Exposure to Endocrine Disrupting Chemicals and Risk of Breast Cancer


 and
and
Abstract
1. Introduction
2. Breast Cancer, the Most Common Cancer in Women Sill on the Rise
2.1. Specificities of the Mammary Gland and Its Windows of Susceptibility
2.2. Complementary Classifications of Breast Cancer
2.3. A Complex Combination of Risk Factors
3. Difficult Identification of the Mode of Action of Endocrine Disrupting Chemicals
3.1. A Recent Concept in Constant Evolution
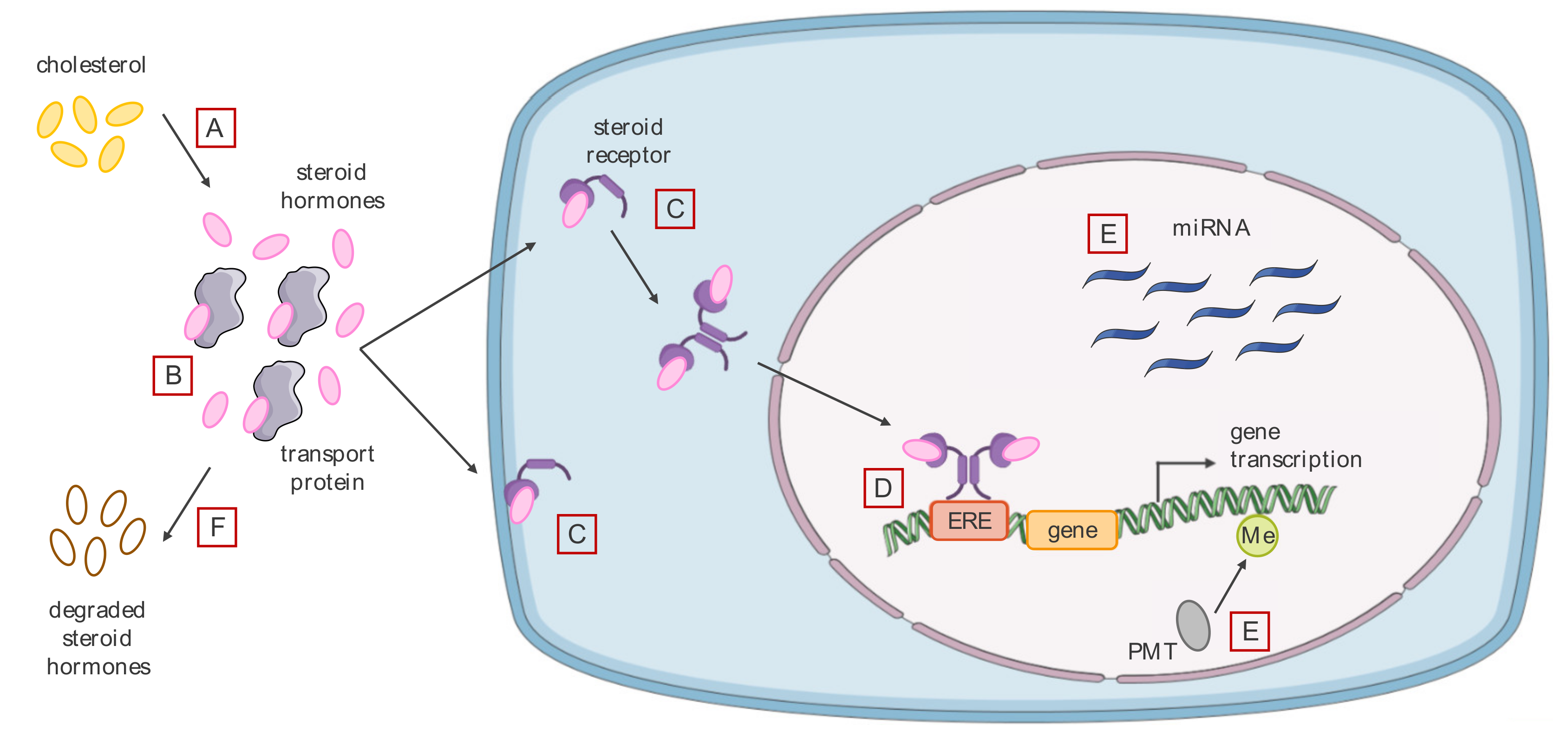
3.2. Effects of Endocrine Disrupting Chemicals on Hormone Signaling
3.2.1. Agonistic or Antagonistic Action on Different Hormone Receptors
3.2.2. Modification of the Level of Bioavailable Endogenous Hormones
3.2.3. Alteration of the Cell Epigenome
4. Case Studies of Endocrine Disrupting Chemicals Linked with Increased Risk of Breast Cancer
4.1. Endocrine Disrupting Chemicals and Medicine: Diethylstilbestrol in Pregnant Women
4.1.1. Diethylstilbestrol, a Synthetic Estrogen Inducing Significant Epigenetic Changes
4.1.2. Diethylstilbestrol during Pregnancy: Three Generations Impacted
4.1.3. Indication and Use of Diethylstilbestrol after the Tragedy
4.2. Massive Use of Dichlorodiphenyltrichloroethane: Awareness of Environmental Pollution by Toxic Endocrine Disruptor Chemicals
4.2.1. The Dual Mode of Action of Dichlorodiphenyltrichloroethane, Both Estrogenic Agonist and Androgenic Antagonist
4.2.2. Prepubertal Exposure to Dichlorodiphenyltrichloroethane and Breast Cancer Occurrence
4.2.3. Partial Ban of Dichlorodiphenyltrichloroethane and Current Regulations
4.3. Industrial Accident and Release of Toxic Dioxins into the Environment: Current Exposure and Risks
4.3.1. 2,3,7,8-tetrachlorodibenzo-p-dioxin, a Potent Agonist of the Aryl Hydrocarbon Receptor
4.3.2. Exposure to Toxic Dioxins and Breast Cancer Risk: Heterogeneous Results
4.3.3. Industrial Risk Management and Control of Dioxin Release
4.4. Bisphenol A: Difficulty in Tracing Exposure to a Synthetic Estrogen Ubiquitously Present in the Environment
4.4.1. Bisphenol A, a Synthetic Estrogen Similar to Diethylstilbestrol
4.4.2. A Ubiquitous Presence Making Epidemiological Studies Difficult
4.4.3. The Precautionary Principle behind Bisphenol A Legislation in Some Countries
5. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADI | Acceptable Daily Intake |
| AhR | Aryl hydrocarbon Receptor |
| AR | Androgen Receptor |
| BC | Breast Cancer |
| BMI | Body Mass Index |
| BPA | Bisphenol A |
| BRCA1/2 | Breast Cancer type 1/2 susceptibility protein |
| DDE | Dichlorodiphenyldichloroethylene |
| DDT | Dichlorodiphenyltrichloroethane |
| DES | Diethylstilbestrol or stilbestrol |
| DNMTs | DNA methyltransferases |
| EDCs | Endocrine Disrupting Chemicals |
| EPA | Environmental Protection Agency |
| ERα/β | Estrogen Receptor α/β |
| ERE | Estrogen Response Elements |
| ERRγ | Estrogen-Related Receptor γ |
| FDA | Food and Drug Administration |
| GPR30 | G Protein-coupled Receptor 30 |
| HER2 | Human Epidermal Growth Factor Receptor 2 |
| HRT | Hormone Replacement Therapy |
| IARC | International Agency for Research on Cancer |
| LOAEL | Lowest Observed Adverse Effect Level |
| miRNA | microRNA |
| mRNA | messenger RNA |
| NOAEL | No Observable Adverse Effect Level |
| PDCD4 | Programmed Cell Death Protein 4 |
| POP | Persistent Organic Pollutant |
| PR | Progesterone Receptor |
| SR | Steroid Receptor |
| TCDD | 2,3,7,8-tetrachlorodibenzo-p-dioxin |
| TEB | Terminal End Buds |
| TEQ | Toxic Equivalent |
| TNBC | Triple Negative Breast Cancer |
| WHO | World Health Organization |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- American Cancer Society. Breast Cancer Facts & Figures 2019–2020; American Cancer Society, Inc.: Atlanta, GA, USA, 2019; pp. 1–44. [Google Scholar]
- Breen, N.; Gentleman, J.F.; Schiller, J.S. Update on mammography trends: Comparisons of rates in 2000, 2005, and 2008. Cancer 2011, 117, 2209–2218. [Google Scholar] [CrossRef] [PubMed]
- Winters, S.; Martin, C.; Murphy, D.; Shokar, N.K. Breast Cancer Epidemiology, Prevention, and Screening. Prog. Mol. Biol. Transl. Sci. 2017, 151. [Google Scholar] [CrossRef]
- Ravdin, P.M.; Cronin, K.A.; Howlader, N.; Berg, C.D.; Chlebowski, R.T.; Feuer, E.J.; Edwards, B.K.; Berry, D.A. The decrease in breast-cancer incidence in 2003 in the United States. N. Engl. J. Med. 2007, 356, 1670–1674. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Cancer Tomorrow—Breast. 2018. Available online: https://gco.iarc.fr/tomorrow/graphic-line?type=0&type_sex=0&mode=population&sex=0&populations=900&cancers=39&age_group=value&apc_male=0&apc_female=0&single_unit=500000&print=0 (accessed on 15 September 2020).
- International Agency for Research on Cancer (IARC). Cancers Attributable to the Lifestyle and Environment in Metropolitan France; IARC: Lyon, France, 2018.
- Tung, N.; Battelli, C.; Allen, B.; Kaldate, R.; Bhatnagar, S.; Bowles, K.; Timms, K.; Garber, J.E.; Herold, C.; Ellisen, L. Frequency of mutations in individuals with breast cancer referred for BRCA1 and BRCA2 testing using next-generation sequencing with a 25-gene panel. Cancer 2015, 121, 25–33. [Google Scholar] [CrossRef]
- Hankinson, S.E. Endogenous hormones and risk of breast cancer in postmenopausal women. Breast Dis. 2006, 24, 3–15. [Google Scholar] [CrossRef]
- Beral, V.; Peto, R.; Pirie, K.; Reeves, G. Type and timing of menopausal hormone therapy and breast cancer risk: Individual participant meta-analysis of the worldwide epidemiological evidence. Lancet 2019, 394, 1159–1168. [Google Scholar] [CrossRef]
- Ji, L.W.; Jing, C.X.; Zhuang, S.L.; Pan, W.C.; Hu, X.P. Effect of age at first use of oral contraceptives on breast cancer risk: An updated meta-analysis. Medicine 2019, 98, e15719. [Google Scholar] [CrossRef]
- Ziaei, S.; Halaby, R. Dietary Isoflavones and Breast Cancer Risk. Medicines 2017, 4, 18. [Google Scholar] [CrossRef]
- Rodgers, K.M.; Udesky, J.O.; Rudel, R.A.; Brody, J.G. Environmental chemicals and breast cancer: An updated review of epidemiological literature informed by biological mechanisms. Environ. Res. 2018, 160, 152–182. [Google Scholar] [CrossRef]
- Johnson Foundation. National Life Science Education Summit: Report of the Wingspread Conference; Johnson Foundation: Racine, WI, USA, 1991. [Google Scholar]
- Colborn, T.; vom Saal, F.S.; Soto, A.M. Developmental effects of endocrine-disrupting chemicals in wildlife and humans. Environ. Health Perspect. 1993, 101, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Soto, A.M.; Sonnenschein, C. Environmental causes of cancer: Endocrine disruptors as carcinogens. Nat. Rev. Endocrinol. 2010, 6, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Diamanti-Kandarakis, E.; Bourguignon, J.P.; Giudice, L.C.; Hauser, R.; Prins, G.S.; Soto, A.M.; Zoeller, R.T.; Gore, A.C. Endocrine-disrupting chemicals: An Endocrine Society scientific statement. Endocr. Rev. 2009, 30, 293–342. [Google Scholar] [CrossRef] [PubMed]
- Kiyama, R.; Wada-Kiyama, Y. Estrogenic endocrine disruptors: Molecular mechanisms of action. Environ. Int. 2015. [Google Scholar] [CrossRef]
- Macias, H.; Hinck, L. Mammary gland development. Wiley Interdiscip. Rev. Dev. Biol. 2012, 1, 533–557. [Google Scholar] [CrossRef]
- Rudel, R.A.; Fenton, S.E.; Ackerman, J.M.; Euling, S.Y.; Makris, S.L. Environmental exposures and mammary gland development: State of the science, public health implications, and research recommendations. Environ. Health Perspect. 2011, 119, 1053–1061. [Google Scholar] [CrossRef]
- Malhotra, G.K.; Zhao, X.; Band, H.; Band, V. Histological, molecular and functional subtypes of breast cancers. Cancer Biol. Ther. 2010, 10, 955–960. [Google Scholar] [CrossRef]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Rakha, E.A.; Green, A.R. Molecular classification of breast cancer: What the pathologist needs to know. Pathology 2017, 49, 111–119. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef]
- Howlader, N.; Altekruse, S.F.; Li, C.I.; Chen, V.W.; Clarke, C.A.; Ries, L.A.G.; Cronin, K.A. US incidence of breast cancer subtypes defined by joint hormone receptor and HER2 status. J. Natl. Cancer Inst. 2014, 106, dju055. [Google Scholar] [CrossRef] [PubMed]
- American Cancer Society. Treating Breast Cancer. 2019. Available online: https://www.cancer.org/cancer/breast-cancer/treatment.html (accessed on 15 September 2020).
