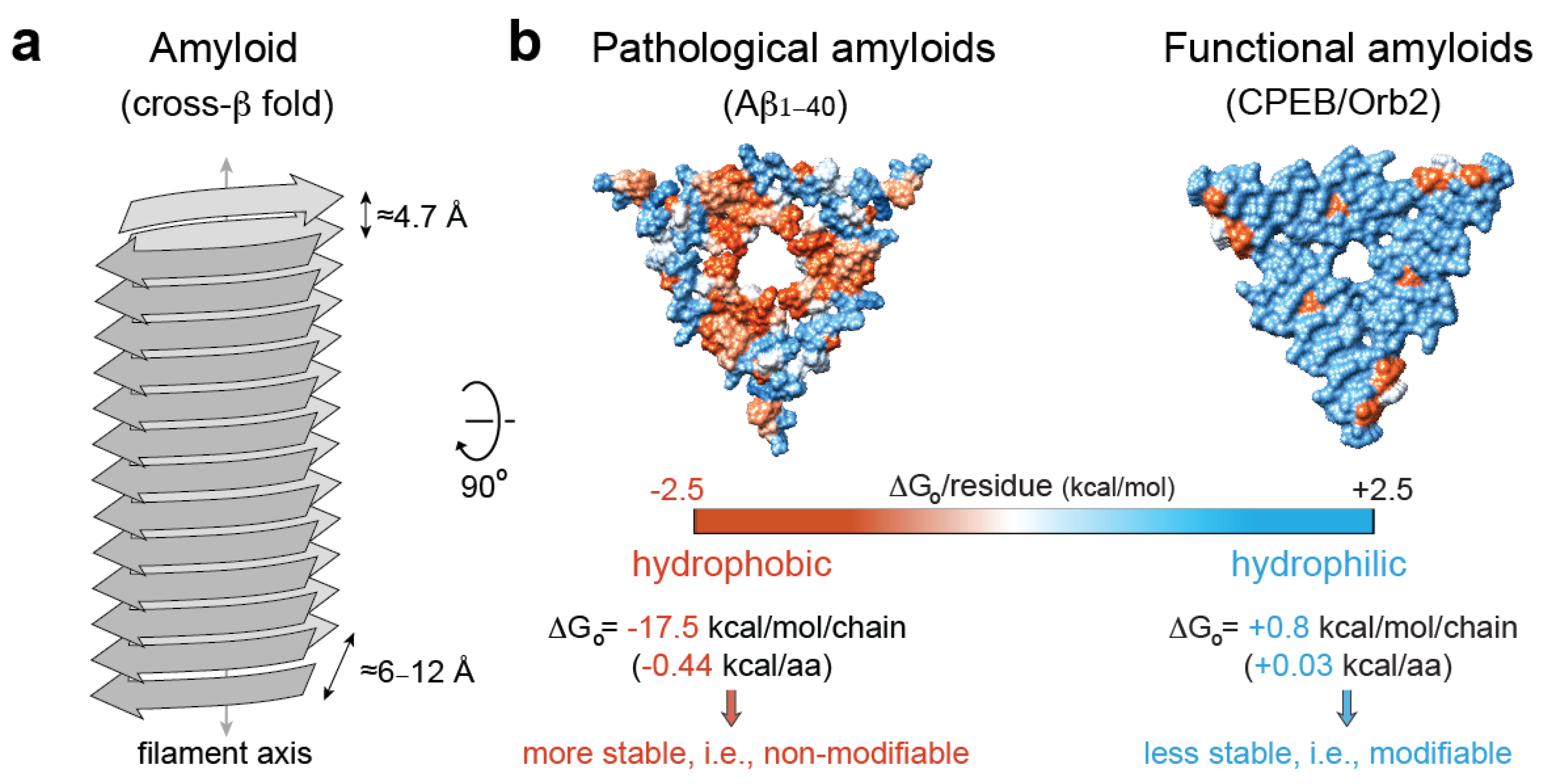
5. Pathological vs. Functional Amyloids
- (i)
- Scaffolding systems, such as curli in microbial biofilm, providing a scaffold to protect bacteria and promote adherence to host cells [128].
- (ii)
- Storage systems, such as Pmel17, which allows for the sequestration and condensation of melanin in the lumen of melanosomes [129]; vicilin, a 7S globulin in garden pea Pisum sativum L. seeds, which plays a crucial role in seed longevity [130]; peptide hormones for storage and release of certain hormones in the pituitary secretory granules [8]; Xvelo in Xenopus or Bucky Ball in zebrafish, which form a non-membranous compartment termed the Balbiani body to store germline-specific maternal mRNAs [131].
- (iii)
- (iv)
- Translation regulator systems, such as the yeast prion [PSI+], a general translational terminator that provides heritable phenotypic variability [134]; Rim4, a translation inhibitor of cyclin mRNA that controls gametogenesis [135]; or Orb2, the Drosophila member of the mRNA-binding cytoplasmic polyadenylation element family of proteins [136,137].
- (v)
- Enzymatic systems, such as the membrane-associated protein Herzog (Hzg), which is required for the proper establishment of segment polarity in Drosophila embryo through an amyloid-like assembly that activates a phosphatase that is crucial for proper development [138].
6. Interplay between Chaperones and Amyloids
7. Aberrant Chaperone Complexes Have Deleterious Consequences
8. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PN | Proteostasis network |
| LLPS | Liquid–liquid phase separation |
| Hsp | Heat-shock protein |
| sHsp | Small Hsp |
| NBD | Nucleotide-binding domain (Hsp70) |
| SBD | Substrate-binding domain (Hsp70) |
| TPR | Tetratricopeptide repeat |
| Hop | Hsp-organizing protein |
| NTD | N-terminal domain (Hsp90) |
| MD | Middle domain (Hsp90) |
| CTD | C-terminal domain (Hsp90) |
| NEF | Nucleotide exchange factor |
| ATP | Adenosine triphosphate |
| TTR | Transthyretin |
| SOD1 | Superoxide dismutase 1 |
| ALS | Amyotrophic lateral sclerosis |
| IDP | Intrinsically disordered protein |
| HTT | Huntingtin |
| IAPP | Islet amyloid polypeptide |
| TDP-43 | TAR DNA binding protein of 43 kDa |
| NMR | Nuclear magnetic resonance |
| Aβ | Amyloid-β |
| RNP | Ribonucleoprotein particle |
| SG | Stress granule |
| Hzg | Herzog |
| NMNAT | Nicotinamide mononucleotide adenylyl transferase |
| PPIase | Peptidyl-proline isomerase |
| USP19 | Ubiquitin-specific protease 19 |
References
- Powers, E.T.; Morimoto, R.I.; Dillin, A.; Kelly, J.W.; Balch, W.E. Biological and chemical approaches to diseases of proteostasis deficiency. Annu. Rev. Biochem. 2009, 78, 959–991. [Google Scholar] [CrossRef]
- Labbadia, J.; Morimoto, R.I. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Brehme, M.; Voisine, C.; Rolland, T.; Wachi, S.; Soper, J.H.; Zhu, Y.; Orton, K.; Vilella, A.; Garza, D.; Vidal, M.; et al. A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Rep. 2014, 9, 1135–1150. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein misfolding, amyloid formation and human disease: A summary of progress over the last decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A. Amyloidosis: A convoluted story. Br. J. Haemat. 2001, 114, 529–538. [Google Scholar] [CrossRef]
- Si, K.; Lindquist, S.; Kandel, E.R. A neuronal isoform of the Aplysia CPEB has prion-like properties. Cell 2003, 115, 879–891. [Google Scholar] [CrossRef]
- Maji, S.K.; Perrin, M.H.; Sawaya, M.R.; Jessberger, S.; Vadodaria, K.; Rissman, R.A.; Singru, P.S.; Nilsson, K.P.R.; Simon, R.; Schubert, D.; et al. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science 2009, 325, 328–332. [Google Scholar] [CrossRef]
- Hervás, R.; Rau, M.J.; Park, Y.; Zhang, W.; Murzin, A.G.; Fitzpatrick, J.A.J.; Scheres, S.H.W.; Si, K. Cryo-EM structure of a neuronal functional amyloid implicated in memory persistence in Drosophila. Science 2020, 367, 1230–1234. [Google Scholar] [CrossRef]
- Arnold, E.S.; Ling, S.-C.; Huelga, S.C.; Lagier-Tourenne, C.; Polymenidou, M.; Ditsworth, D.; Kordasiewicz, H.B.; McAlonis-Downes, M.; Platoshyn, O.; Parone, P.A.; et al. ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc. Natl. Acad. Sci. USA 2013, 110, E736–E745. [Google Scholar] [CrossRef]
- Gordon, D.; Dafinca, R.; Scaber, J.; Alegre-Abarrategui, J.; Farrimond, L.; Scott, C.; Biggs, D.; Kent, L.; Oliver, P.L.; Davies, B.; et al. Single-copy expression of an amyotrophic lateral sclerosis-linked TDP-43 mutation (M337V) in BAC transgenic mice leads to altered stress granule dynamics and progressive motor dysfunction. Neurobiol. Dis. 2019, 121, 148–162. [Google Scholar] [CrossRef] [PubMed]
- Hervás, R.; Oroz, J.; Galera-Prat, A.; Goñi, O.; Valbuena, A.; Vera, A.M.; Gómez-Sicilia, A.; Losada-Urzáiz, F.; Uversky, V.N.; Menéndez, M.; et al. Common features at the start of the neurodegeneration cascade. PLoS Biol. 2012, 10, e1001335. [Google Scholar] [CrossRef] [PubMed]
- Escusa-Toret, S.; Vonk, W.I.M.; Frydman, J. Spatial sequestration of misfolded proteins by a dynamic chaperone pathway enhances cellular fitness during stress. Nat. Cell Biol. 2013, 15, 1231–1243. [Google Scholar] [CrossRef]
- Bolognesi, B.; Faure, A.J.; Seuma, M.; Schmiedel, J.M.; Tartaglia, G.G.; Lehner, B. The mutational landscape of a prion-like domain. Nat. Commun. 2019, 10, 4162. [Google Scholar] [CrossRef] [PubMed]
- Alberti, S.; Hyman, A.A. Are aberrant phase transitions a driver of cellular aging? Bioessays 2016, 38, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Mateju, D.; Franzmann, T.M.; Patel, A.; Kopach, A.; Boczek, E.E.; Maharana, S.; Lee, H.O.; Carra, S.; Hyman, A.A.; Alberti, S. An aberrant phase transition of stress granules triggered by misfolded protein and prevented by chaperone function. EMBO J. 2017, 36, 1669–1687. [Google Scholar] [CrossRef] [PubMed]
- Oroz, J.; Laurents, D.V. RNA binding proteins: Diversity from microsurgeons to cowboys. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 194398. [Google Scholar] [CrossRef] [PubMed]
