Abstract
Focal adhesion kinase (FAK) and Wnt signaling pathways are important contributors to tumorigenesis in several cancers. While most results come from studies investigating these pathways individually, there is increasing evidence of a functional crosstalk between both signaling pathways during development and tumor progression. A number of FAK–Wnt interactions are described, suggesting an intricate, context-specific, and cell type-dependent relationship. During development for instance, FAK acts mainly upstream of Wnt signaling; and although in intestinal homeostasis and mucosal regeneration Wnt seems to function upstream of FAK signaling, FAK activates the Wnt/β-catenin signaling pathway during APC-driven intestinal tumorigenesis. In breast, lung, and pancreatic cancers, FAK is reported to modulate the Wnt signaling pathway, while in prostate cancer, FAK is downstream of Wnt. In malignant mesothelioma, FAK and Wnt show an antagonistic relationship: Inhibiting FAK signaling activates the Wnt pathway and vice versa. As the identification of effective Wnt inhibitors to translate in the clinical setting remains an outstanding challenge, further understanding of the functional interaction between Wnt and FAK could reveal new therapeutic opportunities and approaches greatly needed in clinical oncology. In this review, we summarize some of the most relevant interactions between FAK and Wnt in different cancers, address the current landscape of Wnt- and FAK-targeted therapies in different clinical trials, and discuss the rationale for targeting the FAK–Wnt crosstalk, along with the possible translational implications.
Keywords:
FAK; Wnt; cell signaling; cancer; malignant mesothelioma; clinical trials; combinatorial therapy 1. Introduction
Focal adhesion kinase (FAK) is a key regulator of growth factor receptor and integrin-mediated signaling that coordinates multiple fundamental processes such as migration, invasion, angiogenesis, cell survival, or epithelial–mesenchymal transition (EMT), in both normal and cancer cells. Due to this prominent role in cancer, a vast number of preclinical and clinical studies have explored different strategies to block FAK signaling for therapeutic purposes. Molecular and cellular mechanisms of FAK action on both tumor and stromal cell biology, regulation of its activity, and FAK-targeted anticancer therapeutic strategies that are, nowadays, under preclinical and clinical development have been extensively reviewed in different excellent review articles [1,2,3,4]. However, the complex crosstalk and regulatory loops modulated by FAK, in particular concerning the Wnt signaling pathway, have not been explored and reviewed in detail.
Wnt signaling it is one of the most fundamental and conserved regulatory systems in animals. This pathway controls many critical aspects of development and adult tissue homeostasis by regulating cell proliferation, differentiation, migration, genetic stability, and apoptosis, as well as by maintaining adult stem cells in a pluripotent state. Dysregulation of this pathway is associated with a broad spectrum of human diseases, ranging from neurodegenerative disorders to fibrosis, osteoporosis, and cancer. Consequently, Wnt-related research has become a major area for targeted drug discovery and therapeutic development [5,6]. For a long time, the Wnt pathway was considered undruggable, due its complexity and multiple sub-branches. However, more recently, new emerging insights into the pathway mechanisms and crosstalk with other major pathways has led to the development of multiple inhibitory compounds targeting this pathway at different levels. Several Wnt-signaling inhibitors are under early clinical testing with promising outcomes; however, no Wnt-specific drugs have been approved for clinical usage so far [7].
A crosstalk between pathways occurs when the combinatorial signal from both pathways produce a different response than the one triggered by each individual pathway [8]. It must be considered, however, that perturbations to a signaling network, such as the inhibition of a small molecule or pathogenic mutations in one pathway, may affect different tissues or cell types in different ways, highlighting the importance of the tissue-specific context [9]. Feedback loops and crosstalk between signaling pathways can significantly impact the efficacy of cancer therapeutics and cause resistance to targeted agents, thereby representing a major barrier to effective treatments [10]. Understanding the existing interactions between pathways represents a major challenge but may ultimately contribute to identify new, yet unexplored, therapeutic options. In this review we summarize recent findings and elaborate general considerations of the FAK and Wnt signaling pathways and current insights of the crosstalk existing between both pathways with emphasis on their impact on development and most importantly in cancer. We also provide an overview of current strategies and clinical trials that aim to antagonize FAK and Wnt signaling in cancer and the major challenges that are associated with such approaches. Furthermore, we analyze the potential therapeutic benefits of a combinatorial therapy targeting the crosstalk FAK–Wnt.
2. The Focal Adhesion Kinase (FAK) Signaling Pathway
FAK is a non-receptor protein tyrosine kinase that mediates growth factor- and adhesion-dependent signaling through several downstream pathways leading to cell migration, invasion, cell cycle progression, and survival [11,12,13], during both development and malignancy. The human gene encoding FAK, named PTK2, is located on chromosome 8q24.3 [14] and is highly conserved with over 90% amino acid sequence identity across different species, including human, mouse, chicken, and Xenopus [15,16,17,18]. Its structure is composed of four major domains: (1) a central kinase domain, (2) flanked by an N-terminal four-point-one ezrin–radixin–moesin (FERM) domain, (3) proline-rich regions, and (4) a focal adhesion targeting (FAT) C-terminal domain [3]. The FAK FERM domain represents a critical element in FAK activation. It has a scaffolding role, mediating protein–protein and protein–lipid interactions, and regulates the subcellular localization of FAK [2]. The kinase domain of FAK is localized in the middle of the molecule and contains the activation loop and important tyrosine (Y) residues, such as Y397, Y576, and Y577. FAK tyrosine Y397 serves as the major site of autophosphorylation [3,19], as well as the binding site for various interacting partners, including Src family kinases (SFKs) and the p85 subunit of phosphatidylinositol 3 kinase (PI3-K) [20,21]. Activated FAK–Src promotes cell motility, cell cycle progression, and cell survival, leading to tumor growth and metastasis [22]. For instance, Src–FAK signaling promotes E-cadherin internalization during cancer progression, thus facilitating EMT and enhancing tumor cell motility [23].
Although FAK itself has not been demonstrated to be an oncogene, FAK overexpression and activation have been reported in tumors of broad tissue origin [12], especially in invasive and metastatic tumors [24], including thyroid [25], prostate [26], colorectal [27], and ovary [28] cancers, as well as in malignant mesothelioma [29]. In addition, several studies have associated FAK overexpression with poorer clinical prognosis [30]. However, the exact molecular mechanisms responsible for the increase in FAK expression are not fully deciphered. One of the proposed mechanisms of FAK overexpression is via FAK amplification; although increased FAK expression also occurs independently of FAK gene amplification [31], indicating that transcriptional and/or post-transcriptional mechanisms may also play a role.
FAK signaling is activated by several different mechanisms that include interaction with integrins, growth factor receptors, G protein–coupled receptors, or cytokine receptors [32,33]. The most frequently described mechanism involves engagement of integrins with the extracellular matrix and the subsequent association of proteins, such as talin and paxillin, with the cytoplasmic tail of integrin β subunits [34]. This leads to the recruitment of FAK to sites of integrin clustering via interactions with integrin-associated proteins, leading to FAK activation [3]. Its activity is mediated through kinase-dependent and kinase-independent (scaffolding) mechanisms [1]. FAK kinase-dependent functions are often associated with integrin-related signaling at focal adhesions, where FAK plays an important role in cellular migration and adhesion in both normal and cancer cells [35,36]. FAK also functions as a scaffold protein and participates in protein–protein interactions through its kinase-independent functions. Interestingly, FAK is also able to translocate to the nucleus, where it interacts directly with p53 to promote cell proliferation and survival through p53 degradation [11,37] in a kinase-independent manner. In addition, nuclear FAK has been described to control other transcriptional networks such as the inflammatory signaling pathway, immune escape, and angiogenesis; however, the mechanisms regulating FAK in the nucleus remains unclear [38].
3. The Wnt Signaling Pathway
The Wnt signaling pathway is an ancient and highly conserved pathway that regulates crucial aspects of cell fate determination, cell migration, cell polarity, neural patterning, and organogenesis during embryonic development [39]. It has also determinant roles in tissue regeneration and adult homeostasis. Given its importance it is not surprising that mutations in the Wnt pathway are frequently observed in cancer, most notably in tissues that normally depend on Wnt for high self-renewal or repair [40].
The Wnt family consist of 19 secreted glycoproteins that transduce signals by binding to frizzled (Fzd) receptor complexes [41]. Based on the dependence of its key mediator β-catenin, the pathway is in turn subdivided in canonical (β-catenin-dependent) and non-canonical (β-catenin-independent) signaling pathways [42]. The canonical Wnt signaling pathway is driven by β-catenin, a scaffold protein, linking the cytoplasmic tail of classical cadherins via α-catenin to the actin cytoskeleton. In the absence of Wnt stimulation, cytoplasmic β-catenin levels are maintained low by proteasome-mediated degradation controlled by a multiprotein complex consisting of Axin, Adenomatous polyposis coli (APC), Casein kinase1a (CK1a), and Glycogen synthase kinase 3-β (GSK3-β) that bind to β-catenin [43]. The non-canonical Wnt pathways include the planar cell polarity (PCP) and the Wnt/Ca2+ pathways. In the Wnt/Ca2+ pathway, Wnt-Fzd binding activates phospholipase C (PLC) via G proteins, leading to diacylglycerol (DAG) production and an increase of intracellular Ca2+ levels, subsequently activating downstream effectors that include protein kinase C (PKC) [44]. The increased intracellular Ca2+ levels also activate calcineurin and calmodulin-dependent protein kinase II (CaMKII) [45]. The PCP pathway, also referred as the Wnt/Jun-N-terminal kinase (JNK) pathway, activates small GTPases, including RhoA, Rac, Cdc42, and JNK [46]. This pathway controls cell polarity, cytoskeletal remodeling, directional cell migration and c-Jun-dependent transcription. As these two pathways are involved in cytoskeletal changes and cell migration, their activation has been associated with cancer cell invasion and metastasis [47,48]. In addition to mutational activation, the Wnt pathway can be aberrantly activated by overexpression of its components, such as Wnts or their Fzd receptors [49].
4. FAK–Wnt Pathways Crosstalk in Development
FAK is crucial during development, tissue regeneration and wound healing. Deletion of FAK during mouse development results in lethality at embryonic day 8.5 and a block in cell proliferation. FAK activity is also essential for developmental processes controlling blood vessel formation, cell motility and polarity [32]. FAK-deficient embryos display a general defect of mesoderm development and cells from these embryos have a reduced mobility in vitro [50], while FAK-null embryos present impairment in angiogenesis and defects in the proper development of the heart and blood vessels [51]. In neurons, FAK functions downstream of netrins to promote neurite outgrowth and axon guidance [52]. Several studies have also described FAK playing a role in the development of the placenta, as well as in the musculoskeletal, genitourinary, and respiratory organ systems [53].
Beyond development, Wnt signaling exerts crucial roles in adult tissues and organs, in daily processes of tissue homeostasis and regeneration, in organ repair after injury, and in adult neurogenesis [54]. The best-characterized function of Wnt signaling in adult tissues is the maintenance of stem cell homeostasis, where Wnt ligands promote proliferation and self-renewal of tissue-specific stem cells [55] in the intestine, stomach, skin, liver, and the mammary gland [54].