- Heimes, A.S.; Schmidt, M. Atezolizumab for the treatment of triple-negative breast cancer. Expert Opin. Investig. Drugs 2019, 28, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Barnard, M.E.; Boeke, C.E.; Tamimi, R.M. Established breast cancer risk factors and risk of intrinsic tumor subtypes. Biochim. Biophys. Acta Rev. Cancer 2015, 1856, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Hamajima, N.; Hirose, K.; Tajima, K.; Rohan, T.; Friedenreich, C.M.; Calle, E.E.; Fukao, A. Menarche, menopause, and breast cancer risk: Individual participant meta-analysis, including 118 964 women with breast cancer from 117 epidemiological studies. Lancet Oncol. 2012, 13, 1141–1151. [Google Scholar] [CrossRef]
- Clavel-Chapelon, F.; Gerber, M. Reproductive factors and breast cancer risk. Do they differ according to age at diagnosis? Breast Cancer Res. Treat. 2002, 72, 107–115. [Google Scholar] [CrossRef]
- Unar-Munguía, M.; Torres-Mejía, G.; Colchero, M.A.; González de Cosío, T. Breastfeeding Mode and Risk of Breast Cancer: A Dose-Response Meta-Analysis. J. Hum. Lact. 2017, 33, 422–434. [Google Scholar] [CrossRef]
- Reeves, G.K.; Pirie, K.; Beral, V.; Green, J.; Spencer, E.; Bull, D. Cancer incidence and mortality in relation to body mass index in the Million Women Study: Cohort study. Br. Med. J. 2007, 335, 1134–1145. [Google Scholar] [CrossRef]
- Abdelwahab Yousef, A.J. Male Breast Cancer: Epidemiology and Risk Factors. Semin. Oncol. 2017, 44, 267–272. [Google Scholar] [CrossRef]
- Boyd, N.F.; Byng, J.W.; Jong, R.A.; Fishell, E.K.; Little, L.E.; Miller, A.B.; Lockwood, G.A.; Tritchler, D.L.; Yaffe, M.J. Quantitative classification of mammographic densities and breast cancer risk: Results from the Canadian National Breast Screening Study. J. Natl. Cancer Inst. 1995, 87, 670–675. [Google Scholar] [CrossRef]
- Cheng, Y.; Huang, Z.; Liao, Q.; Yu, X.; Jiang, H.; He, Y.; Yao, S.; Nie, S.; Liu, L. Risk of second primary breast cancer among cancer survivors: Implications for prevention and screening practice. PLoS ONE 2020, 15, e0232800. [Google Scholar] [CrossRef]
- Brewer, H.R.; Jones, M.E.; Schoemaker, M.J.; Ashworth, A.; Swerdlow, A.J. Family history and risk of breast cancer: An analysis accounting for family structure. Breast Cancer Res. Treat. 2017, 165, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Parmigiani, G. Meta-analysis of BRCA1 and BRCA2 penetrance. J. Clin. Oncol. 2007, 25, 1329–1333. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Wei, W.; Zhan, L. Red and processed meat intake and risk of breast cancer: A meta-analysis of prospective studies. Breast Cancer Res. Treat. 2015, 151, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Macacu, A.; Autier, P.; Boniol, M.; Boyle, P. Active and passive smoking and risk of breast cancer: A meta-analysis. Breast Cancer Res. Treat. 2015, 154, 213–224. [Google Scholar] [CrossRef]
- Hamajima, N.; Hirose, K.; Tajima, K.; Rohan, T.; Calle, E.E.; Heath, C.W.; Coates, R.J.; Liff, J.M.; Talamini, R.; Chantarakul, N.; et al. Alcohol, tobacco and breast cancer—Collaborative reanalysis of individual data from 53 epidemiological studies, including 58 515 women with breast cancer and 95 067 women without the disease. Br. J. Cancer 2002, 87, 1234–1245. [Google Scholar] [CrossRef]
- McTiernan, A.; Kooperberg, C.; White, E.; Wilcox, S.; Coates, R.; Adams-Campbell, L.L.; Woods, N.; Ockene, J. Recreational Physical Activity and the Risk of Breast Cancer in Postmenopausal Women: The Women’s Health Initiative Cohort Study. J. Am. Med. Assoc. 2003, 290, 1331–1336. [Google Scholar] [CrossRef]
- Wegrzyn, L.R.; Tamimi, R.M.; Rosner, B.A.; Brown, S.B.; Stevens, R.G.; Eliassen, A.H.; Laden, F.; Willett, W.C.; Hankinson, S.E.; Schernhammer, E.S. Rotating Night-Shift Work and the Risk of Breast Cancer in the Nurses’ Health Studies. Am. J. Epidemiol. 2017, 186, 532–540. [Google Scholar] [CrossRef]
- Travis, L.B.; Hill, D.; Dores, G.M.; Gospodarowicz, M.; van Leeuwen, F.E.; Holowaty, E.; Glimelius, B.; Andersson, M.; Pukkala, E.; Lynch, C.F.; et al. Cumulative absolute breast cancer risk for young women treated for Hodgkin lymphoma. J. Natl. Cancer Inst. 2005, 97, 1428–1437. [Google Scholar] [CrossRef]
- International Programme on Chemical Safety. Global Assessment of the State of the Science of Endocrine Disruptors; World Health Organization (WHO): Geneva, Switzerland, 2002. [Google Scholar]
- Food and Drug Administration (FDA). Endocrine Disruptor Knowledge Base. 2019. Available online: https://www.fda.gov/science-research/bioinformatics-tools/endocrine-disruptor-knowledge-base (accessed on 15 September 2020).
- Soto, A.M.; Justicia, H.; Wray, J.W.; Sonnenschein, C. p-Nonyl-phenol: An estrogenic xenobiotic released from “modified” polystyrene. Environ. Health Perspect. 1991, 92, 167–173. [Google Scholar] [CrossRef]
- Environmental Protection Agency (EPA). Fact Sheet: Nonylphenols and Nonylphenol Ethoxylates. 2016. Available online: https://www.epa.gov/assessing-and-managing-chemicals-under-tsca/fact-sheet-nonylphenols-and-nonylphenol-ethoxylates#risks (accessed on 15 September 2020).
- Agence Nationale de Sécurité Sanitaire de l’alimentation de l’environnement et du Travail (ANSES). Caractérisation des Dangers et des Expositions du 4-Nonylphénol, 15–17. 2015. Available online: https://www.anses.fr/fr/system/files/SUBCHIM2009SA0331-109.pdf (accessed on 15 September 2020).
- Fedak, K.M.; Bernal, A.; Capshaw, Z.A.; Gross, S. Applying the Bradford Hill criteria in the 21st century: How data integration has changed causal inference in molecular epidemiology. Emerg. Themes Epidemiol. 2015, 12, 14. [Google Scholar] [CrossRef]
- International Agency for Research on Cancer (IARC). Agents Classified by the IARC Monographs. 2020; Volumes 1–127. Available online: https://monographs.iarc.fr/agents-classified-by-the-iarc/ (accessed on 15 September 2020).
- Hill, A.B. The Environment and Disease: Association or Causation? Proc. R. Soc. Med. 1965, 58, 295–300. [Google Scholar] [CrossRef]
- Guyton, K.Z.; Rusyn, I.; Chiu, W.A.; Corpet, D.E.; van den Berg, M.; Ross, M.K.; Christiani, D.C.; Beland, F.A.; Smith, M.T. Application of the key characteristics of carcinogens in cancer hazard identification. Carcinogenesis 2018, 39, 614–622. [Google Scholar] [CrossRef] [PubMed]
- La Merrill, M.A.; Vandenberg, L.N.; Smith, M.T.; Goodson, W.; Browne, P.; Patisaul, H.B.; Guyton, K.Z.; Kortenkamp, A.; Cogliano, V.J.; Woodruff, T.J.; et al. Consensus on the key characteristics of endocrine-disrupting chemicals as a basis for hazard identification. Nat. Rev. Endocrinol. 2020, 16, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Safe, S.; Wormke, M. Inhibitory aryl hydrocarbon receptor-estrogen receptor alpha cross-talk and mechanisms of action. Chem. Res. Toxicol. 2003, 16, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Esser, C. The arylhydrocarbon receptor: More than a tox story. Biol. Chem. 2006. [Google Scholar] [CrossRef]
- Tarnow, P.; Tralau, T.; Luch, A. Chemical activation of estrogen and aryl hydrocarbon receptor signaling pathways and their interaction in toxicology and metabolism. Expert Opin. Drug Metab. Toxicol. 2019, 15, 219–229. [Google Scholar] [CrossRef]
- Kelce, W.R.; Stone, C.R.; Laws, S.C.; Gray, L.E.; Kemppainen, J.A.; Wilson, E.M. Persistent DDT metabolite p,p′-DDE is a potent androgen receptor antagonist. Nature 1995, 375, 581–585. [Google Scholar] [CrossRef]
- Paul-Friedman, K.; Martin, M.; Crofton, K.M.; Hsu, C.W.; Sakamuru, S.; Zhao, J.; Xia, M.; Huang, R.; Stavreva, D.A.; Soni, V.; et al. Limited Chemical Structural Diversity Found to Modulate Thyroid Hormone Receptor in the Tox21 Chemical Library. Environ. Health Perspect. 2019, 127, 97009. [Google Scholar] [CrossRef]
- You, S.H.; Gauger, K.J.; Bansal, R.; Zoeller, R.T. 4-Hydroxy-PCB106 acts as a direct thyroid hormone receptor agonist in rat GH3 cells. Mol. Cell. Endocrinol. 2006, 257, 26–34. [Google Scholar] [CrossRef]
- Vandenberg, L.N.; Colborn, T.; Hayes, T.B.; Heindel, J.J.; Jacobs, D.R.; Lee, D.H.; Shioda, T.; Soto, A.M.; vom Saal, F.S.; Welshons, W.V.; et al. Hormones and Endocrine-Disrupting Chemicals: Low-Dose Effects and Nonmonotonic Dose Responses. Endocr. Rev. 2012, 33, 378–455. [Google Scholar] [CrossRef]
- Land, C.E.; Tokunaga, M.; Koyama, K.; Soda, M.; Preston, D.L.; Nishimori, I.; Tokuoka, S. Incidence of female breast cancer among atomic bomb survivors, Hiroshima and Nagasaki, 1950–1990. Radiat. Res. 2003, 160, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Troisi, R.; Hatch, E.E.; Titus-Ernstoff, L.; Hyer, M.; Palmer, J.R.; Robboy, S.J.; Strohsnitter, W.C.; Kaufman, R.; Herbst, A.L.; Hoover, R.N. Cancer risk in women prenatally exposed to diethylstilbestrol. Int. J. Cancer 2007, 121, 356–360. [Google Scholar] [CrossRef] [PubMed]
- Safe, S.; Lee, S.O.; Jin, U.H. Role of the aryl hydrocarbon receptor in carcinogenesis and potential as a drug target. Toxicol. Sci. 2013, 135, 1–16. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO); Food and Agriculture Organization of the United Nations. Principles and Methods for the Risk Assessment of Chemicals in Food. 2009. Available online: https://apps.who.int/iris/bitstream/handle/10665/44065/WHO_EHC_240_8_eng_Chapter5.pdf;jsessionid=604C61125599E48F60D2EDF18BC65A61?sequence=8 (accessed on 15 September 2020).