- Inda, M.C.; Joshi, S.; Wang, T.; Bolaender, A.; Gandu, S.; Koren, J., III; Che, A.Y.; Taldone, T.; Yan, P.; Sun, W.; et al. The epichaperome is a mediator of toxic hippocampal stress and leads to protein connectivity-based dysfunction. Nat. Commun. 2020, 11, 319. [Google Scholar] [CrossRef]
- Ritossa, F. Discovery of the heat shock response. Cell Stress Chaperones 1996, 1, 97–98. [Google Scholar] [CrossRef]
- Haslbeck, M.; Franzmann, T.; Weinfurtner, D.; Buchner, J. Some like it hot: The structure and function of small heat-shock proteins. Nat. Struct. Mol. Biol. 2005, 12, 842–846. [Google Scholar] [CrossRef]
- Kim, Y.E.; Hipp, M.S.; Bracher, A.; Hayer-Hartl, M.; Hartl, F.U. Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 2013, 82, 323–355. [Google Scholar] [CrossRef] [PubMed]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Molec. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Taipale, M.; Jarosz, D.F.; Lindquist, S. HSP90 at the hub of protein homeostasis: Emerging mechanistic insights. Nat. Rev. Molec. Cell Biol. 2010, 11, 515–528. [Google Scholar] [CrossRef]
- Mayer, M.P.; Gierasch, L.M. Recent advances in the structural and mechanistic aspects of Hsp70 molecular chaperones. J. Biol. Chem. 2019, 294, 2085–2097. [Google Scholar] [CrossRef] [PubMed]
- Oroz, J.; Kim, J.H.; Chang, B.J.; Zweckstetter, M. Mechanistic basis for the recognition of a misfolded protein by the molecular chaperone Hsp90. Nat. Struct. Mol. Biol. 2017, 24, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Oroz, J.; Chang, B.J.; Wysoczanski, P.; Lee, C.-T.; Pérez-Lara, A.; Chakraborty, P.; Hofele, R.V.; Baker, J.D.; Blair, L.J.; Biernat, J.; et al. Structure and pro-toxic mechanism of the human Hsp90/PPIase/Tau complex. Nat. Commun. 2018, 9, 4532. [Google Scholar] [CrossRef] [PubMed]
- Kampinga, H.H.; Craig, E.A. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 2010, 11, 579–592. [Google Scholar] [CrossRef]
- Faust, O.; Abayev-Avraham, M.; Wentik, A.S.; Maurer, M.; Nillegoda, N.B.; London, N.; Bukau, B.; Rosenzweig, R. HSP40 proteins use class-specific regulation to drive HSP70 functional diversity. Nature 2020, 587, 489–494. [Google Scholar] [CrossRef]
- Dean, M.E.; Johnson, J.L. Human Hsp90 cochaperones: Perspectives on tissue-specific expression and identification of cochaperones with similar in vivo functions. Cell Stress Chaper. 2020, 1–11. Available online: https://link.springer.com/article/10.1007/s12192-020-01167-0 (accessed on 30 November 2020). [CrossRef]
- Peterson, L.B.; Blagg, B.S.J. To fold or not to fold: Modulation and consequences of Hsp90 inhibition. Future Med. Chem. 2009, 1, 267–283. [Google Scholar] [CrossRef]
- Karamanos, T.; Tugarinov, V.; Clore, G.M. Unraveling the structure and dynamics of the human DNAJB6b chaperone by NMR reveals insights into Hsp40-mediated proteostasis. Proc. Natl. Acad. Sci. USA 2019, 116, 21529–21538. [Google Scholar] [CrossRef]
- Kabani, M.; Martineau, C.N. Multiple Hsp70 isoforms in the eukayoric cytosol: Mere redundancy or functional specificity? Curr. Genomics 2008, 9, 338–348. [Google Scholar] [CrossRef] [PubMed]
- English, C.A.; Sherman, W.; Meng, W.; Gierasch, L.M. The Hsp70 interdomain linker is a dynamic switch that enables allosteric communication between two structured domains. J. Biol. Chem. 2017, 292, 14765–14774. [Google Scholar] [CrossRef] [PubMed]
- Jinwal, U.K.; Akoury, E.; Abisambra, J.F.; O’Leary, J.C., 3rd; Thompson, A.D.; Blair, L.J.; Jin, Y.; Bacon, J.; Nordhues, B.A.; Cockman, M.; et al. Imbalance of Hsp70 family variants fosters tau accumulation. FASEB J. 2013, 27, 1450–1459. [Google Scholar] [CrossRef] [PubMed]
- Lott, A.; Oroz, J.; Zweckstetter, M. Molecular basis of the interaction of Hsp90 with its co-chaperone Hop. Protein Sci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Csermely, P.; Schnaider, T.; Soti, C.; Prohaszka, Z.; Nardai, G. The 90-kDa molecular chaperone family: Structure, function, and clinical applications. A comprehensive review. Pharmacol. Ther. 1998, 79, 129–168. [Google Scholar] [CrossRef]
- Verba, K.A.; Wang, R.Y.-R.; Arakawa, A.; Liu, Y.; Shirouzu, M.; Yokoyama, S.; Agard, D.A. Atomic structure of Hsp90-Cdc37-Cdk4 reveals that Hsp90 traps and stabilizes an unfolded kinase. Science 2016, 352, 1542–1547. [Google Scholar] [CrossRef]
- Daturpalli, S.; Kniess, R.A.; Lee, C.T.; Mayer, M.P. Large rotation of the N-terminal domain of Hsp90 is important for interaction with some but not all client proteins. J. Mol. Biol. 2017, 429, 1406–1423. [Google Scholar] [CrossRef]
- Zhao, R.; Davey, M.; Hsu, Y.C.; Kaplanek, P.; Tong, A.; Parsons, A.B.; Krogan, N.; Cagney, G.; Mai, D.; Greenblatt, J.; et al. Navigating the chaperone network: An integrative map of physical and genetic interactions mediated by the Hsp90 chaperone. Cell 2005, 120, 715–727. [Google Scholar] [CrossRef]
- Meyer, P.; Prodromou, C.; Liao, C.; Hu, B.; Roe, S.M.; Vaughan, C.K.; Vlasic, I.; Panaretou, B.; Piper, P.W.; Pearl, L.H. Structural basis for recruitment of the ATPase activator Aha1 to the Hsp90 chaperone machinery. EMBO J. 2004, 23, 1402–1410. [Google Scholar] [CrossRef]
- Ali, M.M.U.; Roe, S.M.; Vaughan, C.K.; Meyer, P.; Panaretou, B.; Piper, P.W.; Prodromou, C.; Pearl, L.H. Crystal structure of an Hsp90-nucelotide-p23/Sba1 closed chaperone complex. Nature 2006, 440, 1013–1017. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Kadota, Y.; Prodromou, C.; Shirasu, K.; Pearl, L.H. Structural basis for assembly of Hsp90-Sgt1-CHORD protein complexes: Implications for chaperoning of NLR innate immunity receptors. Mol. Cell 2010, 39, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Oroz, J.; Blair, L.J.; Zweckstetter, M. Dynamic Aha1 co-chaperone binding to human Hsp90. Prot. Sci. 2019, 28, 1545–1551. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sun, M.; Myasnikov, A.G.; Elnatan, D.; Delaeter, N.; Nguyenquang, M.; Agard, D.A. Cryo-EM structures reveal a multistep mechanism of Hsp90 activation by co-chaperone Aha1. Biorxiv 2020. [Google Scholar] [CrossRef]