There are many described interactions between FAK and Wnt during embryonic development (reviewed in [56]). FAK and Wnt are involved in controlling apical cell morphogenesis in Drosophila ovarian morphogenesis [57] and in regulating early patterning in the nervous system of Xenopus laevis [58], where FAK regulates Wnt3a gene expression to control cell fate specification in the developing neural plate. Both pathways have been also shown to be implicated in bone remodeling; FAK promotes osteoblast progenitor cell proliferation and differentiation by enhancing Wnt signaling [59]. In addition, FAK was shown to play a pivotal role in promoting BMP9-induced osteogenesis of synovial mesenchymal stem cells via the activation of Wnt and MAPK pathways [60], while another study demonstrated that FAK and BMP-9 synergistically trigger osteogenic differentiation and bone formation of adipose tissue-derived stem cells by enhancing Wnt-β-catenin signaling [61]. FAK and Wnt signaling are also involved in maintaining normal intestinal homeostasis and promoting mucosal regeneration following DNA damage, with FAK required downstream of Wnt signaling for Akt/mTOR activation [62]. More recently it was found that both, the Stat3 pathway and Wnt signaling cooperatively regulate the survival of the epithelial cells in the damaged mucosa and isolated crypts through activation of integrin/FAK signaling [63]. FAK also plays a role in the control of the epidermal stem cells via a mechanism that involves crosstalk with the Wnt/β-catenin pathway [64].
5. FAK–Wnt Pathways Crosstalk in Cancer
Given Wnt’s essential role in embryonic development, tissue homoeostasis, and stem cell biology, this pathway must be tightly regulated; its dysregulation has been associated with many types of cancer. “No man is an island”, and similarly no pathway is modulated without affecting others [5]. Understanding how FAK regulates Wnt transcription and pathway activation during development, and more importantly, during cancer progression, could offer new potential opportunities for cancer treatment [56].
5.1. Colorectal and Intestinal Cancers
Colorectal cancer (CRC) is the second leading cause of cancer morbidity and mortality worldwide [65]. Genetic alterations in Wnt signaling occur in over 90% of human sporadic CRC, among which inactivation of the tumor suppressor adenomatous polyposis coli (APC) occur in 85% of the cases, while activating mutations of the proto-oncogene β-catenin (CTNNB1) occur only in 5% of the cases [66]. Activating mutations of BRAF (mostly V600E mutation), a serine/threonine kinase downstream of KRAS and upstream of MEK (mitogen-activated protein kinase kinase) have been reported to occur in approximately 10% of all CRC patients [67]. While Wnt signaling has been shown to function as tumor suppressor in other cancers [68], activation of this pathway seems a critical step in the tumor initiation and development of CRC [69].
In the absence of Wnt, the canonical function of APC is to form a “destruction complex” with Axin/Axin2, CK1-α, and GSK-3β promoting the sequestration, ubiquitination, and subsequent proteasomal degradation of β-catenin [70]. APC is an essential component of the cytoplasmic protein complex that targets β-catenin for destruction [71]. The loss of functional APC results in less efficient GSK3 (GSK3α and GSK3β)-mediated phosphorylation of β-catenin, leading to reduced degradation of β-catenin, thus mimicking Wnt stimulation. β-catenin then accumulates and enters the nucleus to interact with the transcription factors TCF (T-cell factor)/LEF (lymphoid enhancing factor) to activate the transcription of Wnt target genes [72], including cyclin D1 and c-myc, among others. The tumorigenic consequences of dysregulated β-catenin activity are the stimulation of cellular growth and proliferation, and the disruption of differentiation programs [70].
As mentioned above, GSK3β is a central player in the canonical pathway; it operates by regulating the phosphorylation and degradation of β-catenin, and its activity is tightly controlled [73]. Gao et al. recently reported a novel FAK–Wnt regulation axis: FAK and PYK2 were found to be elevated in adenomas in APCmin/+ mice and in human CRC tissues, in addition they promoted Wnt/β-catenin pathway activation by phosphorylating GSK3β leading to β-catenin accumulation, which in turn initiates and supports intestinal tumorigenesis. Pharmacological inhibition of FAK/PYK2 repressed adenoma formation in APCmin/+ mice and reduced intestinal levels of phospho-GSK3β and β-catenin, indicating that the FAK/PYK2/GSK3β axis is critical in APC-driven intestinal tumorigenesis [74]. Altogether this suggests that drugs inhibiting simultaneously FAK and PYK2 may be an effective treatment for CRC. In line with this, recent studies showed that BRAF inhibitors upregulate the Wnt/β-catenin pathway in BRAFV600E-mutant CRC cell lines through FAK activation-mediated phosphorylation of GSK3. Co-targeting of BRAF/Wnt pathways or BRAF/FAK pathways exerted strong synergistic antitumor effects in cell culture and mouse xenograft models [75]. Given that Wnt/β-catenin pathway activation is one of the causes of CRC resistance to BRAF inhibition, co-targeting the Wnt/β-catenin pathway using FAK inhibitors may represent a new feasible solution to overcome resistance to BRAF inhibitors with great potential to change the landscape for patients with BRAF mutations [76].
Interestingly, FAK inhibition with the small molecule inhibitor Y15 increased DKK1, a known inhibitor of the Wnt pathway that plays an important role in CSC regulation in the metastatic CRC cell line, SW620. Y15 also downregulated Wnt pathway genes, such as LRP5 and FZD2, and upregulated SFRP5, one of several members of the SFRP family that controls Wnt pathway signaling, specifically by preventing it from binding to its receptor [77].
Another link between Wnt and FAK signaling is the finding that the expression of HEF1 (human enhancer of filamentation 1), implicated in progression of CRC, was also shown to modulate both pathways. HEF1 localizes to focal adhesions to coordinate FAK and Src signaling cascades in integrin-dependent adhesion, migration, invasion, and survival [78,79]. In a study evaluating the role of HEF1 in CRC tumorigenesis, HEF1 was identified as a novel Wnt signaling target and classified as a biomarker for tumor aggressiveness; overexpression of HEF1 in CRC cells enhanced cell migration and invasion. In addition, the expression of HEF1 was upregulated by Wnt-3a, β-catenin, and Dvl2 in a dose-dependent manner, and it was suppressed following β-catenin downregulation by shRNA [80].
In conclusion, there is an unequivocal evidence that FAK and Wnt pathways play a role in regulating CRC initiation and progression. These findings suggest that pharmacological inhibition of FAK might be effective in the treatment of CRC [81,82].
5.2. Malignant Mesothelioma and Lung Cancer
An interesting correlation between FAK and Wnt signaling was found in malignant mesothelioma (MM), an aggressive neoplasm that develops from the mesothelial cells lining the pleural, peritoneal, and pericardial cavities [83]. Treatment with the a FAK inhibitor in different MM cell lines strongly activated the Wnt signaling pathway; more specifically, it increased p-JNK T183/Y185 and total JNK levels. Conversely, Wnt inhibition in the same cells led to FAK activation, increasing p-FAK Y397 and total FAK levels; indicating an antagonistic regulation of these two pathways [84]. Simultaneously blocking FAK and Wnt signaling drastically reduced cell proliferation and survival of MM cell lines. Both pathways were already described to independently play a role in MM by promoting different tumorigenic properties; dysregulated Wnt signaling was implicated in invasion and resistance to apoptosis [85,86], while FAK signaling was shown to promote invasion and EMT [29].
A relation between FAK and Wnt signaling was also found in a study evaluating the function and mechanism of FAK in regulating the inflammatory response in the A549 cell line, a model for non-small cell lung cancer (NSCLC). The inhibition of FAK decreased the activation of the Wnt and NF-κB signaling pathways, accompanied by a reduction in inflammatory activity [87]. In another study using the same cell line, FAK was shown to act upstream the Wnt/β-catenin pathway. When A549 cells were treated with Maclurin, a natural organic compound which can be extracted from Morus alba, the c-Src/FAK and ERK pathways were inactivated through the reduction of ROS levels. This in turn activated GSK3β, leading to the inactivation of the β-catenin transcription factor. As a result, migration and invasion of A549 cells were attenuated, demonstrating that the anti-metastatic effect of Maclurin was exerted by antioxidative activity and inhibition of Src/FAK–ERK–β-catenin signaling pathway [88]. Similarly, another study described FAK as an upstream regulator of Wnt signaling, as it modulated cell proliferation and migration through the activation of GSK3β and subsequent decrease of β-catenin and MMPs in the human lung adenocarcinoma CL1 cells after treatment with the leaf extract of Momordica charantia [89].
Both Wnt/β-catenin and FAK pathways were independently found to be implicated in idiopathic pulmonary fibrosis (IPF). Blocking Wnt signaling strongly attenuated and reversed fibrosis in a murine IPF model system [90]. Similarly, FAK inhibition attenuated fibrotic responses to lung injury in vivo [91]. Whether FAK and Wnt signaling pathways are somehow connected in regulating IPF disease progression needs further evaluation [56].
5.3. Ovarian and Breast Cancer
In a model of high-grade serous ovarian cancer (HGSOC) it was recently shown that FAK activity sustained intrinsic and acquired cisplatin resistance via a Wnt/β-catenin-Myc ‘stemness’ pathway [92]. Previous studies have already linked increased activity of FAK with chemoresistance in ovarian carcinoma cells [93]. More recently beta integrin-related protein 1 (ITGBL1), an integrin-like member of the EGF-like protein family rich in cysteine-rich repeats, was found highly expressed in ovarian cancer tissues, promoting ovarian cancer cell migration and adhesion through Wnt/PCP and FAK/Src pathways in vitro [94]. However, the specific mechanisms on how ITGBL1 affects both pathways or if there is a synergistic effect is still not clear and will need further investigation. Different studies support the role of ITGBL1 in the regulation of cancer progression. ITGBL1 was reported to promote breast cancer bone metastasis through TGF-β signaling [95] (being FAK a known mediator of TGF-β signaling [96]) and CRC migration and invasion, possibly also through the FAK signaling pathway [97]. In another study, beta 1 integrin (ITGB1) was identified as a key mediator for Twist-induced EMT, where Twist was shown to induce EMT and cell motility in breast cancer, by regulating a complex signaling network consisting of ITGB1-FAK/ILK axis, PI3K/AKT, MAPK/ERK, Wnt, and P53 signaling [98]. ITGB1 is a component of beta 1 integrin heterodimers, a family of major adhesion receptors for ECM proteins, such as fibronectins, collagens, and laminins, and is a key activator for FAK and ILK pathways [99]. In this context, the activation of ITGB1-FAK/ILK axis seems essential for stimulating Wnt signaling [98]. Similar to CRC, the APC tumor suppressor gene also plays a significant role in breast cancer carcinogenesis. APC is silenced by hypermethylation or mutated in about 70% of human breast cancers [100]. Recently it was reported that APC mutation promoted mammary tumor cell proliferation in a PyMT mouse model through the activation of Src and JNK signaling downstream of FAK [101]. In addition, inhibiting Src or JNK diminished the APC-mediated cell proliferation, suggesting that targeted inhibition of these signaling pathways downstream FAK appears as a promising strategy in APC-mutated breast cancers. In a study on ductal carcinoma in situ (DCIS), Williams et al. showed that CSC-enriched populations were more radio-resistant due to higher levels of FAK activity, and inhibition of FAK both in vitro and in vivo decreased the self-renewal capacity of these cells, accompanied by a reduction in Wnt3a and β-catenin levels. Wnt signaling stimulation after FAK inhibition rescued the self-renewal capacity, indicating once more that Wnt functions downstream of FAK signaling [102]. Similarly, another study showed that pharmacological inhibition of FAK in mouse xenograft models of triple-negative breast cancer (TNBC) reduced the proportion of CSCs in the tumors. In addition, FAK inhibition blocked β-catenin activation and the introduction of an active mutant form of β-catenin reversed the preferential targeting of CSCs by FAK inhibition [103].
5.4. Other Malignancies
While in some cancer types the relationship between FAK and Wnt is already well evident, in other cancers this crosstalk is still elusive. Different studies of hepatocellular carcinoma, melanoma and acute myeloid leukemia suggest that FAK operates upstream of Wnt, while in prostate cancer Wnt seems to acts upstream of FAK. In addition, a study of renal carcinoma reveals that Wnt and FAK might act concomitantly in promoting cancer progression.