- Silva, E.; Rajapakse, N.; Kortenkamp, A. Something from “nothing” eight weak estrogenic chemicals combined at concentrations below NOECs produce significant mixture effects. Environ. Sci. Technol. 2002, 36, 1751–1756. [Google Scholar] [CrossRef] [PubMed]
- Hass, U.; Scholze, M.; Christiansen, S.; Dalgaard, M.; Vinggaard, A.M.; Axelstad, M.; Metzdorff, S.B.; Kortenkamp, A. Combined exposure to anti-androgens exacerbates disruption of sexual differentiation in the rat. Environ. Health Perspect. 2007, 115, 122–128. [Google Scholar] [CrossRef]
- Howdeshell, K.I.; Wilson, V.S.; Furr, J.; Lambright, C.R.; Rider, C.V.; Blystone, C.R.; Hotchkiss, A.K.; Gray, L.E. A mixture of five phthalate esters inhibits fetal testicular testosterone production in the Sprague-Dawley rat in a cumulative, dose-additive manner. Toxicol. Sci. 2008, 105, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Delfosse, V.; Dendele, B.; Huet, T.; Grimaldi, M.; Boulahtouf, A.; Gerbal-Chaloin, S.; Beucher, B.; Roecklin, D.; Muller, C.; Rahmani, R.; et al. Synergistic activation of human pregnane X receptor by binary cocktails of pharmaceutical and environmental compounds. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef]
- Schiffer, L.; Barnard, L.; Baranowski, E.S.; Gilligan, L.C.; Taylor, A.E.; Arlt, W.; Shackleton, C.H.L.; Storbeck, K.H. Human steroid biosynthesis, metabolism and excretion are differentially reflected by serum and urine steroid metabolomes: A comprehensive review. J. Steroid Biochem. Mol. Biol. 2019, 194, 105439. [Google Scholar] [CrossRef]
- Meinhardt, U.; Mullis, P.E. The essential role of the aromatase/p450arom. Semin. Reprod. Med. 2002, 20, 277–284. [Google Scholar] [CrossRef]
- Caron-Beaudoin, É.; Viau, R.; Sanderson, J.T. Effects of neonicotinoid pesticides on promoter-specific aromatase (CYP19) expression in Hs578t breast cancer cells and the role of the VEGF pathway. Environ. Health Perspect. 2018, 126, 047014. [Google Scholar] [CrossRef]
- Van der Spek, A.H.; Fliers, E.; Boelen, A. The classic pathways of thyroid hormone metabolism. Mol. Cell. Endocrinol. 2017, 458, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Wolff, J. Perchlorate and the thyroid gland. Pharmacol. Rev. 1998, 50, 89–105. [Google Scholar] [PubMed]
- Da Silva, M.M.; Gonçalves, C.F.L.; Miranda-Alves, L.; Fortunato, R.S.; Carvalho, D.P.; Ferreira, A.C.F. Inhibition of Type 1 Iodothyronine Deiodinase by Bisphenol A. Horm. Metab. Res. 2019, 51, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Siiteri, P.K.; Murai, J.T.; Hammond, G.L.; Nisker, J.A.; Raymoure, W.J.; Kuhn, R.W. The serum transport of steroid hormones. Recent Prog. Horm. Res. 1982, 38, 457–510. [Google Scholar] [CrossRef]
- Hong, H.; Branham, W.S.; Ng, H.W.; Moland, C.L.; Dial, S.L.; Fang, H.; Perkins, R.; Sheehan, D.; Tong, W. Human sex hormone-binding globulin binding affinities of 125 structurally diverse chemicals and comparison with their binding to androgen receptor, estrogen receptor, and α-Fetoprotein. Toxicol. Sci. 2015, 143, 333–348. [Google Scholar] [CrossRef]
- Kester, M.H.A.; Bulduk, S.; Tibboel, D.; Meinl, W.; Glatt, H.; Falany, C.N.; Coughtrie, M.W.; Bergman, A.; Safe, S.H.; Kuiper, G.G.; et al. Potent inhibition of estrogen sulfotransferase by hydroxylated PCB metabolites: A novel pathway explaining the estrogenic activity of PCB’s. Endocrinology 2000, 141, 1897–1900. [Google Scholar] [CrossRef]
- Walker, C.L. Minireview: Epigenomic Plasticity and Vulnerability to EDC Exposures. Mol. Endocrinol. 2016, 30, 848–855. [Google Scholar] [CrossRef]
- Hermann, A.; Gowher, H.; Jeltsch, A. Biochemistry and biology of mammalian DNA methyltransferases. Cell. Mol. Life Sci. 2004, 61, 2571–2587. [Google Scholar] [CrossRef]
- Yamagata, Y.; Asada, H.; Tamura, I.; Lee, L.; Maekawa, R.; Taniguchi, K.; Taketani, T.; Matsuoka, A.; Tamura, H.; Sugino, N. DNA methyltransferase expression in the human endometrium: Down-regulation by progesterone and estrogen. Hum. Reprod. 2009, 24, 1126–1132. [Google Scholar] [CrossRef]
- Zama, A.M.; Uzumcu, M. Fetal and neonatal exposure to the endocrine disruptor methoxychlor causes epigenetic alterations in adult ovarian genes. Endocrinology 2009, 150, 4681–4691. [Google Scholar] [CrossRef]
- Zhdanov, V.P. Conditions of appreciable influence of microRNA on a large number of target mRNAs. Mol. BioSyst. 2009, 5, 638–643. [Google Scholar] [CrossRef] [PubMed]
- Maillot, G.; Lacroix-Triki, M.; Pierredon, S.; Gratadou, L.; Schmidt, S.; Bénès, V.; Roché, H.; Dalenc, F.; Auboeuf, D.; Millevoi, S.; et al. Widespread estrogen-dependent repression of micrornas involved in breast tumor cell growth. Cancer Res. 2009, 69, 8332–8340. [Google Scholar] [CrossRef] [PubMed]
- Tilghman, S.L.; Bratton, M.R.; Segar, H.C.; Martin, E.C.; Rhodes, L.V.; Li, M.; McLachlan, J.A.; Wiese, T.E.; Nephew, K.P.; Burow, M.E. Endocrine disruptor regulation of microRNA expression in breast carcinoma cells. PLoS ONE 2012, 7, e32754. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, H.; Miyoshi, N.; Miyamoto, Y.; Souda, M.; Umekita, Y.; Yasuda, N.; Yoshida, H. Effects of fetal exposure to diethylstilbestrol on mammary tumorigenesis in rats. J. Vet. Med. Sci. 2009, 71, 1599–1608. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer (IARC). Monographs on the Evaluation of Carcinogenic to Humans: Radiation. IARC Monogr. Eval. Carcinog. Risks Hum. 2012, 75, 103–210. [Google Scholar]
- International Agency for Research on Cancer (IARC). Monographs on the Evaluation of Carcinogenic to Humans: Diethylstilbestrol. IARC Monogr. Eval. Carcinog. Risks Hum. 2012, 21, 175–218. [Google Scholar]
- Dodds, E.C.; Goldberg, L.; Lawson, W.; Robinson, R. OEstrogenic Activity of Certain Synthetic Compounds. Nature 1938, 141, 247–248. [Google Scholar] [CrossRef]
- Korach, K.S.; Metzler, M.; McLachlan, J.A. Estrogenic activity in vivo and in vitro of some diethylstilbestrol metabolites and analogs. Proc. Natl. Acad. Sci. USA 1978, 75, 468–471. [Google Scholar] [CrossRef]
- Watkins Smith, O.; Smith, G.V.S. The influence of diethylstilbestrol on the progress and outcome of pregnancy as based on a comparison of treated with untreated primigravidas. Am. J. Obstet. Gynecol. 1949, 58, 994–1009. [Google Scholar] [CrossRef]
- Watkins Smith, O. Diethylstilbestrol in the prevention and treatment of complications of pregnancy. Am. J. Obstet. Gynecol. 1948, 56, 821–834. [Google Scholar] [CrossRef]
- Food and Drug Administration (FDA). FDA-Approved Drugs. 2020. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=004056 (accessed on 15 September 2020).