- Bertelsen, E.B.; Chang, L.; Gestwicki, J.E.; Zuiderweg, E.R.P. Solution conformation of wild-type E. coli Hsp70 (DnaK) chaperone complexed with ADP and substrate. Proc. Natl. Acad. Sci. USA 2009, 106, 8471–8476. [Google Scholar] [CrossRef]
- Kityk, R.; Kopp, J.; Mayer, M.P. Molecular mechanism of J-domain-triggered ATP hydrolysis by Hsp70 chaperones. Mol. Cell 2018, 69, 227–237. [Google Scholar] [CrossRef]
- Muller, P.; Ruckova, E.; Halada, P.; Coates, P.J.; Hrstka, R.; Lane, D.P.; Vojtesek, B. C-terminal phosphorylation of Hsp70 and Hsp90 regulates alternative binding to co-chaperones CHIP and HOP to determine cellular protein folding/degradation balances. Oncogene 2013, 32, 3101–3110. [Google Scholar] [CrossRef]
- He, M.; Guo, H.; Yang, X.; Zhou, L.; Zhang, X.; Cheng, L.; Zeng, H.; Hu, F.B.; Tanguay, R.M.; Wu, T. Genetic variations in HSPA8 gene associated with coronary heart disease risk in a Chinese population. PLoS ONE 2010, 5, e9684. [Google Scholar] [CrossRef] [PubMed]
- Harms, M.B.; Sommerville, R.B.; Allred, P.; Bell, S.; Ma, D.; Cooper, P.; Lopate, G.; Pestronk, A.; Weihl, C.C.; Baloh, R.H. Exome sequencing reveals DNAJB6 mutations in dominantly-inherited myopathy. Ann. Neurol. 2012, 71, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Blumen, S.C.; Astord, S.; Robin, V.; Vignaud, L.; Toumi, N.; Cieslik, A.; Achiron, A.; Carasso, R.L.; Gurevich, M.; Braverman, I.; et al. A rare recessive distal hereditary motor neuropathy with HSJ1 chaperone mutation. Ann. Neurol. 2012, 71, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Farhan, S.M.K.; Howrigan, D.P.; Abbott, L.E.; Klim, J.R.; Topp, S.D.; Byrnes, A.E.; Churchhouse, C.; Phatnani, H.; Smith, B.N.; Rampersaud, E.; et al. Exome sequencing in amyotrophic lateral sclerosis implicates a novel gene, DNAJC7, encoding a heat-shock protein. Nat. Neurosci. 2019, 22, 1966–1974. [Google Scholar] [CrossRef] [PubMed]
- Sittler, A.; Lurz, R.; Lueder, G.; Priller, J.; Lehrach, H.; Hayer-Hartl, M.K.; Hartl, F.U.; Wanker, E.E. Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of Huntington’s disease. Hum. Mol. Genet. 2001, 10, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Auluck, P.K.; Chan, H.Y.E.; Trojanowski, J.Q.; Lee, V.M.Y.; Bonini, N.M. Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson’s disease. Science 2002, 295, 865–868. [Google Scholar] [CrossRef] [PubMed]
- Hageman, J.; Rujano, M.A.; van Waarde, M.A.W.H.; Kakkar, V.; Dirks, R.P.; Govorukhina, N.; Oosterveld-Hut, H.M.J.; Lubsen, N.H.; Kampinga, H.H. A DNAJB chaperone subfamily with HDAC-dependent activities suppresses toxic protein aggregation. Mol. Cell 2010, 37, 355–369. [Google Scholar] [CrossRef]
- Scior, A.; Buntru, A.; Arnsburg, K.; Ast, A.; Iburg, M.; Juenemann, K.; Pigazzini, M.L.; Mlody, B.; Puchkov, D.; Priller, J.; et al. Complete suppression of Htt fibrillization and disaggregation of Htt fibrils by a trimeric chaperone complex. EMBO J. 2017, 37, 282–298. [Google Scholar] [CrossRef]
- Bersuker, K.; Hipp, M.S.; Calamini, B.; Morimoto, R.I.; Kopito, R.R. Heat shock response activation exacerbates inclusion body formation in a cellular model of Huntington disease. J. Biol. Chem. 2013, 288, 23633–23638. [Google Scholar] [CrossRef]
- Benson, M.D.; Uemichi, T. Transthyretin amyloidosis. Amyloid 1996, 3, 44–56. [Google Scholar] [CrossRef]
- Planté-Bordeneuve, V.; Said, G. Transthyretin related familial amyloid polyneuropathy. Curr. Opin. Neurol. 2000, 13, 569–573. [Google Scholar] [CrossRef]
- Kim, J.H.; Oroz, J.; Zweckstetter, M. Structure of monomeric transthyretin carrying the clinically important T119M mutation. Angew. Chem. Int. Ed. 2016, 55, 16168–16171. [Google Scholar] [CrossRef]
- Santos, S.D.; Magalhães, J.; Saraiva, M.J. Activation of the heat shock response in familial amyloidotic polyneuropathy. J. Neuropathol. Exp. Neurol. 2008, 67, 449–455. [Google Scholar] [CrossRef]
- Masser, A.E.; Kang, W.; Roy, J.; Kaimal, J.M.; Quintana-Cordero, J.; Friedländer, M.R.; Andréasson, C. Cytoplasmic protein misfolding titrates Hsp70 to activate nuclear Hsf1. eLife 2019, 8, e47791. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Funez, P.; Casas-Tinto, S.; Zhang, Y.; Gómez-Velazquez, M.; Morales-Garza, M.A.; Cepeda-Nieto, A.C.; Castilla, J.; Soto, C.; Rincon-Limas, D.E. In vivo generation of neurotoxic prion protein: Role for Hsp70 in accumulation of misfolded isoforms. PLoS Genet. 2009, 5, e1000507. [Google Scholar] [CrossRef] [PubMed]
- Luchinat, E.; Barbieri, L.; Rubino, J.T.; Kozyreva, T.; Cantini, F.; Banci, L. In-cell NMR reveals potential precursor of toxic species from SOD1 fALS mutants. Nat. Commun. 2014, 5, 5502. [Google Scholar] [CrossRef] [PubMed]
- Zetterström, P.; Graffmo, K.S.; Andersen, P.M.; Brännström, T.; Marklund, S.L. Proteins that bind to misfolded mutant superoxide dismutase-1 in spinal cord from transgenic amyotrophic lateral sclerosis (ALS) model mice. J. Biol. Chem. 2011, 286, 20130–20136. [Google Scholar] [CrossRef] [PubMed]
- Claes, F.; Rudyak, S.; Laird, A.S.; Louros, N.; Beerten, J.; Debulpaep, M.; Michiels, E.; van der Kant, R.; Van Durme, J.; De Baets, G.; et al. Exposure of a cryptic Hsp70 binding site determines the cytotoxicity of the ALS-associated SOD1-mutant A4V. Protein Eng. Des. Sel. 2019, 32, 443–457. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Salot, S.; Sengupta, S.; Navalkar, A.; Ghosh, D.; Jacob, R.; Das, S.; Kumar, R.; Jha, N.N.; Sahay, S.; et al. p53 amyloid formation leading to its loss of function: Implications in cancer pathogenesis. Cell Death Differ. 2017, 24, 1784–1798. [Google Scholar] [CrossRef]
- Perez-Canadillas, J.M.; Tidow, H.; Freund, S.M.; Rutherford, T.J.; Ang, H.C.; Fersht, A.R. Solution structure of p53 core domain: Structural basis for its instability. Proc. Natl. Acad. Sci. USA 2006, 103, 2109–2114. [Google Scholar] [CrossRef]
- Boysen, M.; Kityk, R.; Mayer, M.P. Hsp70- and Hsp90-mediated regulation of the conformation of p53 DNA binding domain and p53 cancer variants. Mol. Cell 2019, 74, 831–843. [Google Scholar] [CrossRef]