Deletion of FAK in hepatocytes blocks tumor proliferation and prolongs the survival in a c-Met/β-catenin-driven hepatocellular carcinoma (HCC) mouse model [104], suggesting that FAK is required for c-Met/β-catenin-driven hepatocarcinogenesis. More recently it was demonstrated that overexpressed FAK and β-catenin cooperate to induce HCC in mice [105] and that FAK functionally stimulates Wnt/β-catenin signaling, activates CSC traits, and drives tumorigenicity in HCC cells [106]. The FAK gene is particularly overexpressed in human malignant melanoma cells, leading to constitutive high levels of pVE-cadherin (Y658). This elevated pVE-cadherin (Y658) allows for upregulation of Kaiso-dependent genes in the nucleus (WNT11 and CCDN1), accelerating the vasculogenic mimicry capacity in these cells, which is associated with a high tumor grade, invasion, metastasis, and a short survival [107]. A study on acute myeloid leukemia (AML) also supports a crosstalk between FAK and Wnt signaling. In this case, FAK splice variants are abnormally expressed, and this dysregulation seems to play a key role in maintaining primitive AML cells, by altering Wnt signaling and β-catenin activity [108]. In a study of human renal cell carcinoma (RCC), the proto-oncogene Golgi phosphoprotein 3 (GOLPH3) was able to activate simultaneously the FAK/Raf/MEK pathway and Wnt/β-catenin promoting RCC cell proliferation and malignancy. However, in this scenario, whether FAK and Wnt act independently but concomitantly or interdependently from each other was not addressed [109]. A more intricate interaction between Wnt and FAK was found in a study of prostate cancer where Wnt-induced secreted protein-1 (WISP-1) promoted migration in human prostate cancer cells by downregulating miR-126 expression via αvβ1 integrin, FAK, and p38 signaling pathways [110]. Another study demonstrated that silencing Cripto-1 (CR-1) expression in prostate cancer cells, suppressed proliferation, migration, and invasion of these cells through the inhibition of both FAK/Src/PI3K and Wnt/β-catenin signaling pathways [111].
6. Current Landscape of Wnt- and FAK-Targeted Therapies in Clinical Trials
Despite the increasing number of new targeted anticancer agents available in the clinic, treatment resistance and failure remain major challenges in the management of most advanced solid cancers, including breast [112] and colorectal cancers [113], and malignant mesothelioma [114]. There are multiple compensatory mechanisms that counterbalance the therapeutic effects of targeted-drugs, ultimately leading to treatment failure [115], such as the inactivation of apoptotic factors or enhancement of cell survival pathways [116]. Among promising strategies to improve clinical outcomes are the use of combinatorial therapy to target the distinct adaptive response mechanisms or the disruption of crosstalks existing between different signaling pathways, i.e., targeting downstream proteins or transcriptional factors to reduce the resistance or improve the effects of targeted therapies [117].
In addition to the roles of Wnt signaling in development, dysregulated Wnt also contributes to cell proliferation, chemoresistance and enhanced tumorigenic potential of CSC, all of which are factors involved in tumor recurrence after therapy, resistance to further anticancer therapies, and poor survival [118]. Therefore, targeting Wnt signaling seems theoretically beneficial at multiple levels: inhibition of tumor growth and survival with minimal effects on somatic cells, inhibition of CSC maintenance (and consequently tumor relapse), and prevention (or reversal) of tumor resistance [119]. However, the complexity of this pathway with its multiple sub-branches and the essential role of Wnt in regulating adult homeostasis or bone remodeling underlie the practical difficulties of finding therapeutically relevant and “safe” Wnt-targeting agents. In this section, we describe some of the Wnt and FAK inhibitors that have reached or are currently being tested in early phase clinical trials.
In general, agents targeting Wnt signaling include compounds at the ligand/receptor level (Vantictumab, Ipafricept, Rosmantuzumab, Foxy-5, and OTSA101-DTPA), transcriptional level (CWP232291, PRI-724, and SM08502) and at the level of Wnt secretion (Porcupine inhibitors: WNT974 and ETC-159, RXC004, and CGX1321) (Table 1) (for more details see additional reviews [7,119,120]). In the specific cases of Vantictumab (OMP-18R5; a monoclonal antibody that binds to Fzd receptors [121]) and Ipafricept (OMP-54F28, a recombinant fusion protein that acts as a soluble decoy receptor sequestering secreted Wnts [122]), several clinical trials have evaluated their effects as single agents or in combination with Paclitaxel, Carboplatin, or Gemcitabine in advanced solid tumors. Although both Vantictumab and Ipafricept were in principle well tolerated, they ultimately caused important bone metabolism disorders, i.e., an increased incidence of fractures in patients, therefore restricting the future use of these compounds [123,124]. Given the important role Wnts play in the differentiation of osteoblasts and osteoclasts, it is not surprising that Wnt inhibition affects bone homeostasis [125]. In the case of Rosmantuzumab (OMP-131R10), an anti-R-spondin 3 antibody, there is little information about the outcome of the phase I clinical trial (clinical trial identifier: NCT02482441), being just reported that Rosmantuzumab “failed to provide compelling evidence of clinical benefit” [126]. Regarding Foxy-5, a synthetic Wnt5a-mimicking peptide [127], after having completed a phase I study this compound is currently being tested as neo-adjuvant therapy in a phase II clinical trial aimed at evaluating the anti-metastatic activity of Foxy-5 in subjects with resectable colon cancer (NCT03883802). OTSA101-DTPA, a radiolabeled monoclonal antibody against Fzd10, is currently in phase I testing to assess its safety and pharmacokinetics in patients with relapsed or refractory synovial sarcoma (NCT04176016).

Table 1.
Summary of Wnt signaling inhibitors in clinical trials.
Inhibitors at the transcriptional level include PRI-724, CWP232291 and SM08502. While the small molecule PRI-724 inhibits the recruiting of β-catenin with its coactivator CBP, the small-molecule CWP232291 (CWP291) binds to Sam68, decreasing the expression of β-catenin target genes. In a phase I study, CWP291 was considered safe and demonstrated single-agent activity in acute myeloid leukemia patients, with plans for future combination studies [128]. Regarding PRI-724, despite some adverse events described in different phase I clinical trials that evaluated its effect in advances solid tumors including pancreatic cancer, and chronic myeloid leukemia, this compound was considered overall safe with “modest clinical activity”. Although it seems that it will continue being tested in the setting of fibrosis, no follow-up is foreseen for cancer therapy at the moment [119]. The small molecule SM08502 was shown to reduce Wnt pathway signaling and gene expression through the potent inhibition of CDC-like kinase (CLK) activity [129] and is currently in “recruiting” state in a phase I clinical trial for patients with advanced solid tumors (NCT03355066).
One of the emerging and most promising ways for targeting Wnt signaling is to block Wnt ligand production through the inhibition of the acyl-transferase Porcupine [130]. Wnts need to be coupled to fatty acids to be secreted, this occurs in the Endoplasmic Reticulum by Porcupine. Acylation of Wnts allows binding to Wntless (Wls) in the Golgi apparatus, which in turn facilitates secretion of the mature Wnts [131]. There are currently several competing inhibitors of Porcupine being evaluated in different clinical trials. Wnt974 (LGK974) and ETC-159 are under phase I studies in “recruiting” state (NCT01351103 and NCT02521844, respectively). However, similar to other Wnt inhibitors, skeletal side effects such as impairment of bone mass and increase in bone resorption are described, supporting the notion of adding bone protective agents when treating patients with these type of inhibitors [125]. RXC004 and CGX1321, other potent selective orally bioavailable inhibitors of Porcupine, are also in “recruiting” state in phase I studies in patients with advanced solid tumors (NCT03447470 and NCT02675946, respectively).
Several other small molecules antagonizing the interaction between β-catenin and TCF/LEF1, such as PNU74654, 2,4 diamino-quinazoline, PKF115-584, or CGP 049090, have been identified. Other small molecules include those targeting Dvl (NSC 668036, FJ9, 3289-8625), CK1, AXIN, or Tankyrase 1 and 2 (that promote AXIN stabilization). Natural compounds, such as vitamin D, curcumin, quercetin, or resveratrol, or existing drugs in the market such as Sulindac and Celecoxib are also being investigated in preclinical studies for Wnt signaling inhibition [132].
Taken together, the data and rationale behind using Wnt inhibitors supports them as promising therapeutics. Nevertheless, despite extensive research and some promising outcomes from preclinical and clinical data, enthusiasm about Wnt inhibitors needs to be carefully evaluated. Firstly, the intricate nature of Wnt signaling and crosstalk with other signaling pathways are not fully elucidated. This lack of understanding most probably hinders the development of more efficacious therapeutic strategies. Secondly, we do not know whether the inhibition of Wnt signaling will activate resistance response mechanisms by other signaling pathways. Thirdly, the important role of Wnt signaling in physiological homeostasis and in bone remodeling, and the consequent adverse effects of its inhibition, cannot be dismissed. It is noteworthy to mention that toxicities originated from these treatments may vary dramatically depending on the targeted pathway component [132].
In the case of FAK inhibitors, translation in the clinical setting is more advanced: four adenosine triphosphate (ATP)-competitive, small-molecule inhibitors of FAK (GSK2256098, BI 853520, PF-00562271, and VS-6063), have been proven effective in several preclinical studies [133] and have advanced into phase I and II clinical trials (Table 2). Currently two (GSK2256098 and VS-6063) are in ongoing clinical trials evaluating its use in combination with other chemotherapeutic agents and/or cell signaling/checkpoint inhibitors in order to increase their efficacy.
Table 2.
Summary of FAK signaling inhibitors in clinical trials.
In a phase I study (NCT01138033), GSK2256098 showed clinical activity in patients with malignant mesothelioma, particularly those with merlin (encoded by NF2) loss [134]. With the premise that combined FAK and MEK inhibition may provide greater anticancer effect than FAK monotherapy, another phase I study evaluated GSK2256098 in combination with Trametinib (NCT01938443). Mesothelioma patients with loss of merlin had longer progression-free survival than subjects with wild-type NF2, although support for efficacy with this combination was limited [135]. Recently, another study is attempting to evaluate this combination in patients with advanced pancreatic cancer (NCT02428270).
Two studies have evaluated the effect of BI 853520 monotherapy in advanced or metastatic tumors (NCT01335269 and NCT01905111). These studies have demonstrated that BI 853520 has a manageable and acceptable safety profile and preliminary antitumor activity. However, as single-agent its activity was described as “modest”, therefore it has been proposed to explore this compound in combination with other agents for future studies [136].
VS-6062 was the first FAK inhibitor tested in clinical phase I trials (NCT00666926) but, despite promising results, it was discontinued due to nonlinear pharmacokinetics [137]. Similarly, VS-4718 is also no longer in active clinical development [138]. Almost 10 years ago, Verastem developed VS-6063 (Defactinib), a second-generation compound with a more favorable pharmacokinetics. Defactinib is by far the most studied compound in the clinic. As acceptable phase I safety profiles were obtained in patients with advanced solid tumors [139,140], phase II combinatorial clinical trials with Defactinib are currently under evaluation in different advanced solid tumors. Around sixteen studies have reached phases I and II in different clinical trials, of them, six are currently in “active” or “recruiting” state. Although several attempts have been made in testing the effect of Defactinib in patients with malignant mesothelioma, due in part to the fact that merlin deficiency has been shown to predict FAK-inhibitor sensitivity [141], several trials were discontinued due to insufficient efficacy (NCT02004028, NCT01870609, and NCT02372227). Nevertheless, a phase I study evaluating Defactinib in combination with the immune checkpoint inhibitor Pembrolizumab is planned for patients with pleural malignant mesothelioma (NCT04201145), and another phase II study is currently evaluating the effects of this drug as single agents in patients with NF2 mutations (NCT04439331). Regarding patients with KRAS mutant lung cancer (NCT01951690), Defactinib monotherapy demonstrated modest clinical activity [142]. At present time, other phase I/II studies that are in “active” or “recruiting” phases, are evaluating the effects of Defactinib in combination with Pembrolizumab (NCT03727880 and NCT02758587), Pembrolizumab, and Gemcitabine (NCT02546531), or RAF/MEK inhibitors (NCT03875820) in patients with different advanced solid tumors. A phase I study of Defactinib in combination with Paclitaxel (NCT01778803) in patients with advanced ovarian cancer exhibited signs of modest activity. A phase I/II clinical trial for recurrent platinum-resistant ovarian cancer termed ROCKIF has been initiated (NCT03287271), and will investigate the combination of Defactinib, Carboplatin and Paclitaxel.