- Giusti, R.M.; Iwamoto, K.; Hatch, E.E. Diethylstilbestrol revisited: A review of the long-term health effects. Ann. Intern. Med. 1995, 122, 778–788. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.M.; Waring, R.H. Diethylstilboestrol—A long-term legacy. Maturitas 2012, 72, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Raun, A.; Preston, R. History of diethylstilbestrol use in cattle. J. Anim. Sci. 2002, 1–7. [Google Scholar]
- Dunning, W.F.; Curtis, M.R.; Segaloff, A. Strain differences in response to diethylstilbestrol and the induction of mammary gland, adrenal and bladder cancer in the rat. J. Mich. State Med. Soc. 1948, 47, 511–521. [Google Scholar]
- Richardson, F.L.; Hall, G. Mammary tumors and mammary-gland development in hybrid mice treated with diethylstil bestrol for varying periods. J. Natl. Cancer Inst. 1960, 25, 1023–1039. [Google Scholar]
- Nagasawa, H.; Yanai, R. Long-Term Effects of Neonatal Hormonal Treatments on Plasma Prolactin Levels in Female BALB/cfC3H and BALB/c Mice. Cancer Res. 1978, 38, 942–945. [Google Scholar]
- Bern, H.A.; Edery, M.; Mills, K.T.; Kohrman, A.F.; Mori, T.; Larson, L. Long-Term Alterations in Histology and Steroid Receptor Levels of the Genital Tract and Mammary Gland following Neonatal Exposure of Female BALB/cCrgl Mice to Various Doses of Diethylstilbestrol. Cancer Res. 1987, 47, 4165–4172. [Google Scholar]
- Bern, H.A.; Mills, K.T.; Dorothy, H.L.; Ostrander, P.L.; Iguchi, T. Altered mammary responsiveness to estradiol and progesterone in mice exposed neonatally to diethylstibestrol. Cancer Lett. 1992, 63, 117–124. [Google Scholar] [CrossRef]
- Rothschild, T.C.; Boylan, E.S.; Calhoon, R.E.; Vonderhaar, B.K. Transplacental Effects of Diethylstilbestrol on Mammary Development and Tumorigenesis in Female ACI Rats. Cancer Res. 1987, 47, 4508–4516. [Google Scholar]
- Umekita, Y.; Souda, M.; Hatanaka, K.; Hamada, T.; Yoshioka, T.; Kawaguchi, H.; Tanimoto, A. Gene expression profile of terminal end buds in rat mammary glands exposed to diethylstilbestrol in neonatal period. Toxicol. Lett. 2011, 205, 15–25. [Google Scholar] [CrossRef]
- Doherty, L.F.; Bromer, J.G.; Zhou, Y.; Aldad, T.S.; Taylor, H.S. In Utero Exposure to Diethylstilbestrol (DES) or Bisphenol-A (BPA) Increases EZH2 Expression in the Mammary Gland: An Epigenetic Mechanism Linking Endocrine Disruptors to Breast Cancer. Horm. Cancer 2010, 1, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Erdmann, C.; Chinnaiyan, A.M.; Merajver, S.D.; Kleer, C.G. Identification of EZH2 as a molecular marker for a precancerous state in morphologically normal breast tissues. Cancer Res. 2006, 66, 4095–4099. [Google Scholar] [CrossRef] [PubMed]
- Wormsbaecher, C.; Hindman, A.R.; Avendano, A.; Cortes-Medina, M.; Jones, C.E.; Bushman, A.; Onua, L.; Kovalchin, C.E.; Murphy, A.R.; Helber, H.L.; et al. In utero estrogenic endocrine disruption alters the stroma to increase extracellular matrix density and mammary gland stiffness. Breast Cancer Res. 2020, 22, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Herbst, A.L.; Ulfelder, H.; Poskanzer, D.C. Adenocarcinoma of the Vagina: Association of Maternal Stilbestrol Therapy with Tumor Appearance in Young Women. N. Engl. J. Med. 1971, 284, 878–881. [Google Scholar] [CrossRef] [PubMed]
- Herbst, A.L.; Cole, P.; Colton, T.; Robboy, S.J.; Scully, R.E. Age-incidence and risk of diethylstilbestrol-related clear cell adenocarcinoma of the vagina and cervix. Am. J. Obstet. Gynecol. 1977, 128, 43–50. [Google Scholar] [CrossRef]
- Bibbo, M.; Haenszel, W.M.; Wied, G.L.; Hubby, M.; Herbst, A.L. A Twenty-Five-Year Follow-up Study of Women Exposed to Diethylstilbestrol during Pregnancy. N. Engl. J. Med. 1978, 298, 763–767. [Google Scholar] [CrossRef]
- Greenberg, E.R.; Barnes, A.B.; Resseguie, L.; Barrett, J.A.; Burnside, S.; Lanza, L.L.; Neff, R.K.; Stevens, M.; Young, R.H.; Colton, T. Breast Cancer in Mothers Given Diethylstilbestrol in Pregnancy. N. Engl. J. Med. 1984, 311, 1393–1398. [Google Scholar] [CrossRef]
- Colton, T.; Greenberg, E.R.; Noller, K.; Resseguie, L.; Van Bennekom, C.; Heeren, T.; Zhang, Y. Breast Cancer in Mothers Prescribed Diethylstilbestrol in Pregnancy: Further Follow-up. J. Am. Med. Assoc. 1993, 269, 2096–2100. [Google Scholar] [CrossRef]
- Titus-Ernstoff, L.; Hatch, E.E.; Hoover, R.N.; Palmer, J.; Greenberg, E.R.; Ricker, W.; Kaufman, R.; Noller, K.; Herbst, A.L.; Colton, T.; et al. Long-term cancer risk in women given diethylstilbestrol (DES) during pregnancy. Br. J. Cancer 2001, 84, 126–133. [Google Scholar] [CrossRef]
- Hatch, E.E. Cancer Risk in Women Exposed to Diethylstilbestrol In Utero. JAMA 1998, 280, 630–634. [Google Scholar] [CrossRef]
- Palmer, J.R.; Wise, L.A.; Hatch, E.E.; Troisi, R.; Titus-Ernstoff, L.; Strohsnitter, W.; Kaufman, R.; Herbst, A.L.; Noller, K.L.; Hyer, M.; et al. Prenatal diethylstilbestrol exposure and risk of breast cancer. Cancer Epidemiol. Biomark. Prev. 2006, 15, 1509–1514. [Google Scholar] [CrossRef] [PubMed]
- Verloop, J.; Van Leeuwen, F.E.; Helmerhorst, T.J.M.; Van Boven, H.H.; Rookus, M.A. Cancer risk in DES daughters. Cancer Causes Control 2010, 21, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Hoover, R.N.; Hyer, M.; Pfeiffer, R.M.; Adam, E.; Bond, B.; Cheville, A.L.; Colton, T.; Hartge, P.; Hatch, E.E.; Herbst, A.L.; et al. Adverse health outcomes in women exposed in utero to diethylstilbestrol. N. Engl. J. Med. 2011, 365, 1304–1314. [Google Scholar] [CrossRef] [PubMed]
- Tournaire, M.; Devouche, E.; Espié, M.; Asselain, B.; Levadou, A.; Cabau, A.; Dunbavand, A.; Grosclaude, P.; Epelboin, S. Cancer Risk in Women Exposed to Diethylstilbestrol in Utero. Thérapie 2015, 70, 433–441. [Google Scholar] [CrossRef]
- Troisi, R.; Hatch, E.E.; Titus, L.; Strohsnitter, W.; Gail, M.H.; Huo, D.; Adam, E.; Robboy, S.J.; Hyer, M.; Hoover, R.N.; et al. Prenatal diethylstilbestrol exposure and cancer risk in women. Environ. Mol. Mutagenesis 2019, 60, 395–403. [Google Scholar] [CrossRef]
- Titus, L.; Hatch, E.E.; Drake, K.M.; Parker, S.E.; Hyer, M.; Palmer, J.R.; Strohsnitter, W.C.; Adam, E.; Herbst, A.L.; Huo, D.; et al. Reproductive and hormone-related outcomes in women whose mothers were exposed in utero to diethylstilbestrol (DES): A report from the US National Cancer Institute DES Third Generation Study. Reprod. Toxicol. 2019, 84, 32–38. [Google Scholar] [CrossRef]
- Gill, W.B.; Schumacher, G.F.; Bibbo, M.; Straus, F.H., II; Schoenberg, H.W. Association of diethylstilbestrol exposure in utero with cryptorchidism, testicular hypoplasia and semen abnormalities. J. Urol. 1979, 122, 36–39. [Google Scholar] [CrossRef]
- Reed, C.E.; Fenton, S.E. Exposure to diethylstilbestrol during sensitive life stages: A legacy of heritable health effects. Birth Defects Res. Part C Embryo Today Rev. 2013, 99, 134–146. [Google Scholar] [CrossRef]
- Dieckmann, W.J.; Davis, M.E.; Rynkiewicz, L.M.; Pottinger, R.E. Does the administration of diethylstilbestrol during pregnancy have therapeutic value? Am. J. Obstet. Gynecol. 1953, 66, 1062–1081. [Google Scholar] [CrossRef]
- FDA. Selected item from the FDA drug bulletin-november 1971: Diethylstilbestrol contraindicated in pregnancy. Calif. Med. 1953, 116, 85–86. [Google Scholar]
- Ministère des Solidarités et de la Santé. DISTILBENE 1 mg, Comprimé Enrobé. 2020. Available online: http://base-donnees-publique.medicaments.gouv.fr/extrait.php?specid=68600838 (accessed on 15 September 2020).
- Jarman, W.M.; Ballschmiter, K. From coal to DDT: The history of the development of the pesticide DDT from synthetic dyes till Silent Spring. Endeavour 2012, 36, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Carson, R. Silent Spring; Houghton Mifflin Co.: Boston, MA, USA, 1962. [Google Scholar]
- Agency for Toxic Substances and Disease Registry (ATSDR). Toxicological Profile for DDT, DDE, and DDD. 2020. Available online: https://www.atsdr.cdc.gov/ToxProfiles/tp.asp?id=81&tid=20 (accessed on 13 August 2020).