- Akakura, S.; Yoshida, M.; Yoneda, Y.; Horinouchi, S. A role for Hsc70 in regulating nucleocytoplasmic transport of a temperature-sensitive p53 (p53Val-135). J. Biol. Chem. 2001, 276, 14649–14657. [Google Scholar] [CrossRef]
- Rohde, M.; Daugaard, M.; Jensen, M.H.; Helin, K.; Nylandsted, J.; Jäättelä, M. Members of the heat-shock protein 70 family promote cancer cell growth by distinct mechanisms. Genes Dev. 2005, 19, 570–582. [Google Scholar] [CrossRef]
- Dahiya, V.; Agam, G.; Lawatscheck, J.; Rutz, D.A.; Lamb, D.C.; Buchner, J. Coordinated conformational processing of the tumor suppressor protein p53 by the Hsp70 and Hsp90 chaperone machineries. Mol. Cell 2019, 74, 816–830. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2015, 17, 22–35. [Google Scholar] [CrossRef] [PubMed]
- DiFiglia, M.; Sapp, E.; Chase, K.O.; Davies, S.W.; Bates, G.P.; Vonsattel, J.P.; Aronin, N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997, 277, 1990–1993. [Google Scholar] [CrossRef] [PubMed]
- Monsellier, E.; Redeker, V.; Ruiz-Arlandis, G.; Bousset, L.; Melki, R. Molecular interaction between the chaperone Hsc70 and the N-terminal flank of huntingtin exon 1 modulates aggregation. J. Biol. Chem. 2015, 290, 2560–2576. [Google Scholar] [CrossRef]
- Escobedo, A.; Topal, B.; Kunze, M.B.A.; Aranda, J.; Chiesa, G.; Mungianu, D.; Bernardo-Seisdedos, G.; Eftekharzadeh, B.; Gairí, M.; Pierattelli, R.; et al. Side chain to main chain hydrogen bonds stabilize a polyglutamine helix in a transcription factor. Nat. Commun. 2019, 10, 2034. [Google Scholar] [CrossRef]
- Bongiovanni, M.N.; Aprile, F.A.; Sormanni, P.; Vendruscolo, M. A rationally designed Hsp70 variant rescues the aggregation-associated toxicity of human IAPP in cultured pancreatic islet β-cells. Int. J. Mol. Sci. 2018, 19, 1443. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef]
- Nelson, P.T.; Dickson, D.W.; Trojanowski, J.Q.; Jack, C.R.; Boyle, P.A.; Arfanakis, K.; Rademakers, R.; Alafuzoff, I.; Attems, J.; Brayne, C.; et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): Consensus working group report. Brain 2019, 142, 1503–1527. [Google Scholar] [CrossRef]
- Lin, P.-Y.; Folorunso, O.; Taglialatela, G.; Pierce, A. Overexpression of heat shock factor 1 maintains TAR DNA binding protein 43 solubility via induction of inducible heat shock protein 70 in cultured cells. J. Neurosci. Res. 2016, 94, 671–682. [Google Scholar] [CrossRef]
- Udan-Johns, M.; Bengoechea, R.; Bell, S.; Shao, J.; Diamond, M.I.; True, H.L.; Weihl, C.C.; Baloh, R.H. Prion-like nuclear aggregation of TDP-43 during heat shock is regulated by HSP40/70 chaperones. Hum. Mol. Genet. 2014, 23, 157–170. [Google Scholar] [CrossRef]
- Park, S.J.; Borin, B.N.; Martinez-Yamout, M.A.; Dyson, H.J. The client protein p53 adopts a molten globule-like state in the presence of Hsp90. Nat. Struct. Mol. Biol. 2011, 18, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Kostic, M.; Dyson, H.J. Dynamic interaction of Hsp90 with its client protein p53. J. Mol. Biol. 2011, 411, 158–173. [Google Scholar] [CrossRef] [PubMed]
- Mukrasch, M.D.; Bibow, S.; Korukottu, J.; Jeganathan, S.; Biernat, J.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol. 2009, 7, e34. [Google Scholar] [CrossRef] [PubMed]
- Soto, C.; Castaño, E.M.; Frangione, B.; Inestrosa, N.C. The α -helical to β-strand transition in the amino-terminal fragment of the amyloid β-peptide modulates amyloid formation. J. Biol. Chem. 1995, 270, 3063–3067. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.G.; Wisén, S.; Gestwicki, J.E. Heat shock proteins 70 and 90 inhibit early stages of amyloid β-(1-42) aggregation in vitro. J. Biol. Chem. 2006, 281, 33182–33191. [Google Scholar] [CrossRef] [PubMed]
- Burmann, B.M.; Gerez, J.A.; Matečko-Burmann, I.; Campioni, S.; Kumari, P.; Ghosh, D.; Mazur, A.; Aspholm, E.E.; Šulskis, D.; Wawrzyniuk, M.; et al. α-synuclein regulation by chaperones in mammalian cells. Nature 2020, 577, 127–132. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Del Tredici, K.; Braak, H. 100 years of Lewy pathology. Nat. Rev. Neurol. 2013, 9, 13–24. [Google Scholar] [CrossRef]
- Ulmer, T.S.; Bax, A. Comparison of structure and dynamics of micelle-bound human α-synuclein and Pakinson disease variants. J. Biol. Chem. 2005, 280, 43179–43187. [Google Scholar] [CrossRef]
- He, W.-T.; Xue, W.; Gao, Y.-G.; Hong, J.-Y.; Yue, H.-W.; Jiang, L.-L.; Hu, H.-Y. HSP90 recognizes the N-terminus of huntingtin involved in regulation of huntingtin aggregation by USP19. Sci. Rep. 2017, 7, 14797. [Google Scholar] [CrossRef]
- Jinwal, U.K.; Abisambra, J.F.; Zhang, J.; Dharia, S.; O’Leary, J.C.; Patel, T.; Braswell, K.; Jani, T.; Gestwicki, J.E.; Dickey, C.A. Cdc37/Hsp90 protein complex disruption triggers an autophagic clearance cascade for TDP-43 protein. J. Biol. Chem. 2012, 287, 24814–24820. [Google Scholar] [CrossRef]
- Jinwal, U.K.; Koren, J., 3rd; Dickey, C.A. Reconstructing the Hsp90/Tau machine. Curr. Enzym. Inhib. 2013, 9, 41–45. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Blair, L.J.; Nordhues, B.A.; Hill, S.E.; Scaglione, M.; O’Leary, J.C., 3rd; Fontaine, S.N.; Breydo, L.; Zhang, B.; Li, P.; Wang, L.; et al. Accelerated neurodegeneration through chaperone-mediated oligomerization of tau. J. Clin. Investig. 2013, 123, 4158–4169. [Google Scholar] [CrossRef] [PubMed]
- Blair, L.J.; Baker, J.D.; Sabbagh, J.J.; Dickey, C.A. The emerging role of peptidyl-prolyl isomerase chaperones in tau oligomerization, amyloid processing, and Alzheimer’s disease. J. Neurochem. 2015, 133, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Schwalbe, M.; Ozenne, V.; Bibow, S.; Jaremko, M.; Jaremko, L.; Gajda, M.; Jensen, M.R.; Biernat, J.; Becker, S.; Mandelkow, E.; et al. Predictive atomic resolution descriptions of intrinsically disordered htau40 and α-synuclein in solution from NMR and small angle scattering. Structure 2014, 22, 238–249. [Google Scholar] [CrossRef]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef]
- Handwerger, K.E.; Cordero, J.A.; Gall, J.G. Cajal bodies, nucleoli, and speckles in the Xenopus oocyte nucleus have a low-density, sponge-like structure. Mol. Biol. Cell 2005, 16, 202–211. [Google Scholar] [CrossRef]