Altogether, it seems that the FAK inhibitors examined in different clinical trials have shown manageable toxicity profiles, some efficacy in delaying progression-free survival and maintaining stable disease in different advanced solid tumors; however, they did not produce objective clinical responses. Trials are currently directed to strengthen the efficacy by combining FAK inhibitors with cytotoxic chemotherapy, targeted therapy, or immunotherapy. In the future, prognostic markers could help to select patients who could benefit from FAK inhibitor treatment alone or in combination strategies [143].
7. Potential Benefits of Targeting the Crosstalk Wnt–FAK
An important raising question is whether FAK inhibitors are effective as single agents. Since FAK is activated by so many binding partners and is interconnected with different signaling pathways, a combinatorial therapy seems the most promising approach [4]. A number of preclinical studies have shown that cancer cells adapt to inhibition of signaling pathway circuits by establishing alternative signaling routes through crosstalks in order to evade drug treatments [144]. Hence, it will be important not only to inhibit primary signaling pathways that induce tumorigenesis, but concomitantly, prevent pro-survival pathway crosstalks [115].
The combinatorial therapy using FAK inhibitors with agents blocking other molecules or signaling pathways has already shown effectiveness over the use of single inhibitors in the preclinical setting. In a study of acute lymphoblastic leukemia, FAK downregulation enhanced the inhibitory effect of the mTOR inhibitor Rapamycin on cell growth in vitro, slowed the progression of leukemia in NOD/SCID mice in vivo, and prolonged the median survival time of the mice [145]. Dual inhibition of FAK and EGFR signaling pathways enhanced apoptosis in breast cancers cells [146], while simultaneous inhibition of FAK and Src increased the apoptosis of colon cancer cell lines, involving the blockade of the AKT survival pathway [147]. The connection between PTEN and FAK has also emerged as a potential druggable signaling axis. Loss of PTEN causes the activation of the pro-survival pathway PI3K/AKT/mTOR, but results obtained with specific inhibitors of PI3K/AKT/mTOR are not so encouraging. As reduced PTEN activity often correlated with increased expression of FAK [148], a combinatorial strategy was proposed based on concomitant inhibition of PI3K/AKT and FAK signaling, in advanced metastatic malignancies with altered or reduced PTEN expression [149]. A recent study showed that in vitro downregulation of Calretinin, a calcium-binding protein expressed in malignant mesothelioma [150] and activator of the FAK signaling pathway [29], increases the susceptibility of malignant mesothelioma cells to the FAK inhibitor Defactinib in vitro [84]. However, surviving cells showed increased resistance to chemotherapy due to the activation of the Wnt signaling pathway, demonstrating the induction of a secondary oncogenic pathway conferring resistance to therapy.
The many existing Wnt–FAK crosstalks in different cancers are complex, variable, and cell-type dependent. There is emerging in vitro and in vivo evidence that disrupting these crosstalks at multiple levels has therapeutic benefits. Pharmacological inhibition of FAK/PYK2 repressed adenoma formation in APCmin/+ mice and reduced phospho-GSK3β and β-catenin intestinal levels, decreasing Wnt signaling [74]. Co-targeting the Wnt pathway using FAK inhibitors was proposed as a new approach to overcome resistance to BRAF inhibitors in BRAF-mutated colorectal cancer patients [76]. Indeed, FAK inhibitors in combination with BRAF inhibitors, were shown effective in decreasing tumor growth in a mouse xenograft model [75]. The inhibition of FAK blocks tumor proliferation and prolongs the survival in a c-Met/β-catenin-driven hepatocellular carcinoma (HCC) mouse model [104], suggesting that inhibition of FAK might be beneficial for treating HCC, especially in patients showing activated β-catenin. In various malignant mesothelioma cell lines, the pharmacological inhibition of both pathways decreased dramatically cell proliferation and survival, indicating that the simultaneous targeting of Wnt and FAK might be considered as a prospective strategy for malignant mesothelioma treatment [84].
Nevertheless, despite the many examples of FAK–Wnt interactions described in the literature and summarized here, we are still far away from a comprehensive picture of this crosstalk, especially regarding its different cancer-specific dynamics. Some studies have started to develop mathematical models or network-based computational methods to quantify the signaling crosstalks in order to identify new therapeutic targets and evaluate the most effective drug target combinations [115,151].
8. Conclusions and Future Perspectives
In this review, we have highlighted the current knowledge on Wnt–FAK signaling crosstalks in different cancers (summarized in Figure 1). FAK appears to be an important regulator of the Wnt signaling pathway. However, we found a wide range of FAK–Wnt interactions at different levels that are context- and cell-type dependent; thus, a better understanding of the nature and physiological relevance of this interaction is necessary [56], before devising combinatorial therapeutic strategies.
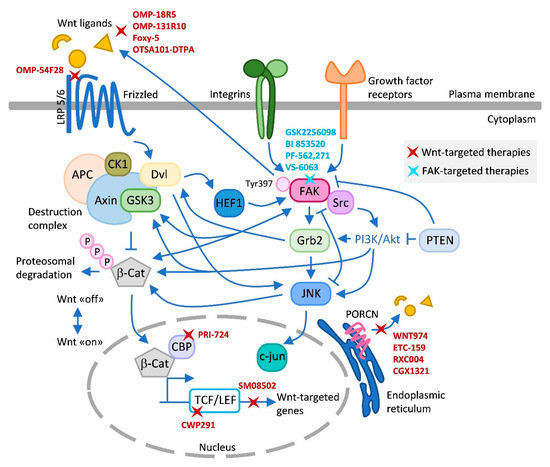
Figure 1.
Wnt–FAK signaling crosstalks and inhibitors. This figure summarizes the main known crosstalks existing between Wnt and FAK that have been described in the text and in the literature, and the different Wnt and FAK inhibitors tested in clinical trials. Upon FAK activation, the FAK/Src complex phosphorylates and recruits several downstream signaling targets, including PI3K/AKT. GSK3 generally acts as a downstream signaling protein molecule of AKT [152]. Grb2 coordinates signaling downstream of integrin/FAK to activate JNK. Grb2 also interacts directly with Dvl [153]. Dvl can stimulate c-Jun-dependent transcription activity and the kinase activity of JNK [154]. Loss of PTEN function causes the activation of PI3K/AKT and JNK pathways [155]. PTEN also controls FAK [148]. FAK and PYK2 promote Wnt/β-catenin pathway activation by phosphorylating GSK3β [74]. This phosphorylation inhibits the activity of GSK3β which otherwise would drive rapid degradation of β-catenin. FAK increases expression of Wnt ligands activating Wnt signaling and CSC self-renewal indirectly or directly by activating β-catenin [102]. In addition, FAK was shown to trigger the β-catenin signaling pathway through nuclear translocation of β-catenin and transcriptional activation of β-catenin target genes [74]. FAK and Wnt have been described to modulate each other antagonistically [84]. HEF1 localizes to focal adhesions to coordinate FAK/Src signaling and is also modulated by Wnt [80]. Wnt-targeted agents include OMP18R5, OMP131R10, Foxy-5, OTSA101-DPTA, and OMP-54F28, which target Wnt signaling at the ligand/receptor level; PRI-724, CWP291, and SM08502 at the transcriptional level; and the Porcupine inhibitors WNT974, ETC-159, RXC004, and CGK1321, which block Wnt ligand secretion. FAK inhibitors include GSK2256098, PF-562,271, and VS-6063, which competitively target the ATP-binding site K454, located in the kinase domain of FAK; and the competitive scaffold inhibitor BI 853520, which binds to the hinge region of the kinase domain of FAK blocking the access of ATP to the ATP binding site [143]. Arrows (↑) indicate activation/induction, and blunt-ended lines (T) indicate inhibition/blockade. LRP, low-density lipoprotein receptor-related protein; Dvl, dishevelled; CK1, casein kinase 1; APC, adenomatous polyposis coli; GSK3, glycogen synthase kinase-3; β-Cat, β-catenin; CBP, CREB binding protein; TCF/LEF, T-cell factor/lymphoid enhancer factor; Grb-2, growth factor receptor-bound protein 2; PTEN, phosphatase and tensin homolog; JNK, c-Jun N-terminal kinase; PORCN, Porcupine.
Clinical trials with Wnt and FAK inhibitors have demonstrated some limitations, in part due to the important roles that both pathways exert in the maintenance of adult homeostasis. The future use of Wnt inhibitors have been restricted as a result of severe side effects; and despite current data that justify the use of FAK inhibitors in solid tumors, single-agent targeting shows still modest clinical activity. Therefore, efforts are nowadays directed in testing both inhibitors in combination with different chemotherapeutic agents and/or cell signaling/checkpoint inhibitors.
Both indirect inhibition of the FAK–Wnt crosstalk by targeting FAK (consequently reducing Wnt activity), or a combinatorial approach targeting key molecules from both pathways (thus disrupting compensatory signaling mechanisms), seem promising strategies. In fact, synergistic drug combinations are generally more specific to particular cellular contexts improving the therapeutically selectivity [156]. The proper design of drug combinations requires a profound understanding of the crosstalks between oncogenic pathways, in addition to other factors, such as intercellular signaling responses in the cancer microenvironment and drug responses in individual cells [157]. Hence, a multidisciplinary approach integrating cancer cell biology, animal and 3D models, biomarker analysis, high-throughput technologies and network-based computational methods seems more and more essential for the discovery of new effective treatments. We believe that additional FAK–Wnt crosstalks will be revealed in the future and that effective and alternative strategies will soon emerge to overcome their signaling crosstalk-mediated effects during tumorigenesis.
Author Contributions
J.W. conceived the study, interpreted the relevant literature and wrote the manuscript. C.R. revised the manuscript for important intellectual content. All authors have read and agreed to the published version of the manuscript.
Funding
Research in our laboratory is presently supported by grants from the Swiss National Science Foundation (31003A_159824), the Swiss Cancer League (KFS 4400-02-2018), the Swiss Heart Foundation (FF18115), Stiftung zur Krebsbekämpfung, the ISREC foundation, Innosuisse and NCCR bioinspired materials.