- Morgan, D.P.; Roan, C.C. The metabolism of DDT in man. In Essays in Toxicology; Elsevier: New York, NY, USA, 1974; pp. 39–97. [Google Scholar]
- Soto, A.M.; Sonnenschein, C.; Chung, K.L.; Fernandez, M.F.; Olea, N.; Olea Serrano, F. The E-SCREEN assay as a tool to identify estrogens: An update on estrogenic environmental pollutants. Environ. Health Perspect. 1995, 103, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Dees, C.; Askari, M.; Foster, J.S.; Ahamed, S.; Wimalasena, J. DDT mimicks estradiol stimulation of breast cancer cells to enter the cell cycle. Mol. Carcinog. 1997, 18, 107–114. [Google Scholar] [CrossRef]
- Bratton, M.R.; Frigo, D.E.; Segar, H.C.; Nephew, K.P.; McLachlan, J.A.; Wiese, T.E.; Burow, M.E. The Organochlorine o,p′-DDT Plays a Role in Coactivator-Mediated MAPK Crosstalk in MCF-7 Breast Cancer Cells. Environ. Health Perspect. 2012, 120, 1291–1296. [Google Scholar] [CrossRef]
- Aubé, M.; Larochelle, C.; Ayotte, P. 1,1-dichloro-2,2-bis(p-chlorophenyl)ethylene (p,p′-DDE) disrupts the estrogen-androgen balance regulating the growth of hormone-dependent breast cancer cells. Breast Cancer Res. 2008, 10, 16–28. [Google Scholar] [CrossRef]
- Han, E.H.; Kim, H.G.; Hwang, Y.P.; Choi, J.H.; Im, J.H.; Park, B.; Yang, J.H.; Jeong, T.C.; Jeong, H.G. The role of cyclooxygenase-2-dependent signaling via cyclic AMP response element activation on aromatase up-regulation by o,p′-DDT in human breast cancer cells. Toxicol. Lett. 2010, 198, 331–341. [Google Scholar] [CrossRef]
- Thompson, L.A.; Ikenaka, Y.; Sobhy Darwish, W.; Nakayama, S.M.M.; Mizukawa, H.; Ishizuka, M. Effects of the organochlorine p,p′-DDT on MCF-7 cells: Investigating metabolic and immune modulatory transcriptomic changes. Environ. Toxicol. Pharmacol. 2019, 72, 103249. [Google Scholar] [CrossRef]
- Kim, D.W.; Gazourian, L.; Quadri, S.A.; Romieu-Mourez, R.; Sherr, D.H.; Sonenshein, G.E. The RelA NF-kappaB subunit and the aryl hydrocarbon receptor (AhR) cooperate to transactivate the c-myc promoter in mammary cells. Oncogene 2000, 19, 5498–5506. [Google Scholar] [CrossRef]
- Skinner, M.K.; Ben Maamar, M.; Sadler-Riggleman, I.; Beck, D.; Nilsson, E.; McBirney, M.; Klukovich, R.; Xie, Y.; Tang, C.; Yan, W. Alterations in sperm DNA methylation, non-coding RNA and histone retention associate with DDT-induced epigenetic transgenerational inheritance of disease. Epigenet. Chromatin 2018, 11, 8. [Google Scholar] [CrossRef]
- Bitman, J.; Cecil, H.C.; Harris, S.J.; Fries, G.F. Estrogenic activity of o,p′-DDT in the mammalian uterus and avian oviduct. Science 1968, 162, 371–372. [Google Scholar] [CrossRef]
- Robison, A.K.; Sirbasku, D.A.; Stancel, G.M. DDT supports the growth of an estrogen-responsive tumor. Toxicol. Lett. 1985, 27, 109–113. [Google Scholar] [CrossRef]
- Brown, N.M.; Lamartiniere, C.A. Xenoestrogens alter mammary gland differentiation and cell proliferation in the rat. Environ. Health Perspect. 1995, 103, 708–713. [Google Scholar] [CrossRef] [PubMed]
- Uppala, P.T.; Roy, S.K.; Tousson, A.; Barnes, S.; Uppala, G.R.; Eastmond, D.A. Induction of cell proliferation, micronuclei and hyperdiploidy/polyploidy in the mammary cells of DDT- and DMBA-treated pubertal rats. Environ. Mol. Mutagenes. 2005, 46, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Kalinina, T.S.; Kononchuk, V.V.; Gulyaeva, L.F. Expression of hormonal carcinogenesis genes and related regulatory microRNAs in uterus and ovaries of DDT-treated female rats. Biochemistry 2017, 82, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Chanyshev, M.D.; Kosorotikov, N.I.; Titov, S.E.; Kolesnikov, N.N.; Gulyaeva, L.F. Expression of microRNAs, CYP1A1 and CYP2B1 in the livers and ovaries of female rats treated with DDT and PAHs. Life Sci. 2014, 103, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Cha, E.S.; Ko, Y.; Hwang, M.S.; Hong, J.H.; Lee, W.J. Exposure to dichlorodiphenyltrichloroethane and the risk of breast cancer: A systematic review and meta-analysis. Osong Public Health Res. Perspect. 2014, 5, 77–84. [Google Scholar] [CrossRef]
- Cohn, B.A.; Wolff, M.S.; Cirillo, P.M.; Scholtz, R.I. DDT and breast cancer in young women: New data on the significance of age at exposure. Environ. Health Perspect. 2007, 115, 1406–1414. [Google Scholar] [CrossRef]
- Cohn, B.A.; La Merrill, M.; Krigbaum, N.Y.; Yeh, G.; Park, J.S.; Zimmermann, L.; Cirillo, P.M. DDT exposure in utero and breast cancer. J. Clin. Endocrinol. Metab. 2015, 100, 2865–2872. [Google Scholar] [CrossRef]
- Cohn, B.A.; Cirillo, P.M.; Terry, M.B. DDT and Breast Cancer: Prospective Study of Induction Time and Susceptibility Windows. J. Natl. Cancer Inst. 2019, 111, 803–810. [Google Scholar] [CrossRef]
- White, A.J.; Teitelbaum, S.L.; Wolff, M.S.; Stellman, S.D.; Neugut, A.I.; Gammon, M.D. Exposure to fogger trucks and breast cancer incidence in the long Island breast cancer study project: A case-control study. Environ. Health 2013, 12, 24–48. [Google Scholar] [CrossRef]
- Niehoff, N.M.; Nichols, H.B.; White, A.J.; Parks, C.G.; D’Aloisio, A.A.; Sandler, D.P. Childhood and adolescent pesticide exposure and breast cancer risk. Epidemiology 2016, 27, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Bachelet, D.; Verner, M.A.; Neri, M.; Duverger, É.C.; Charlier, C.; Arveux, P.; Haddad, S.; Guénel, P. Breast cancer and exposure to organochlorines in the cecile study: Associations with plasma levels measured at the time of diagnosis and estimated during adolescence. Int. J. Environ. Res. Public Health 2019, 16, 271–286. [Google Scholar] [CrossRef] [PubMed]
- Itoh, H.; Iwasaki, M.; Hanaoka, T.; Kasuga, Y.; Yokoyama, S.; Onuma, H.; Nishimura, H.; Kusama, R.; Tsugane, S. Serum organochlorines and breast cancer risk in Japanese women: A case-control study. Cancer Causes Control 2009, 20, 567–580. [Google Scholar] [CrossRef]
- Ingber, S.Z.; Buser, M.C.; Pohl, H.R.; Abadin, H.G.; Edward Murray, H.; Scinicariello, F. DDT/DDE and breast cancer: A meta-analysis. Regul. Toxicol. Pharmacol. 2013, 67, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, B.J.; Christianson, R.E.; Oechsli, F.W. The California Child Health and Development Studies of the School of Public Health, University of California at Berkeley. Paediatr. Perinat. Epidemiol. 1988, 2, 265–282. [Google Scholar] [CrossRef] [PubMed]
- Soto, A.M.; Sonnenschein, C. DDT, endocrine disruption and breast cancer. Nat. Rev. Endocrinol. 2015, 11, 507–508. [Google Scholar] [CrossRef]
- Gammon, M.D.; Neugut, A.I.; Santella, R.M.; Teitelbaum, S.L.; Britton, J.A.; Terry, M.B.; Eng, S.M.; Wolff, M.S.; Stellman, S.D.; Kabat, G.C.; et al. The Long Island Breast Cancer Study Project: Description of a multi-institutional collaboration to identify environmental risk factors for breast cancer. Breast Cancer Res. Treat. 2002, 74, 235–254. [Google Scholar] [CrossRef]
- EU Pesticides Database. DDT. 2016. Available online: https://ec.europa.eu/food/plant/pesticides/eu-pesticides-database/public/?event=activesubstance.detail&language=EN&selectedID=1194 (accessed on 15 September 2020).
- Environmental Protection Agency. DDT Ban Takes Effect. 2016. Available online: https://archive.epa.gov/epa/aboutepa/ddt-ban-takes-effect.html (accessed on 15 September 2020).
- Pesticide Action Network United Kingdom. Which Pesticides are Banned in Europe? Food Fairness Brief. 2008, 1, 1–8. [Google Scholar]
- International Agency for Research on Cancer (IARC). Monographs on the Evaluation of Carcinogenic to Humans: DDT. IARC Monogr. Eval. Carcinog. Risks Hum. 2008, 113, 37–233. [Google Scholar]
- Van den Berg, H.; Manuweera, G.; Konradsen, F. Global trends in the production and use of DDT for control of malaria and other vector-borne diseases. Malar. J. 2017, 16, 401. [Google Scholar] [CrossRef]
- Koureas, M.; Rousou, X.; Haftiki, H.; Mouchtouri, V.A.; Rachiotis, G.; Rakitski, V.; Tsakalof, A.; Hadjichristodoulou, C. Spatial and temporal distribution of p,p′-DDE (1-dichloro-2,2-bis (p-chlorophenyl) ethylene) blood levels across the globe. A systematic review and meta-analysis. Sci. Total Environ. 2019, 686, 440–451. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). The Use of DDT in Malaria Vector Control. 2011. Available online: https://apps.who.int/iris/bitstream/handle/10665/69945/WHO_HTM_GMP_2011_eng.pdf;jsessionid=A4B9406D7E4E3AD840C00B10A404FB60?sequence=1 (accessed on 15 September 2020).
- Azeredo, A.; Torres, J.P.M.; de Freitas Fonseca, M.; Britto, J.L.; Bastos, W.R.; Azevedo E Silva, C.E.; Cavalcanti, G.; Meire, R.O.; Sarcinelli, P.N.; Claudio, L.; et al. DDT and its metabolites in breast milk from the Madeira River basin in the Amazon, Brazil. Chemosphere 2008, 73, S246–S251. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Evaluation of the Carcinogenic Hazards of Food Additive; WHO: Geneva, Switzerland, 1961. [Google Scholar]
- Bouwman, H.; Sereda, B.; Meinhardt, H.M. Simultaneous presence of DDT and pyrethroid residues in human breast milk from a malaria endemic area in South Africa. Environ. Pollut. 2006, 144, 902–917. [Google Scholar] [CrossRef] [PubMed]
- Hecht, D.K. How to make a villain: Rachel Carson and the politics of anti-environmentalism. Endeavour 2012, 36, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Epstein, L. Fifty Years since Silent Spring. Annu. Rev. Phytopathol. 2014, 52, 377–402. [Google Scholar] [CrossRef] [PubMed]
- Marinković, N.; Pašalić, D.; Ferenčak, G.; Gršković, B.; Rukavina, A.S. Dioxins and human toxicity. Arh. Hig. Rada Toksikol. 2010, 61, 445–453. [Google Scholar] [CrossRef]
- Environmental Protection Agency (EPA). Update to An Inventory of Sources and Environmental Releases of Dioxin-Like Compounds in the United States for the Years 1987, 1995, and 2000; EPA: Washington, DC, USA, 2013.
- International Agency for Research on Cancer (IARC). Monographs on the Evaluation of Carcinogenic to Humans: 2,3,7,8-Tetrachlorodibenzo-para-dioxin. IARC Monogr. Eval. Carcinog. Risks Hum. 2012, 69. Available online: https://monographs.iarc.fr/wp-content/uploads/2018/06/mono100F-27.pdf (accessed on 15 September 2020).