- Brangwynne, C.P.; Eckmann, C.R.; Courson, D.S.; Rybarska, A.; Hoege, C.; Gharakhani, J.; Jülicher, F.; Hyman, A.A. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 2009, 324, 1729–1732. [Google Scholar] [CrossRef]
- Alberti, S.; Gladfelter, A.; Mittag, T. Considerations and challenges in studying liquid-liquid phase separation and biomolecular condensates. Cell 2019, 176, 419–434. [Google Scholar] [CrossRef]
- Li, H.-R.; Chiang, W.-C.; Chou, P.-C.; Wang, W.-J.; Huang, J.-R. TAR DNA-binding protein 43 (TDP-43) liquid-liquid phase separation is mediated by just a few aromatic residues. J. Biol. Chem. 2018, 293, 6090–6098. [Google Scholar] [CrossRef]
- Wang, J.; Choi, J.-M.; Holehouse, A.S.; Lee, H.O.; Zhang, X.; Jahnel, M.; Maharana, S.; Lemaitre, R.; Pozniakovsky, A.; Drechsel, D.; et al. A molecular grammar governing the driving forces for phase separation of prion-like RNA binding proteins. Cell 2018, 174, 688–699. [Google Scholar] [CrossRef]
- Martin, E.W.; Holehouse, A.S.; Peran, I.; Farag, M.; Incicco, J.J.; Bremer, A.; Grace, C.R.; Soranno, A.; Pappu, R.V.; Mittag, T. Valence and patterning of aromatic residues determine the phase behavior of prion-like domains. Science 2020, 367, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Molliex, A.; Temirov, J.; Lee, J.; Coughlin, M.; Kanagaraj, A.P.; Kim, H.J.; Mittag, T.; Taylor, J.P. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015, 163, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Ambadipudi, S.; Biernat, J.; Riedel, D.; Mandelkow, E.; Zweckstetter, M. Liquid-liquid phase separation of the microtubule-binding repeats of the Alzheimer-related protein Tau. Nat. Commun. 2017, 8, 275. [Google Scholar] [CrossRef] [PubMed]
- Peskett, T.R.; Rau, F.; O’Driscoll, J.; Patani, R.; Saibil, H.R. A liquid to solid phase transition underlying pathological huntingtin exon1 aggregation. Mol. Cell 2018, 70, 588–601. [Google Scholar] [CrossRef]
- Vogler, T.O.; Wheeler, J.R.; Nguyen, E.D.; Hughes, M.P.; Britson, K.A.; Lester, E.; Rao, B.; Betta, N.D.; Whitney, O.N.; Ewachiw, T.E.; et al. TDP-43 and RNA form amyloid-like myo-granules in regenerating muscle. Nature 2018, 56, 508–513. [Google Scholar] [CrossRef]
- Mazroui, R.; Di Marco, S.; Kaufman, R.J.; Gallouzi, I.-E. Inhibition of the ubiquitin-proteasome system induces stress granule formation. Mol. Biol. Cell 2007, 18, 2603–2618. [Google Scholar] [CrossRef]
- Cherkasov, V.; Hofmann, S.; Druffel-Augustin, S.; Mogk, A.; Tyedmers, J.; Stoecklin, G.; Bukau, B. Coordination of translational control and protein homeostasis during severe heat stress. Curr. Biol. 2013, 23, 2452–2462. [Google Scholar] [CrossRef]
- Kroschwald, S.; Maharana, S.; Mateju, D.; Malinovska, L.; Nüske, E.; Poser, I.; Richter, D.; Alberti, S. Promiscuous interactions and protein disaggregases determine the material state of stress-inducible RNP granules. eLife 2015, 4, e06807. [Google Scholar] [CrossRef]
- Alberti, S.; Mateju, D.; Mediani, L.; Carra, S. Granulostasis: Protein quality control of RNP granules. Front. Mol. Neurosci. 2017, 10, 84. [Google Scholar] [CrossRef]
- Ganassi, M.; Mateju, D.; Bigi, I.; Mediani, L.; Poser, I.; Lee, H.O.; Seguin, S.J.; Morelli, F.F.; Vinet, J.; Leo, G.; et al. A surveillance function of the HSPB8-BAG3-HSP70 chaperone complex ensures stress granule integrity and dynamism. Mol. Cell 2016, 63, 796–810. [Google Scholar] [CrossRef]
- Lechler, M.C.; Crawford, E.D.; Groh, N.; Widmaier, K.; Jung, R.; Kirstein, J.; Trinidad, J.C.; Burlingame, A.L.; David, D.C. Reduced insulin/IGF-1 signaling restores the dynamic properties of key stress granule proteins during aging. Cell Rep. 2017, 18, 454–467. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Minami, M.; Shinozaki, F.; Suzuki, Y.; Abe, K.; Zenno, S.; Matsumoto, S.; Minami, Y. Hsp90 is involved in the formation of P-bodies and stress granules. Biochem. Biophys. Res. Comm. 2011, 407, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.-Y.; Hou, S.-C.; Way, T.-D.; Wong, C.-H.; Wang, I.-F. Heat-shock protein dysregulation is associated with functional and pathological TDP-43 aggregation. Nat. Commun. 2013, 4, 2757. [Google Scholar] [CrossRef] [PubMed]
- Bagatell, R.; Paine-Murrieta, G.D.; Taylor, C.W.; Pulcini, E.J.; Akinaga, S.; Benjamin, I.J.; Whitesell, L. Induction of a heat shock factor 1-dependent stress response alters the cytotoxic activity of hsp90-binding agents. Clin. Cancer. Res. 2000, 6, 3312–3318. [Google Scholar]
- Kudryavtsev, V.A.; Khokhlova, A.V.; Mosina, V.A.; Selivanova, E.I.; Kabakov, A.E. Induction of Hsp70 in tumor cells treated with inhibitors of the Hsp90 activity: A predictive marker and promising target for radiosensitization. PLoS ONE 2017, 12, e0173640. [Google Scholar] [CrossRef]
- Mediani, L.; Galli, V.; Carrà, A.D.; Bigi, I.; Vinet, J.; Ganassi, M.; Antoniani, F.; Tiago, T.; Cimino, M.; Mateju, D.; et al. BAG3 and BAG6 differentially affect the dynamics of stress granules by targeting distinct subsets of defective polypeptides released from ribosomes. Cell Stress Chaper. 2020, 25, 1045–1058. [Google Scholar] [CrossRef]
- Jain, S.; Wheeler, J.R.; Walters, R.W.; Agrawal, A.; Barsic, A.; Parker, R. ATPase-modulated stress granules contain a diverse proteome and substructure. Cell 2016, 164, 487–498. [Google Scholar] [CrossRef]
- Audas, T.E.; Audas, D.E.; Jacob, M.D.; Ho, J.J.D.; Khacho, M.; Wang, M.; Perera, J.K.; Gardiner, C.; Bennett, C.A.; Head, T.; et al. Adaptation to stressors by systemic protein amyloidogenesis. Dev. Cell 2016, 39, 155–168. [Google Scholar] [CrossRef]
- Astbury, W.; Woods, H.; Bragg, W. X-ray studies of the structure of hair, wool and related fibers. II: The molecular structure and elastic properties of hair keratin. Phil. Trans. Roy. Soc. Ser. A 1934, 232, 333–394. [Google Scholar]
- Geddes, A.J.; Parker, K.D.; Atkins, E.D.; Beighton, E. “Cross-beta” conformation in proteins. J. Mol. Biol. 1968, 32, 343–358. [Google Scholar] [CrossRef]