Acknowledgments
We apologize for not being able to cite all published work relevant to this topic due to selective focus of the article and space limitation.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: Mechanistic findings and clinical applications. Nat. Rev. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef]
- Lee, B.Y.; Timpson, P.; Horvath, L.G.; Daly, R.J. FAK signaling in human cancer as a target for therapeutics. Pharmacol. Ther. 2015, 146, 132–149. [Google Scholar] [CrossRef] [PubMed]
- Schaller, M.D. Cellular functions of FAK kinases: Insight into molecular mechanisms and novel functions. J. Cell Sci. 2010, 123, 1007–1013. [Google Scholar] [CrossRef]
- Golubovskaya, V.M. Targeting FAK in human cancer: From finding to first clinical trials. Front. Biosci. 2014, 19, 687–706. [Google Scholar] [CrossRef]
- Kahn, M. Can we safely target the WNT pathway? Nat. Rev. Drug Discov. 2014, 13, 513–532. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R. Wnt signaling. Cold Spring Harb. Perspect. Biol. 2012, 4, 570–581. [Google Scholar] [CrossRef] [PubMed]
- Harb, J.; Lin, P.-J.; Hao, J. Recent Development of Wnt Signaling Pathway Inhibitors for Cancer Therapeutics. Curr. Oncol. Rep. 2019, 21, 12. [Google Scholar] [CrossRef] [PubMed]
- Vert, G.; Chory, J. Crosstalk in Cellular Signaling: Background Noise or the Real Thing? Dev. Cell 2011, 21, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Rowland, M.A.; Greenbaum, J.M.; Deeds, E.J. Crosstalk and the evolvability of intracellular communication. Nat. Commun. 2017, 8, 16009. [Google Scholar] [CrossRef][Green Version]
- Gossage, L.; Eisen, T. Targeting multiple kinase pathways: A change in paradigm. Clin. Cancer Res. 2010, 16, 1973–1978. [Google Scholar] [CrossRef]
- Lim, S.-T.; Chen, X.L.; Lim, Y.; Hanson, D.A.; Vo, T.-T.; Howerton, K.; Larocque, N.; Fisher, S.J.; Schlaepfer, D.D.; Ilic, D. Nuclear FAK Promotes Cell Proliferation and Survival through FERM-Enhanced p53 Degradation. Mol. Cell 2008, 29, 9–22. [Google Scholar] [CrossRef]
- Gabarra-Niecko, V.; Schaller, M.D.; Dunty, J.M. FAK regulates biological processes important for the pathogenesis of cancer. Cancer Metastasis Rev. 2003, 22, 359–374. [Google Scholar] [CrossRef] [PubMed]
- Schaller, M.D. Biochemical signals and biological responses elicited by the focal adhesion kinase. Biochim. Biophys. Acta (BBA) Bioenergy 2001, 1540, 1–21. [Google Scholar] [CrossRef]
- Fiedorek, F.T.; Kay, E.S. Mapping of the focal adhesion kinase (Fadk) gene to mouse chromosome 15 and human chromosome 8. Mamm. Genome 1995, 6, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Schaller, M.D.; Borgman, C.A.; Cobb, B.S.; Vines, R.R.; Reynolds, A.B.; Parsons, J.T. pp125FAK a structurally distinctive protein-tyrosine kinase associated with focal adhesions. Proc. Natl. Acad. Sci. USA 1992, 89, 5192–5196. [Google Scholar] [CrossRef] [PubMed]
- Hanks, S.K.; Calalb, M.B.; Harper, M.C.; Patel, S.K. Focal adhesion protein-tyrosine kinase phosphorylated in response to cell attachment to fibronectin. Proc. Natl. Acad. Sci. USA 1992, 89, 8487–8491. [Google Scholar] [CrossRef]
- Andre, E.; Beckerandre, M. Expression of an N-Terminally Truncated Form of Human Focal Adhesion Kinase in Brain. Biochem. Biophys. Res. Commun. 1993, 190, 140–147. [Google Scholar] [CrossRef]
- Zhang, X.; Wright, C.V.; Hanks, S.K. Cloning of a Xenopus laevis cDNA encoding focal adhesion kinase (FAK) and expression during early development. Gene 1995, 160, 219–222. [Google Scholar] [CrossRef]
- Schaller, M.D.; Hildebrand, J.D.; Shannon, J.D.; Fox, J.W.; Vines, R.R.; Parsons, J.T. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol. Cell. Biol. 1994, 14, 1680–1688. [Google Scholar] [CrossRef]
- Polte, T.R.; Hanks, S.K. Interaction between focal adhesion kinase and Crk-associated tyrosine kinase substrate p130Cas. Proc. Natl. Acad. Sci. USA 1995, 92, 10678–10682. [Google Scholar] [CrossRef]
- Chen, H.C.; Appeddu, P.A.; Isoda, H.; Guan, J.L. Phosphorylation of tyrosine 397 in focal adhesion kinase is required for binding phosphatidylinositol 3-kinase. J. Biol. Chem. 1996, 271, 26329–26334. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.K.; Schlaepfer, D.D. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr. Opin. Cell Biol. 2006, 18, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Avizienyte, E.; Frame, M.C. Src and FAK signalling controls adhesion fate and the epithelial-to-mesenchymal transition. Curr. Opin. Cell Biol. 2005, 17, 542–547. [Google Scholar] [CrossRef]
- Owens, L.V.; Xu, L.; Craven, R.J.; Dent, G.A.; Weiner, T.M.; Kornberg, L.; Liu, E.T.; Cance, W.G. Overexpression of the focal adhesion kinase (p125FAK) in invasive human tumors. Cancer Res. 1995, 55, 2752–2755. [Google Scholar] [PubMed]
- Owens, L.V.; Xu, L.; Dent, G.A.; Yang, X.; Sturge, G.C.; Craven, R.J.; Cance, W. Focal adhesion kinase as a marker of invasive potential in differentiated human thyroid cancer. Ann. Surg. Oncol. 1996, 3, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, L.; Hauck, W.; Aprikian, A.G.; Begin, L.R.; Chapdelaine, A.; Chevalier, S. Focal adhesion kinase (pp125FAK) expression, activation and association with paxillin and p50CSK in human metastatic prostate carcinoma. Int. J. Cancer 1996, 68, 164–171. [Google Scholar] [CrossRef]
- Lark, A.L.; Livasy, C.A.; Calvo, B.; Caskey, L.; Moore, D.T.; Yang, X.; Cance, W.G. Overexpression of focal adhesion kinase in primary colorectal carcinomas and colorectal liver metastases: Immunohistochemistry and real-time PCR analyses. Clin. Cancer Res. 2003, 9, 215–222. [Google Scholar] [PubMed]
- Judson, P.L.; He, X.; Cance, W.G.; Van Le, L. Overexpression of focal adhesion kinase, a protein tyrosine kinase, in ovarian carcinoma. Cancer 1999, 86, 1551–1556. [Google Scholar] [CrossRef]
- Wörthmüller, J.; Blum, W.; Pecze, L.; Salicio, V.; Schwaller, B. Calretinin promotes invasiveness and EMT in malignant mesothelioma cells involving the activation of the FAK signaling pathway. Oncotarget 2018, 9, 36256–36272. [Google Scholar] [CrossRef]
- Yom, C.K.; Noh, D.-Y.; Kim, W.H.; Kim, H.S. Clinical significance of high focal adhesion kinase gene copy number and overexpression in invasive breast cancer. Breast Cancer Res. Treat. 2011, 128, 647–655. [Google Scholar] [CrossRef]
- Canel, M.; Secades, P.; Rodrigo, J.-P.; Cabanillas, R.; Herrero, A.; Suarez, C.; Chiara, M.-D. Overexpression of Focal Adhesion Kinase in Head and Neck Squamous Cell Carcinoma Is Independent of fak Gene Copy Number. Clin. Cancer Res. 2006, 12, 3272–3279. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.T.; Chen, X.L.; Tomar, A.; Miller, N.L.G.; Yoo, J.; Schlaepfer, D.D. Knock-in mutation reveals an essential role for focal adhesion kinase activity in blood vessel morphogenesis and cell motility-polarity but not cell proliferation. J. Biol. Chem. 2010, 285, 21526–21536. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.K.; Hanson, D.A.; Schlaepfer, D.D. Focal adhesion kinase: In command and control of cell motility. Nat. Rev. Mol. Cell Biol. 2005, 6, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Brami-Cherrier, K.; Gervasi, N.; Arsenieva, D.; Walkiewicz, K.; Boutterin, M.; Ortega, A.; Leonard, P.G.; Seantier, B.; Gasmi, L.; Bouceba, T.; et al. FAK dimerization controls its kinase-dependent functions at focal adhesions. EMBO J. 2014, 33, 356–370. [Google Scholar] [CrossRef]
- Cary, L.A.; Chang, J.F.; Guan, J.L. Stimulation of cell migration by overexpression of focal adhesion kinase and its association with Src and Fyn. J. Cell Sci. 1996, 109, 1787–1794. [Google Scholar]
- Parsons, J.T. Focal adhesion kinase: The first ten years. J. Cell Sci. 2003, 116, 1409–1416. [Google Scholar] [CrossRef]
- Golubovskaya, V.M.; Finch, R.; Cance, W.G. Direct Interaction of the N-terminal Domain of Focal Adhesion Kinase with the N-terminal Transactivation Domain of p53. J. Biol. Chem. 2005, 280, 25008–25021. [Google Scholar] [CrossRef]
- Zhou, J.; Yi, Q.; Tang, L. The roles of nuclear focal adhesion kinase (FAK) on Cancer: A focused review. J. Exp. Clin. Cancer Res. 2019, 38, 1–11. [Google Scholar] [CrossRef]
- Komiya, Y.; Habas, R. Wnt signal transduction pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef]
- Clevers, H.; Nusse, R. Wnt/beta-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef]
- Ilyas, M. Wnt signalling and the mechanistic basis of tumour development. J. Pathol. 2005, 205, 130–144. [Google Scholar] [CrossRef] [PubMed]
- Florian, M.C.; Nattamai, K.J.; Dörr, K.; Marka, G.; Überle, B.; Vas, V.; Eckl, C.; Andrä, I.; Schiemann, M.; Oostendorp, R.A.J.; et al. A canonical to non-canonical Wnt signalling switch in haematopoietic stem-cell ageing. Nat. Cell Biol. 2013, 503, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Gordon, M.D.; Nusse, R. Wnt Signaling: Multiple Pathways, Multiple Receptors, and Multiple Transcription Factors. J. Biol. Chem. 2006, 281, 22429–22433. [Google Scholar] [CrossRef] [PubMed]
- Luna-Ulloa, L.B.; Hernández-Maqueda, J.G.; Castañeda-Patlán, M.C.; Robles-Flores, M. Protein kinase C in Wnt signaling: Implications in cancer initiation and progression. IUBMB Life 2011, 63, 915–921. [Google Scholar] [CrossRef]
- Lim, X.; Nusse, R. Wnt Signaling in Skin Development, Homeostasis, and Disease. Cold Spring Harb. Perspect. Biol. 2012, 5, a008029. [Google Scholar] [CrossRef]
- Simons, M.; Mlodzik, M. Planar Cell Polarity Signaling: From Fly Development to Human Disease. Annu. Rev. Genet. 2008, 42, 517–540. [Google Scholar] [CrossRef]
- Kurayoshi, M.; Oue, N.; Yamamoto, H.; Kishida, M.; Inoue, A.; Asahara, T.; Yasui, W.; Kikuchi, A. Expression of Wnt-5a Is Correlated with Aggressiveness of Gastric Cancer by Stimulating Cell Migration and Invasion. Cancer Res. 2006, 66, 10439–10448. [Google Scholar] [CrossRef]