- Young, A.L.; Reggiani, G.M. Historical overview of the controversy surrounding Agent Orange. In Agent Orange and Its Associated Dioxin: Assessment of a Controversy; Elservier: New York, NY, USA, 1988; pp. 31–76. [Google Scholar]
- Lucier, G.W.; McDaniel, O.S.; Hook, G.E.; Fowler, B.A.; Sonawane, B.R.; Faeder, E. TCDD-induced changes in rat liver microsomal enzymes. Environ. Health Perspect. 1973, 5, 199–209. [Google Scholar] [CrossRef]
- Tukey, R.H.; Hannah, R.R.; Negishi, M.; Nebert, D.W.; Eisen, H.J. The Ah locus: Correlation of intranuclear appearance of inducer-receptor complex with induction of cytochrome P1-450 mRNA. Cell 1982, 31, 275–284. [Google Scholar] [CrossRef]
- Yoon, C.Y.; Park, M.; Kim, B.H.; Park, J.Y.; Park, M.S.; Jeong, Y.K.; Kwon, H.; Jung, H.K.; Kang, H.; Lee, Y.S.; et al. Gene expression profile by 2,3,7,8-tetrachlorodibenzo-p-dioxin in the liver of wild-type (AhR+/+) and aryl hydrocarbon receptor-deficient (AhR−/−) mice. J. Vet. Med. Sci. 2006, 68, 663–668. [Google Scholar] [CrossRef]
- Bekki, K.; Vogel, H.; Li, W.; Ito, T.; Sweeney, C.; Haarmann-Stemmann, T.; Matsumura, F.; Vogel, C.F.A. The aryl hydrocarbon receptor (AhR) mediates resistance to apoptosis induced in breast cancer cells. Pestic. Biochem. Physiol. 2015, 120, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Ahn, N.S.; Hu, H.; Park, J.S.; Park, J.S.; Kim, J.S.; An, S.; Kong, G.; Aruoma, O.I.; Lee, Y.S.; Kang, K.S. Molecular mechanisms of the 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced inverted U-shaped dose responsiveness in anchorage independent growth and cell proliferation of human breast epithelial cells with stem cell characteristics. Mutat. Res. 2005, 579, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, P.; Safe, S. Growth inhibitory and antimitogenic activity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in T47D human breast cancer cells. Toxicol. Lett. 1992, 61, 185–197. [Google Scholar] [CrossRef]
- Marquez-Bravo, L.G.; Gierthy, J.F. Differential expression of estrogen receptor alpha (ERalpha) protein in MCF-7 breast cancer cells chronically exposed to TCDD. J. Cell. Biochem. 2008, 103, 636–647. [Google Scholar] [CrossRef]
- Hockings, J.K.; Thorne, P.A.; Kemp, M.Q.; Morgan, S.S.; Selmin, O.; Romagnolo, D.F. The ligand status of the aromatic hydrocarbon receptor modulates transcriptional activation of BRCA-1 promoter by estrogen. Cancer Res. 2006, 66, 2224–2232. [Google Scholar] [CrossRef]
- Brunnberg, S.; Andersson, P.; Poellinger, L.; Hanberg, A. The constitutively active Ah receptor (CA-AhR) mouse as a model for dioxin exposure—Effects in reproductive organs. Chemosphere 2011, 85, 1701–1706. [Google Scholar] [CrossRef]
- Chen, Y.J.; Hung, C.M.; Kay, N.; Chen, C.C.; Kao, Y.H.; Yuan, S.S. Progesterone receptor is involved in 2,3,7,8-tetrachlorodibenzo-p-dioxin-stimulated breast cancer cells proliferation. Cancer Lett. 2012, 319, 223–231. [Google Scholar] [CrossRef]
- Brown, N.M.; Manzolillo, P.A.; Zhang, J.X.; Wang, J.; Lamartiniere, C.A. Prenatal TCDD and predisposition to mammary cancer in the rat. Carcinogenesis 1998, 19, 1623–1629. [Google Scholar] [CrossRef]
- Lewis, B.C.; Hudgins, S.; Lewis, A.; Schorr, K.; Sommer, R.; Peterson, R.E.; Flaws, J.A.; Furth, P.A. In utero and lactational treatment with 2,3,7,8-tetrachlorodibenzo-p-dioxin impairs mammary gland differentiation but does not block the response to exogenous estrogen in the postpubertal female rat. Toxicol. Sci. 2001, 62, 46–53. [Google Scholar] [CrossRef]
- Fenton, S.E.; Hamm, J.T.; Birnbaum, L.S.; Youngblood, G.L. Persistent abnormalities in the rat mammary gland following gestational and lactational exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Toxicol. Sci. 2002, 67, 63–74. [Google Scholar] [CrossRef]
- Jenkins, S.; Rowell, C.; Wang, J.; Lamartiniere, C.A. Prenatal TCDD exposure predisposes for mammary cancer in rats. Reprod. Toxicol. 2007, 23, 391–396. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Papoutsis, A.J.; Selmin, O.I.; Borg, J.L.; Romagnolo, D.F. Gestational exposure to the AhR agonist 2,3,7,8-tetrachlorodibenzo-p-dioxin induces BRCA-1 promoter hypermethylation and reduces BRCA-1 expression in mammary tissue of rat offspring: Preventive effects of resveratrol. Mol. Carcinog. 2015, 54, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Collins, L.L.; Lew, B.J.; Lawrence, B.P. TCDD exposure disrupts mammary epithelial cell differentiation and function. Reprod. Toxicol. 2009, 28, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Takeda, T.; Fujii, M.; Izumoto, W.; Hattori, Y.; Matsushita, T.; Yamada, H.; Ishii, Y. Gestational dioxin exposure suppresses prolactin-stimulated nursing in lactating dam rats to impair development of postnatal offspring. Biochem. Pharmacol. 2020, 178, 114106. [Google Scholar] [CrossRef] [PubMed]
- Stellman, J.M.; Stellman, S.D. Agent orange during the Vietnam war: The lingering issue of its civilian and military health impact. Am. J. Public Health 2018, 108, 726–728. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, A.; Silano, V.; Viviano, G.; Zapponi, G. Accidental release of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) at Sèveso, Italy: II. TCDD distribution in the soil surface layer. Ecotoxicol. Environ. Saf. 1980, 121, 71–84. [Google Scholar] [CrossRef]
- Revich, B.; Aksel, E.; Ushakova, T.; Ivanova, I.; Zhuchenko, N.; Klyuev, N.; Brodsky, B.; Sotskov, Y. Dioxin exposure and public health in Chapaevsk, Russia. Chemosphere 2001, 43, 951–966. [Google Scholar] [CrossRef]
- Warner, M.; Eskenazi, B.; Mocarelli, P.; Gerthoux, P.M.; Samuels, S.; Needham, L.; Patterson, D.; Brambilla, P. Serum dioxin concentrations and breast cancer risk in the Seveso Women’s Health Study. Environ. Health Perspect. 2002, 110, 625–628. [Google Scholar] [CrossRef]
- Warner, M.; Mocarelli, P.; Samuels, S.; Needham, L.; Brambilla, P.; Eskenazi, B. Dioxin exposure and cancer risk in the seveso women’s health study. Environ. Health Perspect. 2011, 119, 1700–1705. [Google Scholar] [CrossRef]
- Pesatori, A.C.; Consonni, D.; Rubagotti, M.; Grillo, P.; Bertazzi, P.A. Cancer incidence in the population exposed to dioxin after the “seveso accident”: Twenty years of follow-up. Environ. Health Glob. Access Sci. Source 2009, 8, 39–50. [Google Scholar] [CrossRef]
- Danjou, A.M.N.; Fervers, B.; Boutron-Ruault, M.C.; Philip, T.; Clavel-Chapelon, F.; Dossus, L. Estimated dietary dioxin exposure and breast cancer risk among women from the French E3N prospective cohort. Breast Cancer Res. 2015, 17, 39. [Google Scholar] [CrossRef] [PubMed]
- Danjou, A.M.N.; Coudon, T.; Praud, D.; Lévêque, E.; Faure, E.; Salizzoni, P.; le Romancer, M.; Severi, G.; Mancini, F.R.; Leffondré, K.; et al. Long-term airborne dioxin exposure and breast cancer risk in a case-control study nested within the French E3N prospective cohort. Environ. Int. 2019, 124, 236–248. [Google Scholar] [CrossRef]
- VoPham, T.; Bertrand, K.A.; Jones, R.R.; Deziel, N.C.; DuPré, N.C.; James, P.; Liu, Y.; Vieira, V.M.; Tamimi, R.M.; Hart, J.E.; et al. Dioxin exposure and breast cancer risk in a prospective cohort study. Environ. Res. 2020, 186, 109516. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Ye, Y.; Huang, F.; Chen, H.; Wu, H.; Huang, J.; Hu, J.; Xia, D.; Wu, Y. Association between dioxin and cancer incidence and mortality: A meta-analysis. Sci. Rep. 2016, 6, 38012. [Google Scholar] [CrossRef] [PubMed]
- Den Hond, E.; Roels, H.A.; Hoppenbrouwers, K.; Nawrot, T.; Thijs, L.; Vandermeulen, C.; Winneke, G.; Vanderschueren, D.; Staessen, J.A. Sexual maturation in relation to polychlorinated aromatic hydrocarbons: Sharpe and Skakkebaek’s hypothesis revisited. Environ. Health Perspect. 2002, 110, 771–776. [Google Scholar] [CrossRef]
- Leijs, M.M.; Koppe, J.G.; Olie, K.; van Aalderen, W.M.C.; de Voogt, P.; Vulsma, T.; Westra, M.; ten Tusscher, G.W. Delayed initiation of breast development in girls with higher prenatal dioxin exposure; a longitudinal cohort study. Chemosphere 2008, 73, 999–1004. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Assessment of the Health Risk of Dioxins: Re-Evaluation of the Tolerable Daily Intake (TDI); WHO: Geneva, Switzerland, 1998. [Google Scholar]
- Food and Drug Administration (FDA). Dioxins & PCBs. 2019. Available online: https://www.fda.gov/food/chemicals/dioxins-pcbs (accessed on 15 September 2020).
- European Commission. Council Directive 82/501/EEC. 1982. Available online: https://eur-lex.europa.eu/eli/dir/1982/501/oj (accessed on 15 September 2020).
- European Commission. Council Directive 2012/18/EU. 2012. Available online: https://eur-lex.europa.eu/eli/dir/2012/18/oj (accessed on 15 September 2020).
- European Commission. Commission Regulation (EU) No 1259/2011. 2011. Available online: https://eur-lex.europa.eu/eli/reg/2011/1259/oj (accessed on 19 November 2020).
- European Commission. Regulation (EU) 2015/848. 2015. Available online: https://eur-lex.europa.eu/eli/reg/2015/848/oj (accessed on 19 November 2020).
- Salian, K.; Strezov, V.; Evans, T.J.; Taylor, M.; Nelson, P.F. Application of national pollutant inventories for monitoring trends on dioxin emissions from stationary industrial sources in Australia, Canada and European Union. PLoS ONE 2019, 14, e0224328. [Google Scholar] [CrossRef]
- Zincke, T. Ueber die Einwirkung von Brom und von Chlor auf Phenole: Substitutionsproducte, Pseudobromide und Pseudochloride. Justus Liebigs Ann. Chem. 1905, 343, 75–99. [Google Scholar] [CrossRef]
- Dodds, E.C.; Lawson, W. Synthetic œstrogenic agents without the phenanthrene nucleus. Nature 1936, 137, 996. [Google Scholar] [CrossRef]
- Castan, P. Process for the manufacture of thermosetting synthetic resins by the polymerization of alkylene oxide derivatives. United States Pat. Off. 1948. [Google Scholar] [CrossRef]
- Global Industry Analysts, Inc. Bisphenol A Global Market Trajectory & Analytics. 2020. Available online: https://www.researchandmarkets.com/reports/1227819/bisphenol_a_global_market_trajectory_and#rela2-4613546 (accessed on 15 September 2020).
- Environmental Protection Agency (EPA). Risk Management for Bisphenol A. 2017. Available online: https://www.epa.gov/assessing-and-managing-chemicals-under-tsca/risk-management-bisphenol-bpa (accessed on 15 September 2020).
