Insights into Transcriptional Repression of the Homologous Toxin-Antitoxin Cassettes yefM-yoeB and axe-txe


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
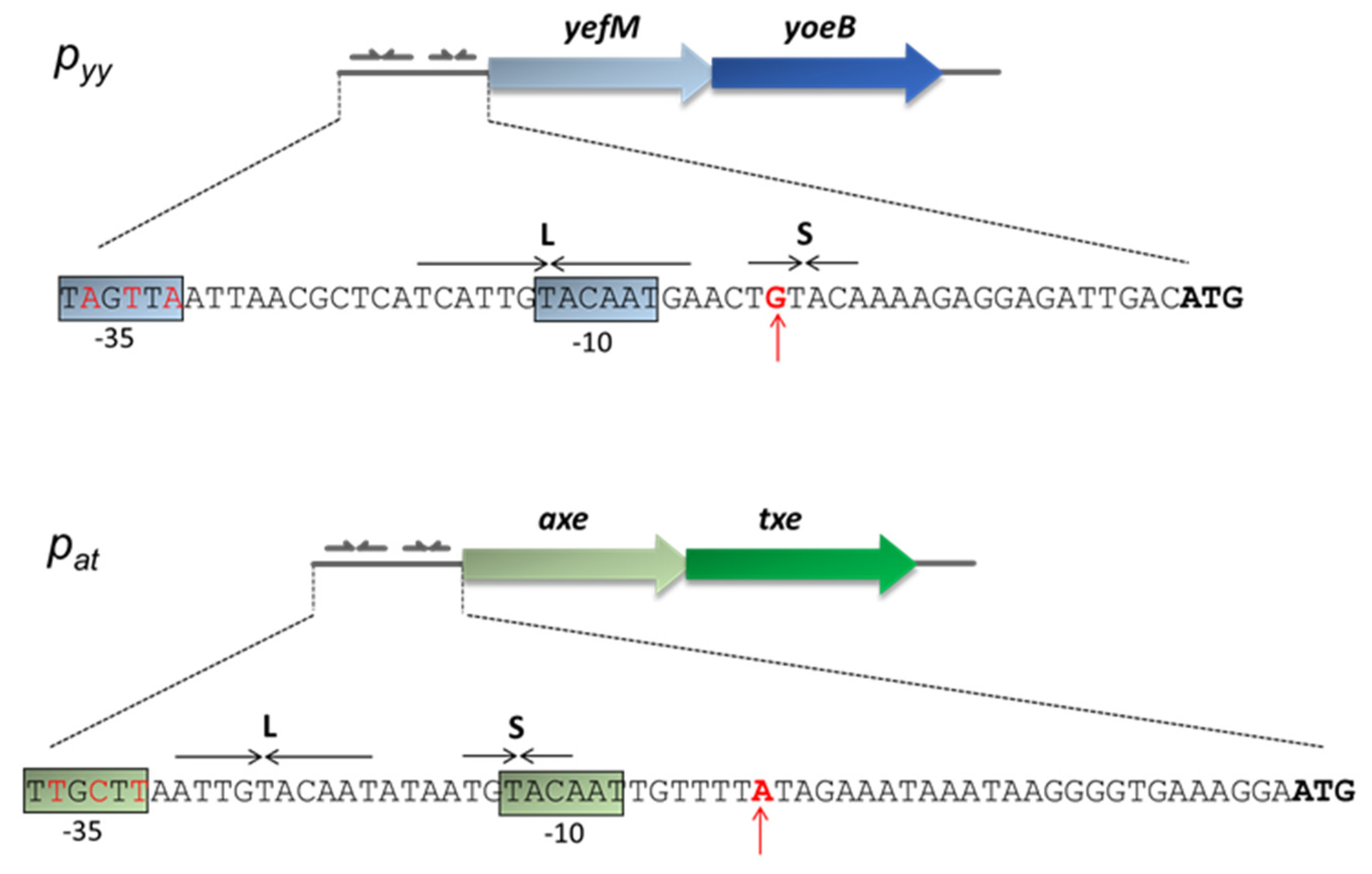
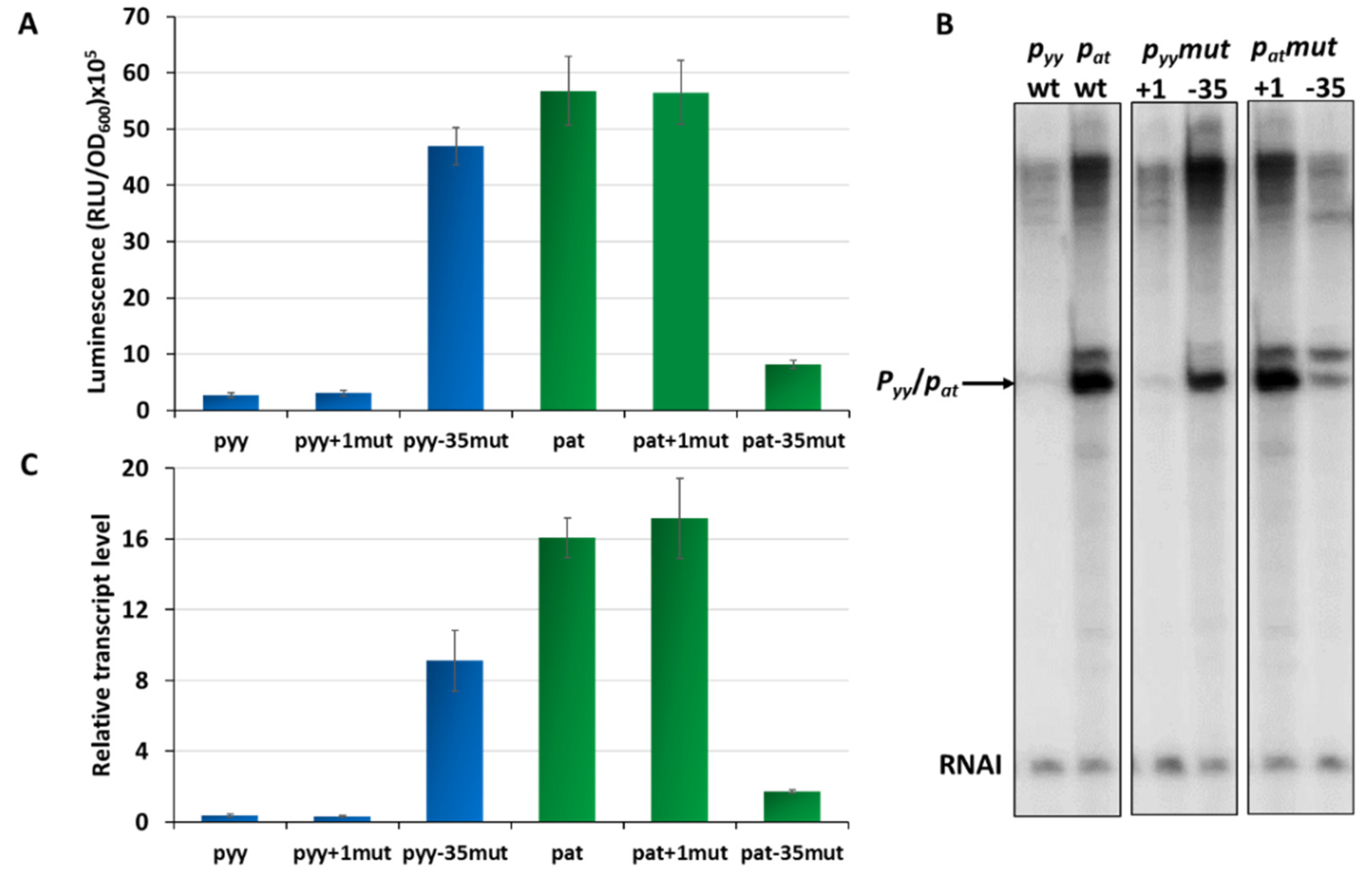
2.1. Differences in the −35 Box