- Corda, G.; Sala, G.; Lattanzio, R.; Iezzi, M.; Sallese, M.; Fragassi, G.; Lamolinara, A.; Mirza, H.; Barcaroli, D.; Ermler, S.; et al. Functional and prognostic significance of the genomic amplification of frizzled 6 (FZD6) in breast cancer. J. Pathol. 2017, 241, 350–361. [Google Scholar] [CrossRef]
- Koval, A.; Katanaev, V.L. Dramatic dysbalancing of the Wnt pathway in breast cancers. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Ilić, D.; Furuta, Y.; Kanazawa, S.; Takeda, N.; Sobue, K.; Nakatsuji, N.; Nomura, S.; Fujimoto, J.; Okada, M.; Yamamoto, T.; et al. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nat. Cell Biol. 1995, 377, 539–544. [Google Scholar] [CrossRef]
- Shen, T.L.; Park, A.Y.; Alcaraz, A.; Peng, X.; Jang, I.; Koni, P.; Flavell, R.A.; Gu, H.; Guan, J.L. Conditional knockout of focal adhesion kinase in endothelial cells reveals its role in angiogenesis and vascular development in late embryogenesis. J Cell Biol. 2005, 169, 941–952. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Lee, J.; Vikis, H.G.; Lee, S.H.; Liu, G.; Aurandt, J.; Shen, T.L.; Fearon, E.R.; Guan, J.L.; Han, M.; et al. Activation of FAK and Src are receptor-proximal events required for netrin signaling. Nat. Neurosci. 2004, 7, 1213–1221. [Google Scholar] [CrossRef] [PubMed]
- Chatzizacharias, N.A.; Kouraklis, G.P.; Theocharis, S.E. The role of focal adhesion kinase in early development. Histol. Histopathol. 2010, 25, 1039–1055. [Google Scholar] [PubMed]
- Steinhart, Z.; Angers, S. Wnt signaling in development and tissue homeostasis. Development 2018, 145, dev146589. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.; Loh, K.M.; Nusse, R. Stem cell signaling. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science 2014, 346, 1248012. [Google Scholar] [CrossRef] [PubMed]
- Fonar, Y.; Frank, D. FAK and WNT Signaling: The Meeting of Two Pathways in Cancer and Development. Anti-Cancer Agents Med. Chem. 2011, 11, 600–606. [Google Scholar] [CrossRef]
- Cohen, E.D.; Mariol, M.-C.; Wallace, R.M.; Weyers, J.; Kamberov, Y.G.; Pradel, J.; Wilder, E.L. DWnt4 Regulates Cell Movement and Focal Adhesion Kinase during Drosophila Ovarian Morphogenesis. Dev. Cell 2002, 2, 437–448. [Google Scholar] [CrossRef]
- Fonar, Y.; Gutkovich, Y.E.; Root, H.; Malyarova, A.; Aamar, E.; Golubovskaya, V.M.; Elias, S.; Elkouby, Y.M.; Frank, D. Focal adhesion kinase protein regulates Wnt3a gene expression to control cell fate specification in the developing neural plate. Mol. Biol. Cell 2011, 22, 2409–2421. [Google Scholar] [CrossRef]
- Sun, C.; Yuan, H.; Wang, L.; Wei, X.; Williams, L.; Krebsbach, P.H.; Guan, J.-L.; Liu, F. FAK Promotes Osteoblast Progenitor Cell Proliferation and Differentiation by Enhancing Wnt Signaling. J. Bone Miner. Res. 2016, 31, 2227–2238. [Google Scholar] [CrossRef]
- Zheng, W.; Gu, X.; Sun, X.; Wu, Q.; Dan, H. FAK mediates BMP9-induced osteogenic differentiation via Wnt and MAPK signaling pathway in synovial mesenchymal stem cells. Artif. Cells Nanomed. Biotechnol. 2019, 47, 2641–2649. [Google Scholar] [CrossRef]
- Yuan, C.; Gou, X.; Deng, J.; Dong, Z.; Ye, P.; Hu, Z. FAK and BMP-9 synergistically trigger osteogenic differentiation and bone formation of adipose derived stem cells through enhancing Wnt-beta-catenin signaling. Biomed. Pharm. 2018, 105, 753–757. [Google Scholar] [CrossRef] [PubMed]
- Ashton, G.H.; Morton, J.P.; Myant, K.; Phesse, T.J.; Ridgway, R.A.; Marsh, V.; Wilkins, J.A.; Athineos, D.; Muncan, V.; Kemp, R.; et al. Focal adhesion kinase is required for intestinal regeneration and tumorigenesis downstream of Wnt/c-Myc signaling. Dev. Cell. 2010, 19, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Oshima, H.; Kok, S.-Y.; Nakayama, M.; Murakami, K.; Voon, D.C.-C.; Kimura, T.; Oshima, M. Stat3 is indispensable for damage-induced crypt regeneration but not for Wnt-driven intestinal tumorigenesis. FASEB J. 2019, 33, 1873–1886. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, R.A.; Serrels, B.; Mason, S.; Kinnaird, A.; Muir, M.; Patel, H.; Muller, W.J.; Sansom, O.J.; Brunton, V.G. Focal adhesion kinase is required for beta-catenin-induced mobilization of epidermal stem cells. Carcinogenesis 2012, 33, 2369–2376. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Siegel, R.; Ward, E.; Murray, T.; Xu, J.; Smigal, C.; Thun, M.J. Cancer Statistics, 2006. CA: A Cancer J. Clin. 2006, 56, 106–130. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Chien, A.J.; Moore, E.C.; Lonsdorf, A.S.; Kulikauskas, R.M.; Rothberg, B.G.; Berger, A.J.; Major, M.B.; Hwang, S.T.; Rimm, D.L.; Moon, R.T. Activated Wnt/beta-catenin signaling in melanoma is associated with decreased proliferation in patient tumors and a murine melanoma model. Proc. Natl. Acad. Sci. USA 2009, 106, 1193–1198. [Google Scholar] [CrossRef]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef]
- Kwong, L.N.; Dove, W.F. APC and Its Modifiers in Colon Cancer. Adv. Exp. Med. Biol. 2009, 656, 85–106. [Google Scholar] [CrossRef]
- Parker, T.W.; Neufeld, K.L. APC controls Wnt-induced beta-catenin destruction complex recruitment in human colonocytes. Sci. Rep. 2020, 10, 2957. [Google Scholar] [CrossRef] [PubMed]
- Li, V.S.; Ng, S.S.; Boersema, P.J.; Low, T.Y.; Karthaus, W.R.; Gerlach, J.P.; Mohammed, S.; Heck, A.J.; Maurice, M.M.; Mahmoudi, T.; et al. Wnt signaling through inhibition of beta-catenin degradation in an intact Axin1 complex. Cell 2012, 149, 1245–1256. [Google Scholar] [CrossRef] [PubMed]
- Tejeda-Muñoz, N.; Gonzalez-Aguilar, H.; Santoyo-Ramos, P.; Castañeda-Patlan, M.C.; Robles-Flores, M. Glycogen Synthase Kinase 3beta Is Positively Regulated by Protein Kinase Czeta-Mediated Phosphorylation Induced by Wnt Agonists. Mol. Cell Biol. 2015, 36, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Chen, G.; Kuan, S.F.; Zhang, D.H.; Schlaepfer, D.D.; Hu, J. FAK/PYK2 promotes the Wnt/beta-catenin pathway and intestinal tumorigenesis by phosphorylating GSK3beta. Elife 2015, 4, e10072. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Gao, C.; Gao, X.; Zhang, D.H.; Kuan, S.F.; Burns, T.F.; Hu, J. Wnt/beta-Catenin Pathway Activation Mediates Adaptive Resistance to BRAF Inhibition in Colorectal Cancer. Mol. Cancer Ther. 2018, 17, 806–813. [Google Scholar] [CrossRef]
- Gao, C.; Hu, J. Targeting parallel bypass signaling to combat adaptive resistance to BRAF inhibition in colorectal Cancer. Oncoscience 2018, 5, 57–58. [Google Scholar] [CrossRef]
- Heffler, M.; Golubovskaya, V.M.; Conroy, J.; Liu, S.; Wang, D.; Cance, W.G.; Dunn, K.M.B. FAK and HAS inhibition synergistically decrease colon cancer cell viability and affect expression of critical genes. Anti-Cancer Agents Med. Chem. 2013, 13, 584–594. [Google Scholar] [CrossRef][Green Version]
- van Seventer, G.A.; Salmen, H.J.; Law, S.F.; .O’Neill, G.M.; Mullen, M.M.; Franz, A.M.; Kanner, S.B.; Golemis, E.A.; van Seventer, J.M. Focal adhesion kinase regulates beta1 integrin-dependent T cell migration through an HEF1 effector pathway. Eur. J. Immunol. 2001, 31, 1417–1427. [Google Scholar] [CrossRef]
- Natarajan, M.; Stewart, J.E.; Golemis, E.A.; Pugacheva, E.N.; Alexandropoulos, K.; Cox, B.D.; Wang, W.; Grammer, J.R.; Gladson, C.L. HEF1 is a necessary and specific downstream effector of FAK that promotes the migration of glioblastoma cells. Oncogene 2005, 25, 1721–1732. [Google Scholar] [CrossRef]
- Li, Y.; Bavarva, J.H.; Wang, Z.; Guo, J.; Qian, C.; Thibodeau, S.N.; Golemis, E.A.; Liu, W. HEF1, a novel target of Wnt signaling, promotes colonic cell migration and cancer progression. Oncogene 2011, 30, 2633–2643. [Google Scholar] [CrossRef]
- Morton, J.P.; Myant, K.B.; Sansom, O.J. A FAK-PI-3K-mTOR axis is required for Wnt-Myc driven intestinal regeneration and tumorigenesis. Cell Cycle 2011, 10, 173–175. [Google Scholar] [CrossRef]
- McLean, G.W.; Carragher, N.O.; Avizienyte, E.; Evans, J.A.; Brunton, V.G.; Frame, M.C. The role of focal-adhesion kinase in cancer—A new therapeutic opportunity. Nat. Rev. Cancer 2005, 5, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Testa, J.R.; Carbone, M. Mesothelioma Epidemiology, Carcinogenesis, and Pathogenesis. Curr. Treat. Options Oncol. 2008, 9, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Wörthmüller, J.; Salicio, V.; Oberson, A.; Blum, W.; Schwaller, B. Modulation of Calretinin Expression in Human Mesothelioma Cells Reveals the Implication of the FAK and Wnt Signaling Pathways in Conferring Chemoresistance towards Cisplatin. Int. J. Mol. Sci. 2019, 20, 5391. [Google Scholar] [CrossRef]
- Uematsu, K.; Kanazawa, S.; You, L.; He, B.; Xu, Z.; Li, K.; Peterlin, B.M.; McCormick, F.; Jablons, D.M. Wnt pathway activation in mesothelioma: Evidence of Dishevelled overexpression and transcriptional activity of beta-catenin. Cancer Res. 2003, 63, 4547–4551. [Google Scholar]
- Lee, A.Y.; He, B.; You, L.; Dadfarmay, S.; Xu, Z.; Mazieres, J.; Mikami, I.; McCormick, F.; Jablons, D.M. Expression of the secreted frizzled-related protein gene family is downregulated in human mesothelioma. Oncogene 2004, 23, 6672–6676. [Google Scholar] [CrossRef]
- Bai, D.; Cong, S.; Zhu, L.P. Attenuation of Focal Adhesion Kinase Reduces Lipopolysaccharide-Induced Inflammation Injury through Inactivation of the Wnt and NF-kappaB Pathways in A549 Cells. Biochemistry 2017, 82, 446–453. [Google Scholar]
- Ku, M.J.; Kim, J.H.; Lee, J.; Cho, J.Y.; Chun, T.; Lee, S.Y. Maclurin suppresses migration and invasion of human non-small-cell lung cancer cells via anti-oxidative activity and inhibition of the Src/FAK-ERK-beta-catenin pathway. Mol. Cel. Biochem. 2015, 402, 243–252. [Google Scholar] [CrossRef]