- Im, J.; Löffler, F.E. Fate of Bisphenol A in Terrestrial and Aquatic Environments. Environ. Sci. Technol. 2016, 50, 8403–8416. [Google Scholar] [CrossRef]
- Goodson, A.; Robin, H.; Summerfield, W.; Cooper, I. Migration of bisphenol A from can coatings—Effects of damage, storage conditions and heating. Food Addit. Contam. 2004, 21, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Stahlhut, R.W.; Welshons, W.V.; Swan, S.H. Bisphenol A data in NHANES suggest longer than expected half-life, substantial nonfood exposure, or both. Environ. Health Perspect. 2009, 117, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Calafat, A.M.; Ye, X.; Wong, L.Y.; Reidy, J.A.; Needham, L.L. Exposure of the U.S. population to Bisphenol A and 4-tertiary-octylphenol: 2003–2004. Environ. Health Perspect. 2008, 116, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Olsen, C.M.; Meussen-Elholm, E.T.M.; Samuelsen, M.; Holme, J.A.; Hongslo, J.K. Effects of the environmental oestrogens bisphenol A, tetrachlorobisphenol A, tetrabromobisphenol A, 4-hydroxybiphenyl and 4,4′-dihydroxybiphenyl on oestrogen receptor binding, cell proliferation and regulation of oestrogen sensitive proteins in the human. Pharmacol. Toxicol. 2003, 92, 180–188. [Google Scholar] [CrossRef]
- Lee, H.S.; Park, E.J.; Oh, J.H.; Moon, G.; Hwang, M.S.; Kim, S.Y.; Shin, M.K.; Koh, Y.H.; Suh, J.H.; Kang, H.S.; et al. Bisphenol A exerts estrogenic effects by modulating CDK1/2 and p38 MAP kinase activity. Biosci. Biotechnol. Biochem. 2014, 78, 1371–1375. [Google Scholar] [CrossRef]
- Zhang, X.L.; Wang, H.S.; Liu, N.; Ge, L.C. Bisphenol A stimulates the epithelial mesenchymal transition of estrogen negative breast cancer cells via FOXA1 signals. Arch. Biochem. Biophys. 2015, 585, 10–16. [Google Scholar] [CrossRef]
- Okada, H.; Tokunaga, T.; Liu, X.; Takayanagi, S.; Matsushima, A.; Shimohigashi, Y. Direct evidence revealing structural elements essential for the high binding ability of bisphenol A to human estrogen-related receptor-gamma. Environ. Health Perspect. 2008, 116, 32–38. [Google Scholar] [CrossRef]
- Song, H.; Zhang, T.; Yang, P.; Li, M.; Yang, Y.; Wang, Y.; Du, J.; Pan, K.; Zhang, K. Low doses of bisphenol A stimulate the proliferation of breast cancer cells via ERK1/2/ERRγ signals. Toxicol. Vitr. 2015, 30, 521–528. [Google Scholar] [CrossRef]
- Zhang, X.L.; Liu, N.; Weng, S.F.; Wang, H.S. Bisphenol A Increases the Migration and Invasion of Triple-Negative Breast Cancer Cells via Oestrogen-related Receptor Gamma. Basic Clin. Pharmacol. Toxicol. 2016, 119, 389–395. [Google Scholar] [CrossRef]
- Deb, P.; Bhan, A.; Hussain, I.; Ansari, K.I.; Bobzean, S.A.; Pandita, T.K.; Perrotti, L.I.; Mandal, S.S. Endocrine disrupting chemical, bisphenol-A, induces breast cancer associated gene HOXB9 expression in vitro and in vivo. Gene 2016, 590, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Terasaka, S.; Kiyama, R. Bisphenol A induces a rapid activation of Erk1/2 through GPR30 in human breast cancer cells. Environ. Pollut. 2011, 159, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, H.S.; Moon, W.K. Comparison of transcriptome expression alterations by chronic exposure to low-dose bisphenol A in different subtypes of breast cancer cells. Toxicol. Appl. Pharmacol. 2019, 385, 114814. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.Y.; Zaha, H.; Nagano, R.; Yoshinaga, J.; Yonemoto, J.; Sone, H. Xenoestrogens down-regulate aryl-hydrocarbon receptor nuclear translocator 2 mRNA expression in human breast cancer cells via an estrogen receptor alpha-dependent mechanism. Toxicol. Lett. 2011, 206, 152–157. [Google Scholar] [CrossRef]
- Kitamura, S.; Suzuki, T.; Sanoh, S.; Kohta, R.; Jinno, N.; Sugihara, K.; Yoshihara, S.; Fujimoto, N.; Watanabe, H.; Ohta, S. Comparative study of the endocrine-disrupting activity of bisphenol A and 19 related compounds. Toxicol. Sci. 2005, 84, 249–259. [Google Scholar] [CrossRef]
- Markey, C.M.; Luque, E.H.; De Toro, M.M.; Sonnenschein, C.; Soto, A.M. In utero exposure to bisphenol A alters the development and tissue organization of the mouse mammary gland. Biol. Reprod. 2001, 65, 1215–1223. [Google Scholar] [CrossRef]
- Muñoz-de-Toro, M.; Markey, C.M.; Wadia, P.R.; Luque, E.H.; Rubin, B.S.; Sonnenschein, C.; Soto, A.M. Perinatal exposure to bisphenol-A alters peripubertal mammary gland development in mice. Endocrinology 2005, 146, 4138–4147. [Google Scholar] [CrossRef]
- Vandenberg, L.N.; Maffini, M.V.; Wadia, P.R.; Sonnenschein, C.; Rubin, B.S.; Soto, A.M. Exposure to environmentally relevant doses of the xenoestrogen bisphenol-A alters development of the fetal mouse mammary gland. Endocrinology 2007, 148, 116–127. [Google Scholar] [CrossRef]
- Wadia, P.R.; Cabaton, N.J.; Borrero, M.D.; Rubin, B.S.; Sonnenschein, C.; Shioda, T.; Soto, A.M. Low-Dose BPA Exposure Alters the Mesenchymal and Epithelial Transcriptomes of the Mouse Fetal Mammary Gland. PLoS ONE 2013, 8, e63902. [Google Scholar] [CrossRef]
- Durando, M.; Kass, L.; Piva, J.; Sonnenschein, C.; Soto, A.M.; Luque, E.H.; Muñoz-De-Toro, M. Prenatal bisphenol a exposure induces preneoplastic lesions in the mammary gland in wistar rats. Environ. Health Perspect. 2006, 115, 80–86. [Google Scholar] [CrossRef]
- Murray, T.J.; Maffini, M.V.; Ucci, A.A.; Sonnenschein, C.; Soto, A.M. Induction of mammary gland ductal hyperplasias and carcinoma in situ following fetal bisphenol A exposure. Reprod. Toxicol. 2007, 23, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Dhimolea, E.; Wadia, P.R.; Murray, T.J.; Settles, M.L.; Treitman, J.D.; Sonnenschein, C.; Shioda, T.; Soto, A.M. Prenatal exposure to BPA alters the epigenome of the rat mammary gland and increases the propensity to neoplastic development. PLoS ONE 2014, 9, e99800. [Google Scholar] [CrossRef] [PubMed]
- Tharp, A.P.; Maffini, M.V.; Hunt, P.A.; VandeVoort, C.A.; Sonnenschein, C.; Soto, A.M. Bisphenol A alters the development of the rhesus monkey mammary gland. Proc. Natl. Acad. Sci. USA 2012, 109, 8190–8195. [Google Scholar] [CrossRef] [PubMed]
- Corrales, J.; Kristofco, L.A.; Baylor Steele, W.; Yates, B.S.; Breed, C.S.; Spencer Williams, E.; Brooks, B.W. Global assessment of bisphenol a in the environment: Review and analysis of its occurrence and bioaccumulation. Dose-Response 2015, 13, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.E.; Kendig, E.L.; Belcher, S.M. Assessment of bisphenol A released from reusable plastic, aluminium and stainless steel water bottles. Chemosphere 2011, 85, 943–947. [Google Scholar] [CrossRef] [PubMed]
- Biedermann, S.; Tschudin, P.; Grob, K. Transfer of bisphenol A from thermal printer paper to the skin. Anal. Bioanal. Chem. 2010, 398, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Lang, I.A.; Galloway, T.S.; Scarlett, A.; Henley, W.E.; Depledge, M.; Wallace, R.B.; Melzer, D. Association of Urinary Bisphenol A concentration with medical disorders and laboratory abnormalities in adults. J. Am. Med. Assoc. 2008, 300, 1303–1310. [Google Scholar] [CrossRef]
- Trabert, B.; Falk, R.T.; Figueroa, J.D.; Graubard, B.I.; Garcia-Closas, M.; Lissowska, J.; Peplonska, B.; Fox, S.D.; Brinton, L.A. Urinary bisphenol A-glucuronide and postmenopausal breast cancer in Poland. Cancer Causes Control 2014, 25, 1587–1593. [Google Scholar] [CrossRef]
- Yang, M.; Ryu, J.H.; Jeon, R.; Kang, D.; Yoo, K.Y. Effects of bisphenol A on breast cancer and its risk factors. Arch. Toxicol. 2009, 83, 281–285. [Google Scholar] [CrossRef]
- Sprague, B.L.; Trentham-Dietz, A.; Hedman, C.J.; Wang, J.; Hemming, J.D.C.; Hampton, J.M.; Buist, D.S.; Bowles, E.J.A.; Sisney, G.S.; Burnside, E.S. Circulating serum xenoestrogens and mammographic breast density. Breast Cancer Res. 2013, 15, R45–R53. [Google Scholar] [CrossRef]
- Binder, A.M.; Corvalan, C.; Pereira, A.; Calafat, A.M.; Ye, X.; Shepherd, J.; Michels, K.B. Prepubertal and Pubertal Endocrine-Disrupting Chemical Exposure and Breast Density among Chilean Adolescents. Cancer Epidemiol. Biomark. Prev. 2018, 27, 1491–1499. [Google Scholar] [CrossRef] [PubMed]
- Vom Saal, F.S.; Hughes, C. An extensive new literature concerning low-dose effects of bisphenol A shows the need for a new risk assessment. Environ. Health Perspect. 2005, 113, 926–933. [Google Scholar] [CrossRef] [PubMed]
- European Food Safety Authority (EFSA). BPA Update: Working Group to Start Reviewing New Studies. 2018. Available online: https://www.efsa.europa.eu/en/press/news/180904 (accessed on 15 September 2020).
- European General Court (EGC). PlasticsEurope c/ECHA, Case T-636/17. 2019. Available online: http://curia.europa.eu/juris/document/document.jsf?docid=217994&text=&dir=&doclang=EN&part=1&occ=first&mode=DOC&pageIndex=0&cid=2178886 (accessed on 15 September 2020).
- Food and Drug Administration (FDA). Bisphenol A. 2018. Available online: https://www.fda.gov/food/food-additives-petitions/bisphenol-bpa (accessed on 15 September 2020).
- Mesnage, R.; Phedonos, A.; Arno, M.; Balu, S.; Corton, J.C.; Antoniou, M.N. Transcriptome profiling reveals bisphenol a alternatives activate estrogen receptor alpha in human breast cancer cells. Toxicol. Sci. 2017, 158, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Fenton, S.E.; Reed, C.; Newbold, R.R. Perinatal environmental exposures affect mammary development, function, and cancer risk in adulthood. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 455–479. [Google Scholar] [CrossRef]
- Environmental Protection Agency (EPA). U.S. Environmental Protection Agency Endocrine Disruptor Screening Program Comprehensive Management Plan. 2012. Available online: https://www.epa.gov/sites/production/files/2015-08/documents/edsp-comprehensive-management-plan-2012.pdf (accessed on 15 September 2020).
- Gore, A.C.; Chappell, V.A.; Fenton, S.E.; Flaws, J.A.; Nadal, A.; Prins, G.S.; Toppari, J.; Zoeller, R.T. Executive Summary to EDC-2: The Endocrine Society’s second Scientific Statement on endocrine-disrupting chemicals. Endocr. Rev. 2015, 36, 593–602. [Google Scholar] [CrossRef]
- Kortenkamp, A.; Demeneix, B.; Slama, R.; Bard, E.; Bergman, A.; Ehrlich, P.R.; Grandjean, P.; Mann, M.; Myers, J.P.; Oreskes, N.; et al. Let’s stop the Manipulation of Science. Le Monde. Available online: https://www.lemonde.fr/idees/article/2016/11/29/let-s-stop-the-manipulation-of-science_5039867_3232.html (accessed on 29 November 2016).