- Baldwin, A.J.; Knowles, T.P.J.; Tartaglia, G.G.; Fitzpatrick, A.W.; Devlin, G.L.; Shammas, S.L.; Waudby, C.A.; Mossuto, M.F.; Meehan, S.; Gras, S.L.; et al. Metastability of native proteins and the phenomenon of amyloid formation. J. Am. Chem. Soc. 2011, 133, 14160–14163. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.; Sawaya, M.R.; Balbirnie, M.; Madsen, A.; Riekel, C.; Grothe, R.; Eisenberg, D. Structure of the cross-β spine of amyloid-like fibrils. Nature 2005, 435, 773–778. [Google Scholar] [CrossRef]
- Bolton, D.C.; Mckinley, M.P.; Prusiner, S.B. Identification of a protein that purifies with the scrapie prion. Science 1982, 218, 1309–1311. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Ihara, Y.; Salazar, F.J. Alzheimer’s disease: Insolubility of partially purified paired helical filaments in sodium dodecyl sulfate and urea. Science 1982, 215, 1243–1245. [Google Scholar] [CrossRef] [PubMed]
- Sipe, J.D.; Benson, M.D.; Buxbaum, J.N.; Ikeda, S.I.; Merlini, G.; Saraiva, M.J.M.; Westermark, P. Nomenclature 2014: Amyloid fibril proteins and clinical classification of the amyloidosis. Amyloid 2014, 21, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Lin, T.Y.; Chang, D.; Guo, Z. Thioflavin T as an amyloid dye: Fibril quantification, optimal concentration and effect on aggregation. R. Soc. Open Sci. 2017, 4, 160696. [Google Scholar] [CrossRef]
- Chapman, M.R.; Robinson, L.S.; Pinkner, J.S.; Roth, R.; Heuser, J.; Hammar, M.; Normark, S.; Hultgren, S.J. Role of Escherichia coli curli operons in directing amyloid fiber formation. Science 2002, 295, 851–855. [Google Scholar] [CrossRef]
- Fowler, D.M.; Koulov, A.V.; Alory-Jost, C.; Marks, M.S.; Balch, W.E.; Kelly, J.W. Functional amyloid formation within mammalian tissue. PLoS Biol. 2005, 4, e6. [Google Scholar] [CrossRef]
- Antonets, K.S.; Belousov, M.V.; Sulatskaya, A.I.; Belousova, M.E.; Kosolapova, A.O.; Sulatsky, M.I.; Andreeva, E.A.; Zykin, P.A.; Malovichko, Y.V.; Shtark, O.Y.; et al. Accumulation of storage proteins in plant seeds is mediated by amyloid formation. PLoS Biol. 2020, 18, e3000564. [Google Scholar] [CrossRef]
- Boke, E.; Ruer, M.; Wühr, M.; Coughlin, M.; Lemaitre, R.; Gygi, S.P.; Alberti, S.; Drechsel, D.; Hyman, A.A.; Mitchison, T.J. Amyloid-like self-assembly of a cellular compartment. Cell 2016, 166, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; McQuade, T.; Siemer, A.B.; Napetschnig, J.; Moriwaki, K.; Hsiao, Y.S.; Damko, E.; Moquin, D.; Walz, T.; McDermott, A.; et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 2012, 150, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Saupe, S.J.; Daskalov, A. The [Het-s] prion, an amyloid fold as a cell death activation trigger. PLoS Pathog. 2012, 8, e1002687. [Google Scholar] [CrossRef] [PubMed]
- True, H.L.; Lindquist, S.L. A yeast prion provides a mechanism for genetic variation and phenotypic diversity. Nature 2000, 407, 477–483. [Google Scholar] [CrossRef]
- Berchowitz, L.E.; Kabachinski, G.; Walker, M.R.; Carlile, T.M.; Gilbert, W.V.; Schwartz, T.U.; Amon, A. Regulated formation of an amyloid-like translational repressor governs gametogenesis. Cell 2015, 163, 406–418. [Google Scholar] [CrossRef]
- Keleman, K.; Krüttner, S.; Alenius, M.; Dickson, B.J. Function of the Drosophila CPEB protein Orb2 in long-term courtship memory. Nat. Neurosci. 2007, 10, 1587–1593. [Google Scholar] [CrossRef]
- Majumdar, A.; Cesario, W.C.; White-Grindley, E.; Jiang, H.; Ren, F.; Khan, M.R.; Li, L.; Choi, E.M.L.; Kannan, K.; Guo, F.; et al. Critical role of amyloid-like oligomers of Drosophila Orb2 in the persistence of memory. Cell 2012, 148, 515–529. [Google Scholar] [CrossRef]
- Nil, Z.; Hervás, R.; Gerbich, T.; Leal, P.; Yu, Z.; Saraf, A.; Sardiu, M.; Lange, J.J.; Yi, K.; Unruh, J.; et al. Amyloid-like assembly activates a phosphatase in the developing Drosophila embryo. Cell 2019, 179, 801. [Google Scholar] [CrossRef]
- Arakhamia, T.; Lee, C.E.; Carlomagno, Y.; Duong, D.M.; Kundinger, S.R.; Wang, K.; Williams, D.; DeTure, M.; Dickson, D.W.; Cook, C.N.; et al. Posttranslational modifications mediate the structural diversity of tauopathy strains. Cell 2020, 180, 633–644. [Google Scholar] [CrossRef]
- Fitzpatrick, A.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef]
- Falcon, B.; Zhang, W.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Ghetti, B.; Scheres, S.H.W.; Goedert, M. Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 2018, 561, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Falcon, B.; Zivanov, J.; Zhang, W.; Murzin, A.G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Newell, K.L.; Ghetti, B.; Goedert, M.; et al. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature 2019, 568, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Tarutani, A.; Newell, K.L.; Murzin, A.G.; Matsubara, T.; Falcon, B.; Vidal, R.; Garringer, H.J.; Shi, Y.; Ikeuchi, T.; et al. Novel tau filament fold in corticobasal degeneration. Nature 2020, 580, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Schweighauser, M.; Shi, Y.; Tarutani, A.; Kametani, F.; Murzin, A.G.; Ghetti, B.; Matsubara, T.; Tomita, T.; Ando, T.; Hasegawa, K.; et al. Structures of α-synuclein filaments from multiple system atrophy. Nature 2020, 585, 464–469. [Google Scholar] [CrossRef]
- Liberta, F.; Loerch, S.; Rennegarbe, M.; Schierhorn, A.; Westermark, P.; Westermark, G.T.; Hazenberg, B.P.C.; Grigorieff, N.; Fändrich, M.; Schmidt, M. Cryo-EM fibril structures from systemic AA amyloidosis reveal the species complementarity of pathological amyloids. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef]
- Hughes, M.P.; Sawaya, M.R.; Boyer, D.R.; Goldschmidt, L.; Rodriguez, J.A.; Cascio, D.; Chong, L.; Gonen, T.; Eisenberg, D.S. Atomic structures of low-complexity protein segments reveal kinked β sheets that assemble networks. Science 2018, 359, 698–701. [Google Scholar] [CrossRef]
- Wasmer, C.; Lange, A.; Van Melckebeke, H.; Siemer, A.B.; Riek, R.; Meier, B.H. Amyloid fibrils of the HET-s(218-289) prion form a β solenoid with a triangular hydrophobic core. Science 2008, 319, 1523–1526. [Google Scholar] [CrossRef]