- Hsu, H.-Y.; Lin, J.-H.; Li, C.-J.; Tsang, S.-F.; Tsai, C.-H.; Chyuan, J.-H.; Chiu, S.-J.; Chuang, S.-E. Antimigratory Effects of the Methanol Extract from Momordica charantia on Human Lung Adenocarcinoma CL1 Cells. Evidence-Based Complement. Altern. Med. 2012, 2012, 1–12. [Google Scholar] [CrossRef]
- Kim, T.H.; Kim, S.H.; Seo, J.; Chung, H.; Kwak, H.J.; Lee, S.K.; Yoon, H.J.; Shin, D.H.; Park, S.S.; Sohn, J.W. Blockade of the Wnt/beta-catenin pathway attenuates bleomycin-induced pulmonary fibrosis. Tohoku J. Exp. Med. 2011, 223, 45–54. [Google Scholar] [CrossRef]
- Vittal, R.; Horowitz, J.C.; Moore, B.B.; Zhang, H.; Martinez, F.J.; Toews, G.B.; Standiford, T.J.; Thannickal, V.J. Modulation of Prosurvival Signaling in Fibroblasts by a Protein Kinase Inhibitor Protects against Fibrotic Tissue Injury. Am. J. Pathol. 2005, 166, 367–375. [Google Scholar] [CrossRef][Green Version]
- Diaz-Osterman, C.J.; Ozmadenci, D.; Kleinschmidt, E.G.; Taylor, K.N.; Barrie, A.M.; Jiang, S.; Bean, L.M.; Sulzmaier, F.J.; Jean, C.; Tancioni, I.; et al. FAK activity sustains intrinsic and acquired ovarian cancer resistance to platinum chemotherapy. eLife 2019, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Villedieu, M.; Deslandes, E.; Duval, M.; Heron, J.-F.; Gauduchon, P.; Poulain, L. Acquisition of chemoresistance following discontinuous exposures to cisplatin is associated in ovarian carcinoma cells with progressive alteration of FAK, ERK and p38 activation in response to treatment. Gynecol. Oncol. 2006, 101, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, D.; Li, X.; Zhang, L.; Zhang, H.; Zhang, Y. Extracellular matrix protein ITGBL1 promotes ovarian cancer cell migration and adhesion through Wnt/PCP signaling and FAK/SRC pathway. Biomed. Pharmacother. 2016, 81, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Q.; Du, X.; Li, D.M.; Kong, P.Z.; Sun, Y.; Liu, P.F.; Wang, Q.S.; Feng, Y.M. ITGBL1 Is a Runx2 Transcriptional Target and Promotes Breast Cancer Bone Metastasis by Activating the TGFbeta Signaling Pathway. Cancer Res. 2015, 75, 3302–3313. [Google Scholar] [CrossRef] [PubMed]
- Leask, A. Focal Adhesion Kinase: A Key Mediator of Transforming Growth Factor Beta Signaling in Fibroblasts. Adv. Wound Care 2013, 2, 247–249. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Feng, J.-R.; Qiu, J.; Liu, L.; Xie, Y.; Zhang, Y.-P.; Liu, J.; Zhao, Q. ITGBL1 promotes migration, invasion and predicts a poor prognosis in colorectal cancer. Biomed. Pharmacother. 2018, 104, 172–180. [Google Scholar] [CrossRef]
- Yang, J.; Hou, Y.; Zhou, M.; Wen, S.; Zhou, J.; Xu, L.; Tang, X.; Du, Y.-E.; Hu, P.; Liu, M. Twist induces epithelial-mesenchymal transition and cell motility in breast cancer via ITGB1-FAK/ILK signaling axis and its associated downstream network. Int. J. Biochem. Cell Biol. 2016, 71, 62–71. [Google Scholar] [CrossRef]
- Yeh, Y.C.; Lin, H.H.; Tang, M.J. A tale of two collagen receptors, integrin beta1 and discoidin domain receptor 1, in epithelial cell differentiation. Am. J. Physiol. Cell Physiol. 2012, C1207–C1217. [Google Scholar] [CrossRef]
- Prosperi, J.R.; Goss, K.H. A Wnt-ow of opportunity: Targeting the Wnt/beta-catenin pathway in breast cancer. Curr. Drug Targets 2010, 11, 1074–1088. [Google Scholar] [CrossRef]
- Prosperi, J.R.; Khramtsov, A.I.; Khramtsova, G.F.; Goss, K.H. Apc mutation enhances PyMT-induced mammary tumorigenesis. PLoS ONE 2011, 6, e29339. [Google Scholar] [CrossRef]
- Williams, K.E.; Bundred, N.J.; Landberg, G.; Clarke, R.B.; Farnie, G. Focal Adhesion Kinase and Wnt Signaling Regulate Human Ductal Carcinoma In Situ Stem Cell Activity and Response to Radiotherapy. Stem Cells 2015, 33, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Kolev, V.N.; Tam, W.F.; Wright, Q.G.; McDermott, S.P.; Vidal, C.M.; Shapiro, I.M.; Xu, Q.; Wicha, M.S.; Pachter, J.A.; Weaver, D.T. Inhibition of FAK kinase activity preferentially targets cancer stem cells. Oncotarget 2017, 8, 51733–51747. [Google Scholar] [CrossRef] [PubMed]
- Shang, N.; Arteaga, M.; Zaidi, A.; Stauffer, J.; Cotler, S.J.; Zeleznik-Le, N.J.; Zhang, J.; Qiu, W. FAK is required for c-Met/beta-catenin-driven hepatocarcinogenesis. Hepatology 2015, 61, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Shang, N.; Wang, H.; Bank, T.; Perera, A.; Joyce, C.; Kuffel, G.; Zilliox, M.J.; Cotler, S.J.; Ding, X.; Dhanarajan, A.; et al. Focal Adhesion Kinase and beta-Catenin Cooperate to Induce Hepatocellular Carcinoma. Hepatology 2019, 70, 1631–1645. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Duan, J.; Wang, L.; Xiao, S.; Li, L.; Yan, X.; Yao, W.; Wu, L.; Zhang, S.; Zhang, Y.; et al. PTK2 promotes cancer stem cell traits in hepatocellular carcinoma by activating Wnt/beta-catenin signaling. Cancer Lett. 2019, 450, 132–143. [Google Scholar] [CrossRef]
- Delgado-Bellido, D.; Fernández-Cortés, M.; Rodríguez, M.I.; Serrano-Saenz, S.; Carracedo, A.; Garcia-Diaz, A.; Oliver, F.J. VE-cadherin promotes vasculogenic mimicry by modulating kaiso-dependent gene expression. Cell Death Differ. 2018, 26, 348–361. [Google Scholar] [CrossRef]
- Despeaux, M.; Chicanne, G.; Rouer, E.; De Toni-Costes, F.; Bertrand, J.; Mansat-De Mas, V.; Vergnolle, N.; Eaves, C.; Payrastre, B.; Girault, J.-A.; et al. Focal Adhesion Kinase Splice Variants Maintain Primitive Acute Myeloid Leukemia Cells Through Altered Wnt Signaling. Stem Cells 2012, 30, 1597–1610. [Google Scholar] [CrossRef]
- He, M.; Wang, Z.; Hu, W.; Wang, K.; Wang, D.; Fang, Z.; Huang, A.; Gao, Y.; Xia, J.; Li, W. Golgi Phosphoprotein 3 Promotes Malignant Phenotypes via FAK/Raf/MEK and Wnt/beta-Catenin Signaling Pathways in Human Renal Cell Carcinoma. J. Biomed. Nanotechnol. 2019, 15, 1812–1823. [Google Scholar] [CrossRef]
- Tai, H.C.; Chang, A.C.; Yu, H.J.; Huang, C.Y.; Tsai, Y.C.; Lai, Y.W.; Sun, H.L.; Tang, C.H.; Wang, S.W. Osteoblast-derived WNT-induced secreted protein 1 increases VCAM-1 expression and enhances prostate cancer metastasis by down-regulating miR-126. Oncotarget 2014, 5, 7589–7598. [Google Scholar] [CrossRef]
- Wu, D.; Shi, Z.; Xu, H.; Chen, R.; Xue, S.; Sun, X. Knockdown of Cripto-1 inhibits the proliferation, migration, invasion, and angiogenesis in prostate carcinoma cells. J. Biosci. 2017, 42, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.K.; Schiff, R. Mechanisms of Endocrine Resistance in Breast Cancer. Annu. Rev. Med. 2011, 62, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Hammond, W.A.; Swaika, A.; Mody, K. Pharmacologic resistance in colorectal cancer: A review. Ther. Adv. Med Oncol. 2015, 8, 57–84. [Google Scholar] [CrossRef]
- Bronte, G.; Incorvaia, L.; Rizzo, S.; Passiglia, F.; Galvano, A.; Rizzo, F.; Rolfo, C.; Fanale, D.; Listì, A.; Natoli, C.; et al. The resistance related to targeted therapy in malignant pleural mesothelioma: Why has not the target been hit yet? Crit. Rev. Oncol. 2016, 107, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, S.; Igea, A.; Arroyo, R.; Alcalde, V.; Canovas, B.; Orozco, M.; Nebreda, A.R.; Aloy, P. Quantification of Pathway Cross-talk Reveals Novel Synergistic Drug Combinations for Breast Cancer. Cancer Res. 2016, 77, 459–469. [Google Scholar] [CrossRef]
- Mohammad, R.; Muqbil, I.; Lowe, L.; Yedjou, C.; Hsu, H.-Y.; Lin, L.-T.; Siegelin, M.D.; Fimognari, C.; Kumar, N.B.; Dou, Q.P.; et al. Broad targeting of resistance to apoptosis in cancer. Semin. Cancer Biol. 2015, 35, S78–S103. [Google Scholar] [CrossRef]
- Sun, X.; Bao, J.; You, Z.; Chen, X.; Cui, J. Modeling of signaling crosstalk-mediated drug resistance and its implications on drug combination. Oncotarget 2016, 7, 63995–64006. [Google Scholar] [CrossRef]
- Martín-Orozco, E.; Sanchez-Fernandez, A.; Ortiz-Parra, I.; Ayala-San Nicolas, M. WNT Signaling in Tumors: The Way to Evade Drugs and Immunity. Front. Immunol. 2019, 10, 2854. [Google Scholar] [CrossRef]
- Shaw, H.V.; Koval, A.; Katanaev, V.L. Targeting the Wnt signalling pathway in cancer: Prospects and perils. Swiss Med Wkly. 2019, 149, w20129. [Google Scholar] [CrossRef]
- Jung, Y.S.; Park, J.I. Wnt signaling in cancer: Therapeutic targeting of Wnt signaling beyond beta-catenin and the destruction complex. Exp. Mol. Med. 2020, 52, 183–191. [Google Scholar] [CrossRef]
- Gurney, A.; Axelrod, F.; Bond, C.J.; Cain, J.; Chartier, C.; Donigan, L.; Fischer, M.; Chaudhari, A.; Ji, M.; Kapoun, A.M.; et al. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 11717–11722. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.M.; Cancilla, B.; Yeung, V.P.; Cattaruzza, F.; Chartier, C.; Murriel, C.L.; Cain, J.; Tam, R.; Cheng, C.-Y.; Evans, J.W.; et al. WNT antagonists exhibit unique combinatorial antitumor activity with taxanes by potentiating mitotic cell death. Sci. Adv. 2017, 3, e1700090. [Google Scholar] [CrossRef]
- Jimeno, A.; Gordon, M.; Chugh, R.; Messersmith, W.A.; Mendelson, D.; Dupont, J.; Stagg, R.; Kapoun, A.M.; Xu, L.; Uttamsingh, S.; et al. A First-in-Human Phase I Study of the Anticancer Stem Cell Agent Ipafricept (OMP-54F28), a Decoy Receptor for Wnt Ligands, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 7490–7497. [Google Scholar] [CrossRef]
- Diamond, J.R.; Becerra, C.; Richards, D.; Mita, A.; Osborne, C.; O’Shaughnessy, J.; Zhang, C.; Henner, R.; Kapoun, A.M.; Xu, L.; et al. Phase Ib clinical trial of the anti-frizzled antibody vantictumab (OMP-18R5) plus paclitaxel in patients with locally advanced or metastatic HER2-negative breast cancer. Breast Cancer Res. Treat. 2020, 184, 53–62. [Google Scholar] [CrossRef]
- Madan, B.; McDonald, M.J.; Foxa, G.E.; Diegel, C.R.; Williams, B.O.; Virshup, D.M. Bone loss from Wnt inhibition mitigated by concurrent alendronate therapy. Bone Res. 2018, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- OncoMed Pharmaceuticals, Inc. Annual Report. Available online: https://sec.report/Document/1302573/000156459019006795/omed-10k_20181231.htm (accessed on 23 November 2020).