{kind=link}
| Risk | Example | Impact | Refs |
|---|---|---|---|
| Reproductive factors | Age at menarche | BC risk decreases by 5% for each year without menstruation between 11 and 17 years of age | [29] |
| Age at menopause | BC risk decreases by 3% for each year without being menopausal between 35 and 55 years of age | ||
| Age at first birth | BC risk increases by 3% before menopause and 5% after menopause for each year that first full-term pregnancy is delayed | [30] | |
| Parity | Each full-term pregnancy decreases BC risk by 3% before menopause and 12% after menopause | ||
| Breastfeeding | Breastfeeding decreases BC risk by 14% before menopause and 11% after menopause | [31] | |
| Exogenous hormones | Combined hormonal replacement therapy (HRT) | BC risk increases by 60% for 1 to 4 years of use and by 108% for more than 5 years of combined HRT use | [10] |
| Hormonal contraception | BC risk increases by 0.7% for each year of contraceptive use | [11] | |
| Anthropometric factors | Body mass index (BMI) | The risk of postmenopausal BC increases by 40% for every 10-point increase in BMI | [32] |
| Sex and age | Sex | Less than 1% of BC develop in men | [33] |
| Age | More than 70% of BC are diagnosed after 50 years of age | [2] | |
| Breast density and personal history of BC | Breast density | A 5% increase in breast density increases BC risk by 5 to 10% | [34] |
| Personal history | Surviving BC increases the risk of developing second primary BC by 74% | [35] | |
| Familial history of BC | First-degree family history | One history of BC increases the risk by 77% Two or more histories of BC increase the risk by 250% | [36] |
| Breast cancer type 1 susceptibility protein mutation | 55% risk of developing BC after 70 years of age | [37] | |
| Breast cancer type 2 susceptibility protein mutation | 47% risk of developing BC after 70 years of age | ||
| Lifestyle | Diet | Consumption of 120 g per day of red meat increases BC risk by 11% | [38] |
| Tobacco | BC risk increases by 0.5% for each year of smoking | [39] | |
| Alcohol | Every unit of alcohol (10 g of alcohol) drunk per day increases BC risk by 7% | [40] | |
| Physical activity | BC risk decreases by 18% with the practice of 1 to 3 h of physical activity per week and 21% for more than 7 h per week | [41] | |
| Occupation | Night shift work | 20 years or more of rotating nightshift work at baseline induce a 2-fold increase in BC risk 20 years or more of cumulative rotating night-shift work increases BC risk by 40% | [42] |
| Exposure to radiation | Hodgkin lymphoma radiation | 29% risk of developing BC after 55 years of age for women who received chest radiation before 25 years of age | [43] |
| Author (Year) | Study Years | Country | Design | Cases/Controls | Exposure Assessment | Results |
|---|---|---|---|---|---|---|
| Bibbo (1978) [108] | 1976–1977 | USA | Prospective | 693/668 | Participants in the 1951 clinical study | No significant increase in BC risk in DES mothers |
| Greenberg (1984) [109] | 1981 | USA | Prospective | 2885/2816 | Obstetric records | Significant increase in BC risk for DES mothers exposed more than 30 years prior the study (RR = 2.5; 95% CI: 1.1–5.8) |
| Colton (1993) [110] | 1986–1989 | USA | Prospective | 2590/2471 | Obstetric records | Significant increase in BC risk for DES mothers after 60 years of age (RR = 1.47; 95% CI: 1.02–2.13) |
| Titus-Ernstoff (2001) [111] | 1992–1994 | USA | Prospective | 2434/2402 | Obstetric records | Significant increase in BC risk for DES mothers exposed less than 40 years prior the study (RR = 1.27; 95% CI: 1.07–1.52) |
| Hatch (1998) [112] | 1978–1994 | USA | Prospective | 3650/1202 | Obstetric records | No significant increase in BC risk in DES daughters (RR = 1.18; 95% CI: 0.56–2.49) |
| Palmer (2006) [113] | 1978–2003 | USA | Prospective | 3812/1637 | Obstetric records | DES daughters have a significantly increased BC risk after 40 years of age (RR = 1.91; 95% CI: 1.09–3.33) and after 50 years of age (RR = 3.00; 95% CI: 1.01–8.98) |
| Troisi (2007) [62] | 1978–2001 | USA | Prospective | 3813/1642 | Obstetric records | DES daughters have a significantly increased BC risk after 40 years of age (RR = 1.83; 95% CI: 1.1–3.2) |
| Hoover (2011) [114] | 1975–2001 | USA | Prospective | 3796/1659 | Obstetric records | DES daughters have a significantly increased BC risk after 40 years of age (HR = 1.82; 95% CI: 1.04–3.18) |
| Troisi (2019) [115] | 1994–2011 | USA | Prospective | 4822/2083 | Obstetric records | DES daughters have a significantly increased BC risk between 40 and 49 years of age (RR = 1.33; 95% CI: 1.05–1.66) |
| Tournaire (2015) [116] | 2013 | France | Prospective | 3436/3256 | Self-report or medical records | DES daughters have a significantly increased BC risk (RR = 2.10; 95% CI: 1.60–2.76) but risk varies with low (RR = 1.63; 95% CI: 0.87–3.08) or high (RR = 2.16; 95% CI: 1.18–3.96) DES dose |
| Verloop (2010) [117] | 1992–2008 | Netherlands | Prospective | 12,091 participants | Self-report or medical records | No significantly increase in BC risk in DES daughters (RR = 1.05; 95% CI: 0.90–1.23) |
| Titus (2019) [118] | 2001–2012 | USA | Prospective | 796/469 | Obstetric records | DES granddaughters have genital malformations and other health problems similar to those of DES daughters |
| Author (Year) | Study Years | Country | Design | Cases/Controls | Exposure Assessment | Results |
|---|---|---|---|---|---|---|
| Cohn (2007) [143] | 2000–2001 | USA | Prospective | 129/129 | Serum (1959–1967) | High DDT serum concentrations are associated with a significant increase in BC risk in women born after 1931 (OR = 5.4; 95% CI: 1.7–17.1) |
| Cohn (2015) [144] | 2010–2013 | USA | Prospective | 103/315 | Serum (1959–1967) | High DDT serum concentrations in mothers are associated with a significant increase in BC risk (OR = 3.7; 95% CI: 1.5–9.0); advanced stage at diagnosis (OR = 4.6; 95% CI: 1.3–16.5); and Human Epidermal Growth Factor Receptor 2 + (HER2+) tumors in daughters (OR = 2.1; 95% CI: 1.0–4.8) |
| Cohn (2019) [145] | 1970–2010 | USA | Prospective | 146/422 | Serum (1959–1967) | Exposure to DDT after 4 years of age significantly increases the risk of BC diagnosed before the age of 54 (OR = 3.70; 95% CI: 1.22–11.26) |
| White (2013) [146] | NA | USA | Retrospective | 1508/1556 | Residential exposure by questionnaire | Women with hormone-dependent BC have a significantly greater risk of having ever seen spreaders (OR = 1.44; 95% CI: 1.08–1.93) Women with Estrogen Receptor + (ER+) or Progesterone Receptor + (PR+) BC have a significantly increased odds of ever seeing a fogger truck (OR = 1.33; 95% CI: 1.11–1.59) |
| Niehoff (2016) [147] | 2003–2009 | USA + Puerto Rico | Prospective | 2134 participants | Residential exposure by questionnaire | No significant association between having ever seen a spreader before DDT ban and BC risk (HR = 1.3; 95% CI: 0.92–1.7) |
| Bachelet (2019) [148] | NA | France | Retrospective | 695/1055 | Serum (2005–2007) | No significant association between high DDE serum concentrations and BC risk (OR = 0.93; 95% CI: 0.73–1.18) |
| Itoh (2009) [149] | NA | Japan | Retrospective | 403/403 | Serum (2001–2005) | No significant association between high DDT serum concentrations and BC risk (OR = 0.58; 95% CI: 0.27–1.25) |
| Ingber (2013) [150] | 2012 | Multi-centric | Meta-analysis | 40 DDT or DDE studies | Serum | No significant association between BC risk and high serum concentrations of DDT (OR = 1.02; 95% CI: 0.92–1.13) or DDE (OR = 1.04; 95% CI: 0.94–1.15) |
| Park (2014) [142] | 2012 | Multi-centric | Meta-analysis | 35 DDE studies | Serum | No significant association between high DDE serum concentrations and BC risk (OR = 1.03; 95% CI: 0.95–1.12) |
| Author (Year) | Study Years | Country | Design | Cases/Controls | Exposure Assessment | Results |
|---|---|---|---|---|---|---|
| Warner (2002) [190] | 1996–1998 | Italy | Prospective | 981 participants | Serum (1976–1981) | A 10-fold increase in TCDD plasma concentrations was associated with an increase in BC risk (HR = 2.1; 95% CI: 1.0–4.6) |
| Warner (2011) [191] | 1996–2008 | Italy | Prospective | 833 participants | Serum (1976–1981) | No association between high TCDD serum concentrations and BC risk (HR = 1.44; 95% CI: 0.89–2.33) |
| Pesatori (2009) [192] | 2006-2009- | Italy | Prospective | 2122 participants | Medical records (1992–1996) | Living near the chemical plant during the accident significantly increases BC risk (RR = 2.57; 95% CI: 1.07–6.20) |
| Revich (2001) [189] | 1997–1998 | Russia | Prospective | 14 participants | Human milk and serum (1997–1998) | BC incidence and mortality are doubled in Chapayevsk compared to the national average |
| Danjou (2015) [193] | 1993–2008 | France | Prospective | 63,830 participants | Dietary exposure | No significant association between higher dietary dioxin exposure and BC risk (HR = 1.00; 95% CI: 0.96–1.05) |
| Danjou (2019) [194] | 1993–2008 | France | Prospective | 429/716 | Airborne exposure | No significant association between higher estimated airborne dioxin exposure and BC risk (OR = 1.124; 95% CI: 0.693–1.824) |
| VoPham (2020) [195] | 1989–2013 | USA | Prospective | 112,397 participants | Airborne exposure | Living less than 10 km from a municipal solid waste incinerator significantly increases BC risk (HR = 1.15; 95% CI: 1.03–1.28) The risk increases again by living less than 5 km away (HR = 1.25; 95% CI: 1.04–1.52) |
| Xu (206) [196] | 2015 | Multi-centric | Meta-analysis | 3 studies | Various | No significant association between higher TCDD exposure and BC risk (RR = 0.99; 95% CI: 0.93–1.06) |
| Author (Year) | Study Years | Country | Design | Cases/Controls | Exposure Assessment | Results |
|---|---|---|---|---|---|---|
| Lang (2008) [237] | 2003–2004 | USA | Retrospective | 1455 participants | Urine (2003–2004) | No significant association between high urinary BPA levels and cancer risk (including BC) (OR = 1.12; 95% CI: 0.85–1.48) |
| Trabert (2014) [238] | 2000–2003 | Poland | Retrospective | 575/575 | Urine (2000–2003) | No significant association between high urinary BPA levels and postmenopausal BC risk (OR = 1.09; 95% CI: 0.73–1.63) |
| Yang (2009) [239] | 2004–2007 | Korea | Prospective | 70/82 | Serum (1994–1997) | Significant association between high serum BPA levels and nulliparity (p < 0.05) No significant association between BPA levels and BC risk (p = 0.42) |
| Sprague (2013) [240] | 2008–2009 | USA | Retrospective | 264 participants | Serum (2008–2009) | Significant association between high serum BPA levels and high breast density (p = 0.01) |
| Binder (2018) [241] | 2006 | Chile | Prospective | 200 participants | Urine (2006) | Significant association between lower and higher urine BPA levels and high breast density (p < 0.01) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eve, L.; Fervers, B.; Le Romancer, M.; Etienne-Selloum, N. Exposure to Endocrine Disrupting Chemicals and Risk of Breast Cancer. Int. J. Mol. Sci. 2020, 21, 9139. https://doi.org/10.3390/ijms21239139
Eve L, Fervers B, Le Romancer M, Etienne-Selloum N. Exposure to Endocrine Disrupting Chemicals and Risk of Breast Cancer. International Journal of Molecular Sciences. 2020; 21(23):9139. https://doi.org/10.3390/ijms21239139
Chicago/Turabian StyleEve, Louisane, Béatrice Fervers, Muriel Le Romancer, and Nelly Etienne-Selloum. 2020. "Exposure to Endocrine Disrupting Chemicals and Risk of Breast Cancer" International Journal of Molecular Sciences 21, no. 23: 9139. https://doi.org/10.3390/ijms21239139
APA StyleEve, L., Fervers, B., Le Romancer, M., & Etienne-Selloum, N. (2020). Exposure to Endocrine Disrupting Chemicals and Risk of Breast Cancer. International Journal of Molecular Sciences, 21(23), 9139. https://doi.org/10.3390/ijms21239139