- Murray, D.T.; Kato, M.; Lin, Y.; Thurber, K.R.; Hung, I.; McKnight, S.L.; Tycko, R. Structure of FUS protein fibrils and its relevance to self-assembly and phase separation of low-complexity domains. Cell 2017, 171, 615–627. [Google Scholar] [CrossRef]
- Luo, F.; Gui, X.; Zhou, H.; Gu, J.; Li, Y.; Liu, X.; Zhao, M.; Li, D.; Li, X.; Liu, C. Atomic structures of FUS LC domain segments reveal bases for reversible amyloid fibril formation. Nat. Struct. Mol. Biol. 2018, 25, 341–346. [Google Scholar] [CrossRef]
- Gui, X.; Luo, F.; Li, Y.; Zhou, H.; Qin, Z.; Liu, Z.; Gu, J.; Xie, M.; Zhao, K.; Dai, B.; et al. Structural basis for reversible amyloids of hnRNPA1 elucidates their role in stress granule assembly. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Lu, J.; Cao, Q.; Hughes, M.P.; Sawaya, M.R.; Boyer, D.R.; Cascio, D.; Eisenberg, D.S. CryoEM structure of the low-complexity domain of hnRNPA2 and its conversion to pathogenic amyloid. Nat. Commun. 2020, 11, 4090. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.X.; Qiang, W.; Yau, W.M.; Schwieters, C.D.; Meredith, S.C.; Tycko, R. Molecular structure of β-amyloid fibrils in alzheimer’s disease brain tissue. Cell 2013, 154, 1257. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://people.mbi.ucla.edu/sawaya/amyloidatlas/ (accessed on 1 December 2020).
- Nachman, E.; Wentink, A.S.; Madiona, K.; Bousset, L.; Katsinelos, T.; Allinson, K.; Kampinga, H.; McEwan, W.A.; Jahn, T.R.; Melki, R.; et al. Disassembly of tau fibrils by the human Hsp70 disaggregation machinery generates small seeding-competent species. J. Biol. Chem. 2020, 295, 9676–9690. [Google Scholar] [CrossRef] [PubMed]
- Shorter, J. The mammalian disaggregase machinery: Hsp110 synergizes with Hsp70 and Hsp40 to catalyze protein disaggregation and reactivation in a cell-free system. PLoS ONE 2011, 6, e26319. [Google Scholar] [CrossRef]
- Gao, X.; Carroni, M.; Nussbaum-Krammer, C.; Mogk, A.; Nillegoda, N.B.; Szlachcic, A.; Guilbride, D.L.; Saibil, H.R.; Mayer, M.P.; Bukau, B. Human Hsp70 disaggregase reverses Parkinson’s-linked α-synuclein amyloid fibrils. Mol. Cell 2015, 59, 781–793. [Google Scholar] [CrossRef] [PubMed]
- Wentik, A.S.; Nillegoda, N.B.; Feufel, J.; Ubartaité, G.; Schneider, C.P.; De Los Rios, P.; Hennig, J.; Barducci, A.; Bukau, B. Molecular dissection of amyloid disaggregation by human HSP70. Nature 2020, 587, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Tittelmeier, J.; Sandhof, C.A.; Ries, H.M.; Druffel-Augustin, S.; Mogk, A.; Bukau, B.; and Nussbaum-Krammer, C. The HSP110/HSP70 disaggregation system generates spreading-competent toxic α-synuclein species. EMBO J. 2020, 39, e103954. [Google Scholar] [CrossRef]
- Carlomagno, Y.; Zhang, Y.; Davis, M.; Lin, W.L.; Cook, C.; Dunmore, J.; Tay, W.; Menkosky, K.; Cao, X.; Petrucelli, L.; et al. Casein kinase II induced polymerization of soluble TDP-43 into filaments is inhibited by heat shock proteins. PLoS ONE 2014, 9, e90452. [Google Scholar] [CrossRef]
- Ali, Y.O.; Allen, H.M.; Yu, L.; Li-Kroeger, D.; Bakhshizadehmahmoudi, D.; Hatcher, A.; McCabe, C.; Xu, J.; Bjorklund, N.; Taglialatela, G.; et al. NMNAT2:HSP90 complex mediates proteostasis in proteinopathies. PLoS Biol. 2016, 14, e1002472. [Google Scholar] [CrossRef]
- Li, L.; Sanchez, C.P.; Slaughter, B.D.; Zhao, Y.; Khan, M.R.; Unruh, J.R.; Rubinstein, B.; Si, K. A putative biochemical engram of long-term memory. Curr. Biol. 2016, 26, 3143–3156. [Google Scholar] [CrossRef]
- Chernoff, Y.O.; Lindquist, S.L.; Ono, B.I.; Inge-Vechtomov, S.G.; Liebman, S.W. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+]. Science 1995, 268, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Pfefferkorn, C.M.; McGlinchey, R.P.; Lee, J.C. Effects of pH on aggregation kinetics of the repeat domain of a functional amyloid, Pmel17. Proc. Natl. Acad. Sci. USA 2010, 107, 21447–21452. [Google Scholar] [CrossRef] [PubMed]
- McGlinchey, R.P.; Shewmaker, F.; Hu, K.; McPhie, P.; Tycko, R.; Wickner, R.B. Repeat domains of melanosome matrix protein Pmel17 orthologs form amyloid fibrils at the acidic melanosomal pH. J. Biol. Chem. 2011, 286, 8385–8393. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhou, Y.; Ren, J.-J.; Hammer, N.D.; Chapman, M.R. Gatekeeper residues in the major curlin subunit modulate bacterial amyloid fiber biogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, L.; Madurga, S.; Pukala, T.; Vilaseca, M.; López-Iglesias, C.; Robinson, C.V.; Giralt, E.; Carulla, N. Ab40 and Ab42 amyloid fibrils exhibit distinct molecular recycling properties. J. Am. Chem. Soc. 2011, 133, 6505–6508. [Google Scholar] [CrossRef] [PubMed]
- Schlecht, R.; Erbse, A.H.; Bukau, B.; Mayer, M.P. Mechanics of Hsp70 chaperones enables differential interaction with client proteins. Nat. Struct. Mol. Biol. 2011, 18, 345–351. [Google Scholar] [CrossRef]
- Nillegoda, N.B.; Bukau, B. Metazoan Hsp70-based protein disaggregases: Emergence and mechanisms. Front. Mol. Biosci. 2015, 2, 57. [Google Scholar] [CrossRef]
- Nillegoda, N.B.; Wentink, A.S.; Bukau, B. Protein disaggregation in multicellular organisms. Trends Biochem. Sci. 2018, 43, 285–300. [Google Scholar] [CrossRef]
- Nillegoda, N.B.; Kirstein, J.; Szlachcic, A.; Berynskyy, M.; Stank, A.; Stengel, F.; Arnsburg, K.; Gao, X.; Scior, A.; Aebersold, R.; et al. Crucial HSP70 co-chaperone complex unlocks metazoan protein disaggregation. Nature 2015, 524, 247–251. [Google Scholar] [CrossRef]
- Poepsel, S.; Sprengel, A.; Sacca, B.; Kaschani, F.; Kaiser, M.; Gatsogiannis, C.; Raunser, S.; Clausen, T.; Ehrmann, M. Determinants of amyloid fibril degradation by the PDZ protease HTRA1. Nat. Chem. Biol. 2015, 11, 862–869. [Google Scholar] [CrossRef]
- Zaarur, N.; Xu, X.; Lestienne, P.; Meriin, A.B.; McComb, M.; Costello, C.E.; Newnam, G.P.; Ganti, R.; Romanova, N.V.; Shanmugasundaram, M.; et al. RuvbL1 and RuvbL2 enhance aggresome formation and disaggregate amyloid fibrils. EMBO J. 2015, 34, 2363–2382. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.D.; Shelton, L.B.; Zheng, D.; Favretto, F.; Nordhues, B.A.; Darling, A.; Sullivan, L.E.; Sun, Z.; Solanki, P.K.; Martin, M.D.; et al. Human cyclophilin 40 unravels neurotoxic amyloids. PLoS Biol. 2017, 15, e2001336. [Google Scholar] [CrossRef] [PubMed]