- Säfholm, A.; Leandersson, K.; Dejmek, J.; Nielsen, C.K.; Villoutreix, B.O.; Andersson, T. A Formylated Hexapeptide Ligand Mimics the Ability of Wnt-5a to Impair Migration of Human Breast Epithelial Cells. J. Biol. Chem. 2005, 281, 2740–2749. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Faderl, S.; Pagel, J.M.; Jung, C.W.; Yoon, S.-S.; Pardanani, A.D.; Becker, P.S.; Lee, H.; Choi, J.; Lee, K.; et al. Phase 1 study of CWP232291 in patients with relapsed or refractory acute myeloid leukemia and myelodysplastic syndrome. Blood Adv. 2020, 4, 2032–2043. [Google Scholar] [CrossRef] [PubMed]
- Tam, B.Y.; Chiu, K.; Chung, H.; Bossard, C.; Nguyen, J.D.; Creger, E.; Eastman, B.W.; Mak, C.C.; Ibanez, M.; Ghias, A.; et al. The CLK inhibitor SM08502 induces anti-tumor activity and reduces Wnt pathway gene expression in gastrointestinal cancer models. Cancer Lett. 2020, 473, 186–197. [Google Scholar] [CrossRef]
- Kadowaki, T.; Wilder, E.; Klingensmith, J.; Zachary, K.; Perrimon, N. The segment polarity gene porcupine encodes a putative multitransmembrane protein involved in Wingless processing. Genes Dev. 1996, 10, 3116–3128. [Google Scholar] [CrossRef]
- Zimmerli, D.; Hausmann, G.; Cantù, C.; Basler, K. Pharmacological interventions in the Wnt pathway: Inhibition of Wnt secretion versus disrupting the protein-protein interfaces of nuclear factors. Br. J. Pharmacol. 2017, 174, 4600–4610. [Google Scholar] [CrossRef]
- Sebio, A.; Kahn, M.; Lenz, H.J. The potential of targeting Wnt/beta-catenin in colon cancer. Expert Opin. Ther. Targets 2014, 18, 611–615. [Google Scholar] [CrossRef] [PubMed]
- Roy-Luzarraga, M.; Hodivala-Dilke, K. Molecular Pathways: Endothelial Cell FAK—A Target for Cancer Treatment. Clin. Cancer Res. 2016, 22, 3718–3724. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.-C.; Gan, H.K.; Blagden, S.P.; Plummer, R.; Arkenau, H.T.; Ranson, M.; Evans, T.R.J.; Zalcman, G.; Bahleda, R.; Hollebecque, A.; et al. A phase I, pharmacokinetic and pharmacodynamic study of GSK2256098, a focal adhesion kinase inhibitor, in patients with advanced solid tumors. Ann. Oncol. 2016, 27, 2268–2274. [Google Scholar] [CrossRef] [PubMed]
- Mak, G.; Soria, J.-C.; Blagden, S.P.; Plummer, R.; Fleming, R.A.; Nebot, N.; Zhang, J.; Mazumdar, J.; Rogan, D.; Gazzah, A.; et al. A phase Ib dose-finding, pharmacokinetic study of the focal adhesion kinase inhibitor GSK2256098 and trametinib in patients with advanced solid tumours. Br. J. Cancer 2019, 120, 975–981. [Google Scholar] [CrossRef]
- De Jonge, M.; Steeghs, N.; Lolkema, M.; Hotte, S.J.; Hirte, H.; Van Der Biessen, D.A.J.; Razak, A.R.A.; De Vos, F.Y.; Verheijen, R.B.; Schnell, D.; et al. Phase I Study of BI 853520, an Inhibitor of Focal Adhesion Kinase, in Patients with Advanced or Metastatic Nonhematologic Malignancies. Target. Oncol. 2019, 14, 43–55. [Google Scholar] [CrossRef]
- Infante, J.R.; Camidge, D.R.; Mileshkin, L.R.; Chen, E.X.; Hicks, R.J.; Rischin, D.; Fingert, H.; Pierce, K.J.; Xu, H.; Roberts, W.G.; et al. Safety, Pharmacokinetic, and Pharmacodynamic Phase I Dose-Escalation Trial of PF-00562271, an Inhibitor of Focal Adhesion Kinase, in Advanced Solid Tumors. J. Clin. Oncol. 2012, 30, 1527–1533. [Google Scholar] [CrossRef]
- Lv, P.-C.; Jiang, A.-Q.; Zhang, W.-M.; Zhu, H.-L. FAK inhibitors in Cancer, a patent review. Expert Opin. Ther. Patents 2018, 28, 139–145. [Google Scholar] [CrossRef]
- Jones, S.F.; Siu, L.L.; Bendell, J.C.; Cleary, J.M.; Razak, A.R.A.; Infante, J.R.; Pandya, S.S.; Bedard, P.L.; Pierce, K.J.; Houk, B.; et al. A phase I study of VS-6063, a second-generation focal adhesion kinase inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2015, 33, 1100–1107. [Google Scholar] [CrossRef]
- Shimizu, T.; Fukuoka, K.; Takeda, M.; Iwasa, T.; Yoshida, T.; Horobin, J.; Keegan, M.; Vaickus, L.; Chavan, A.; Padval, M.; et al. A first-in-Asian phase 1 study to evaluate safety, pharmacokinetics and clinical activity of VS-6063, a focal adhesion kinase (FAK) inhibitor in Japanese patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2016, 77, 997–1003. [Google Scholar] [CrossRef]
- Shapiro, I.M.; Kolev, V.N.; Vidal, C.M.; Kadariya, Y.; Ring, J.E.; Wright, Q.; Weaver, D.T.; Menges, C.; Padval, M.; McClatchey, A.I.; et al. Merlin Deficiency Predicts FAK Inhibitor Sensitivity: A Synthetic Lethal Relationship. Sci. Transl. Med. 2014, 6, 237ra68. [Google Scholar] [CrossRef]
- Gerber, D.E.; Camidge, D.R.; Morgensztern, D.; Cetnar, J.; Kelly, R.; Ramalingam, S.S.; Spigel, D.R.; Jeong, W.; Scaglioni, P.P.; Zhang, S.; et al. Phase 2 study of the focal adhesion kinase inhibitor defactinib (VS-6063) in previously treated advanced KRAS mutant non-small cell lung cancer. Lung Cancer 2020, 139, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, A.; Pharaon, R.R.; Nam, A.; Salgia, S.; Kulkarni, P.; Massarelli, E. FAK-targeted and combination therapies for the treatment of cancer: An overview of phase I and II clinical trials. Expert Opin. Investig. Drugs 2020, 29, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Bernards, R. A Missing Link in Genotype-Directed Cancer Therapy. Cell 2012, 151, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Shi, P.-J.; Xu, L.H.; Lin, K.; Weng, W.-J.; Fang, J. Synergism between the mTOR inhibitor rapamycin and FAK down-regulation in the treatment of acute lymphoblastic leukemia. J. Hematol. Oncol. 2016, 9, 12. [Google Scholar] [CrossRef]
- Golubovskaya, V.; Beviglia, L.; Xu, L.H.; Earp, H.S., 3rd; Craven, R.; Cance, W. Dual inhibition of focal adhesion kinase and epidermal growth factor receptor pathways cooperatively induces death receptor-mediated apoptosis in human breast cancer cells. J. Biol. Chem. 2002, 277, 38978–38987. [Google Scholar] [CrossRef]
- Golubovskaya, V.M.; Gross, S.; Kaur, A.S.; Wilson, R.I.; Xu, L.-H.; Yang, X.H.; Cance, W.G. Simultaneous inhibition of focal adhesion kinase and SRC enhances detachment and apoptosis in colon cancer cell lines. Mol. Cancer Res. 2003, 1, 755–764. [Google Scholar]
- Tamura, M.; Gu, J.; Matsumoto, K.; Aota, S.; Parsons, R.; Yamada, K.M. Inhibition of Cell Migration, Spreading, and Focal Adhesions by Tumor Suppressor PTEN. Science 1998, 280, 1614–1617. [Google Scholar] [CrossRef]
- Alfieri, R.; Giovannetti, E.; Bonelli, M.; Cavazzoni, A. New Treatment Opportunities in Phosphatase and Tensin Homolog (PTEN)-Deficient Tumors: Focus on PTEN/Focal Adhesion Kinase Pathway. Front. Oncol. 2017, 7, 170. [Google Scholar] [CrossRef]
- Doglioni, C.; Dei Tos, A.P.; Laurino, L.; Iuzzolino, P.; Chiarelli, C.; Celio, M.R.; Viale, G. Calretinin: A Novel Immunocytochemical Marker for Mesothelioma. Am. J. Surg. Pathol. 1996, 20, 1037–1046. [Google Scholar] [CrossRef]
- Tang, J.; Karhinen, L.; Xu, T.; Szwajda, A.; Yadav, B.; Wennerberg, K.; Aittokallio, T. Target Inhibition Networks: Predicting Selective Combinations of Druggable Targets to Block Cancer Survival Pathways. PLoS Comput. Biol. 2013, 9, e1003226. [Google Scholar] [CrossRef]
- Ackermann, T.F.; Kempe, D.S.; Lang, F.; Lang, U.E. Hyperactivity and Enhanced Curiosity of Mice Expressing PKB/SGK-resistant Glycogen Synthase Kinase-3 (GSK-3). Cell. Physiol. Biochem. 2010, 25, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Crampton, S.P.; Wu, B.; Park, E.J.; Kim, J.H.; Solomon, C.; Waterman, M.L.; Hughes, C.C. Integration of the beta-catenin-dependent Wnt pathway with integrin signaling through the adaptor molecule Grb2. PLoS ONE 2009, 4, e7841. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yuan, H.; Xie, W.; Mao, J.; Caruso, A.M.; McMahon, A.; Sussman, D.J.; Wu, D. Dishevelled proteins lead to two signaling pathways. Regulation of LEF-1 and c-Jun N-terminal kinase in mammalian cells. J. Biol. Chem. 1999, 274, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Vivanco, I.; Palaskas, N.; Tran, C.; Finn, S.P.; Getz, G.; Kennedy, N.J.; Jiao, J.; Rose, J.; Xie, W.; Loda, M.; et al. Identification of the JNK Signaling Pathway as a Functional Target of the Tumor Suppressor PTEN. Cancer Cell 2007, 11, 555–569. [Google Scholar] [CrossRef]
- Lehar, J.; Krueger, A.S.; Avery, W.; Heilbut, A.M.; Johansen, L.M.; Price, E.R.; Rickles, R.J.; Short 3rd, G.F.; Staunton, J.E.; Jin, X.; et al. Synergistic drug combinations tend to improve therapeutically relevant selectivity. Nat. Biotechnol. 2009, 27, 659–666. [Google Scholar] [CrossRef]
- Chen, S.-H.; Lahav, G. Two is better than one; toward a rational design of combinatorial therapy. Curr. Opin. Struct. Biol. 2016, 41, 145–150. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).