- Ramelot, T.A.; Nicholson, L.K. Phosphorylation-induced structural changes in the amyloid precursor protein cytoplasmic tail detected by NMR. J. Mol. Biol. 2001, 307, 871–884. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Greenwood, A.; Binder, L.; Bigio, E.K.; Denial, S.; Nicholson, L.; Zhou, X.Z.; Lu, K.P. Proline isomer-specific antibodies reveal the early pathogenic tau conformation in Alzheimer’s disease. Cell 2012, 149, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.P.; Zhou, X.Z. The prolyl isomerase PIN1: A pivotal new twist in phosphorylation signaling and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 904–916. [Google Scholar] [CrossRef] [PubMed]
- Rein, T. Peptidylprolylisomerases, protein folders, or scaffolders? The example of FKBP51 and FKBP52. Bioessays 2020, 42, e1900250. [Google Scholar] [CrossRef]
- Bibow, S.; Ozenne, V.; Biernat, J.; Blackledge, M.; Mandelkow, E.; Zweckstetter, M. Structural impact of proline-directed psudophosphorylation at AT8, AT100, and PHF1 epitopes on 441-residue tau. J. Am. Chem. Soc. 2011, 133, 15842–15845. [Google Scholar] [CrossRef]
- Shelton, L.B.; Koren, J., 3rd; Blair, L.J. Imbalances in the Hsp90 chaperone machinery: Implications for tauopathies. Front Neurosci. 2017, 11, 724. [Google Scholar] [CrossRef]
- Spires-Jones, T.L.; Attems, J.; Thal, D.R. Interactions of pathological proteins in neurodegenerative diseases. Acta Neuropathol. 2017, 134, 187–205. [Google Scholar] [CrossRef]
- Chornenkyy, Y.; Fardo, D.W.; Nelson, P.T. Tau and TDP-43 proteinopathies: Kindred pathologic cascades and genetic pleiotropy. Lab. Investig. 2019, 99, 993–1007. [Google Scholar] [CrossRef]
- Park, S.-H.; Kukushkin, Y.; Gupta, R.; Chen, T.; Konagai, A.; Hipp, M.S.; Hayer-Hartl, M.; Hartl, F.U. PolyQ proteins interfere with nuclear degradation of cytosolic proteins by sequestering the Sis1p chaperone. Cell 2013, 154, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.; Shibata, Y.; Shah, B.; Calamini, B.; Lo, D.C.; Morimoto, R.I. Protein aggregation can inhibit clathrin-mediated endocytosis by chaperone competition. Proc. Natl. Acad. Sci. USA 2014, 111, E1481–E1490. [Google Scholar] [CrossRef] [PubMed]
- Pratt, W.B.; Gestwicki, J.E.; Osawa, Y.; Lieberman, A.P. Targeting proteostasis through the protein quality control function of the Hsp90/Hsp70-based chaperone machinery for treatment of adult onset neurodegenerative diseases. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 353–371. [Google Scholar] [CrossRef] [PubMed]
- Waza, M.; Adachi, H.; Katsuno, M.; Minamiyama, M.; Sang, C.; Tanaka, F.; Inukai, A.; Doyu, M.; Sobue, G. 17-AAG, an Hsp90 inhibitor, ameliorates polyglutamine-mediated motor neuron degeneration. Nat. Med. 2005, 11, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Fujikake, N.; Nagai, Y.; Popiel, H.A.; Okamoto, Y.; Yamaguchi, M.; Toda, T. Heat shock transcription factor 1-activating compounds suppress polyglutamine-induced neurodegeneration through induction of multiple molecular chaperones. J. Biol. Chem. 2008, 283, 26188–26197. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.W.; Tsui, Y.T.C.; Wong, T.T.; Cheung, S.K.-K.; Goggins, W.B.; Yi, L.M.; Cheng, K.K.; Baum, L. Effects of 17-allylamino-17-demethoxygeldanamycin (17-AAG) in transgenic mouse models of frontotemporal lobar degeneration and Alzheimer’s disease. Transl. Neurodegenr. 2013, 2, 24. [Google Scholar] [CrossRef]
- Ding, Y.; Adachi, H.; Katsuno, M.; Sahashi, K.; Kondo, N.; Iida, M.; Tohnai, G.; Nakatsuji, H.; Sobue, G. BIIB021, a synthetic Hsp90 inhibitor, induces mutant ataxin-1 degradation through the activation of heat shock factor 1. Neuroscience 2016, 327, 20–31. [Google Scholar] [CrossRef]
- Muchowski, P.J.; Wacker, J.L. Modulation of neurodegeneration by molecular chaperones. Nat. Rev. Neurosci. 2005, 6, 11–22. [Google Scholar] [CrossRef]
- Whitesell, L.; Lindquist, S. Inhibiting the transcription factor HSF1 as an anticancer strategy. Expert Opin. Ther. Targets 2009, 13, 469–478. [Google Scholar] [CrossRef]
- Serlidaki, D.; van Waarde, M.A.W.H.; Rohland, L.; Wentink, A.S.; Dekker, S.L.; Kamphuis, M.J.; Boertien, J.M.; Brunsting, J.F.; Nillegoda, N.B.; Bukau, B.; et al. Functional diversity between HSP70 paralogs caused by variable interactions with specific co-chaperones. J. Biol. Chem. 2020, 295, 7301–7316. [Google Scholar] [CrossRef]
- Aprile, F.A.; Källstig, E.; Limorenko, G.; Vendruscolo, M.; Ron, D.; Hansen, C. The molecular chaperones DNAJB6 and Hsp70 cooperate to suppress α-synuclein aggregation. Sci. Rep. 2017, 7, 9039. [Google Scholar] [CrossRef] [PubMed]
- Shorter, J. Designer protein disaggregases to counter neurodegenerative disease. Curr. Opin. Genet. Dev. 2017, 44, 1–8. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hervás, R.; Oroz, J. Mechanistic Insights into the Role of Molecular Chaperones in Protein Misfolding Diseases: From Molecular Recognition to Amyloid Disassembly. Int. J. Mol. Sci. 2020, 21, 9186. https://doi.org/10.3390/ijms21239186
Hervás R, Oroz J. Mechanistic Insights into the Role of Molecular Chaperones in Protein Misfolding Diseases: From Molecular Recognition to Amyloid Disassembly. International Journal of Molecular Sciences. 2020; 21(23):9186. https://doi.org/10.3390/ijms21239186
Chicago/Turabian StyleHervás, Rubén, and Javier Oroz. 2020. "Mechanistic Insights into the Role of Molecular Chaperones in Protein Misfolding Diseases: From Molecular Recognition to Amyloid Disassembly" International Journal of Molecular Sciences 21, no. 23: 9186. https://doi.org/10.3390/ijms21239186
APA StyleHervás, R., & Oroz, J. (2020). Mechanistic Insights into the Role of Molecular Chaperones in Protein Misfolding Diseases: From Molecular Recognition to Amyloid Disassembly. International Journal of Molecular Sciences, 21(23), 9186. https://doi.org/10.3390/ijms21239186