Sequence are Responsible for Different Strengths of the pat and pyy Promoters
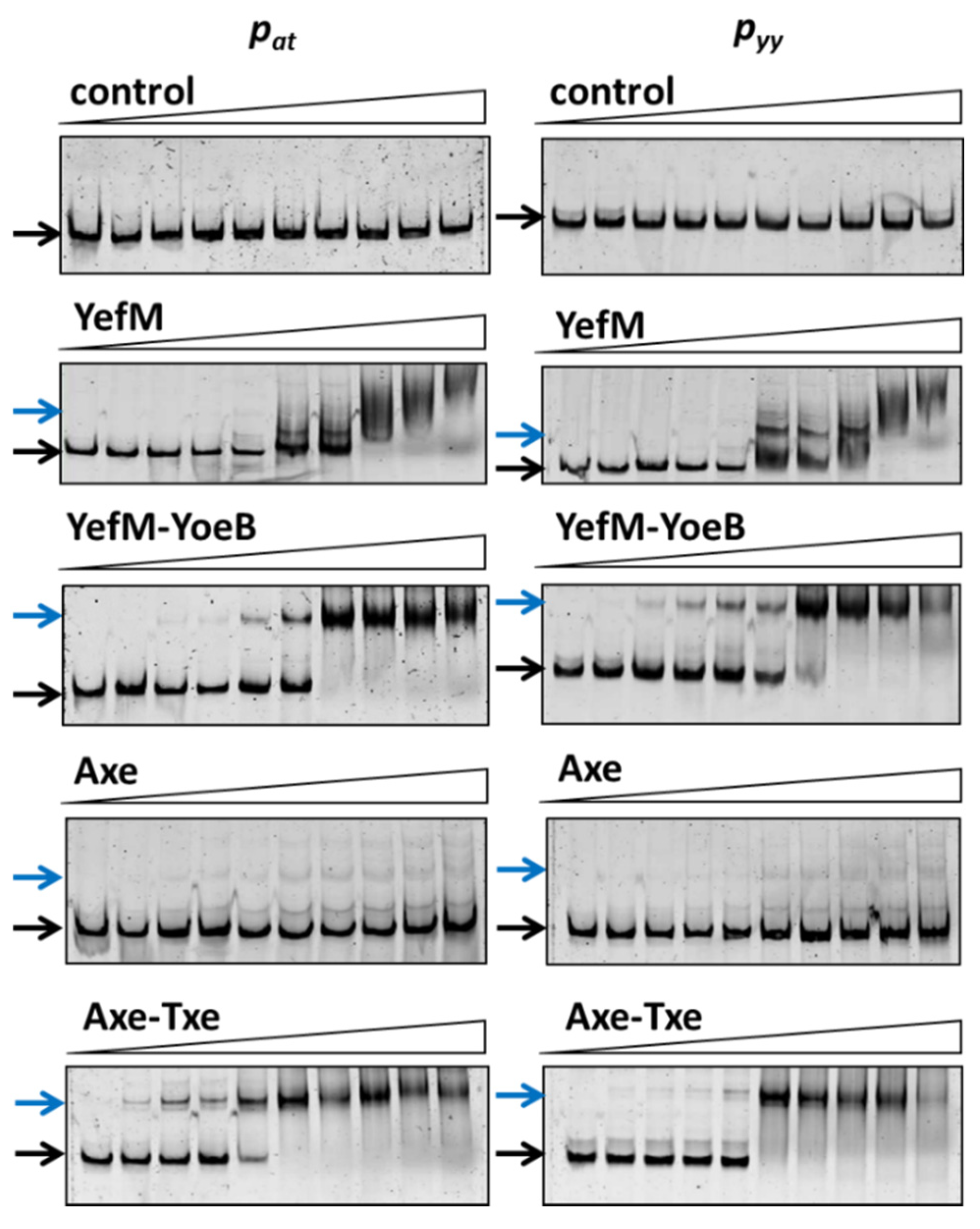
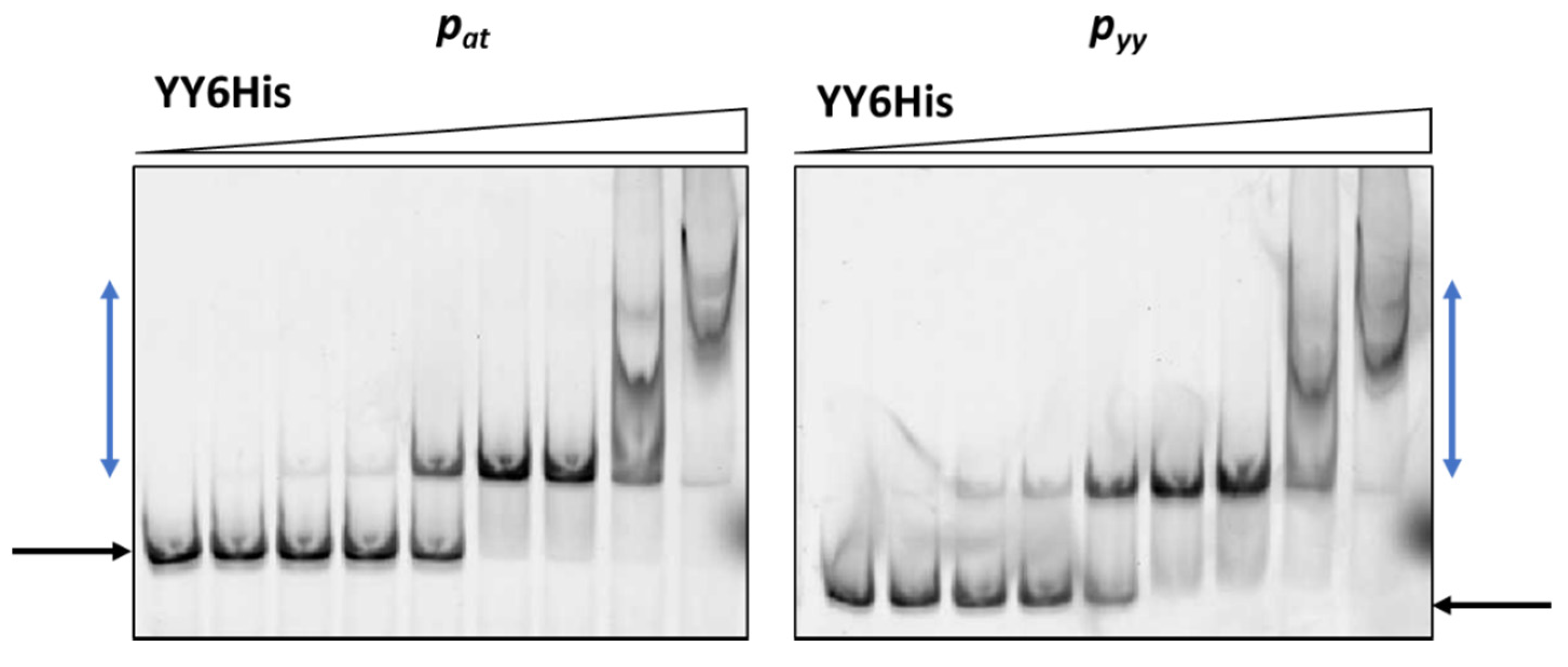
2.2. Axe-Txe and YefM-YoeB Repressor Complexes Exhibit Similar Specificity to Cognate and Non-Cognate Operator Sequences
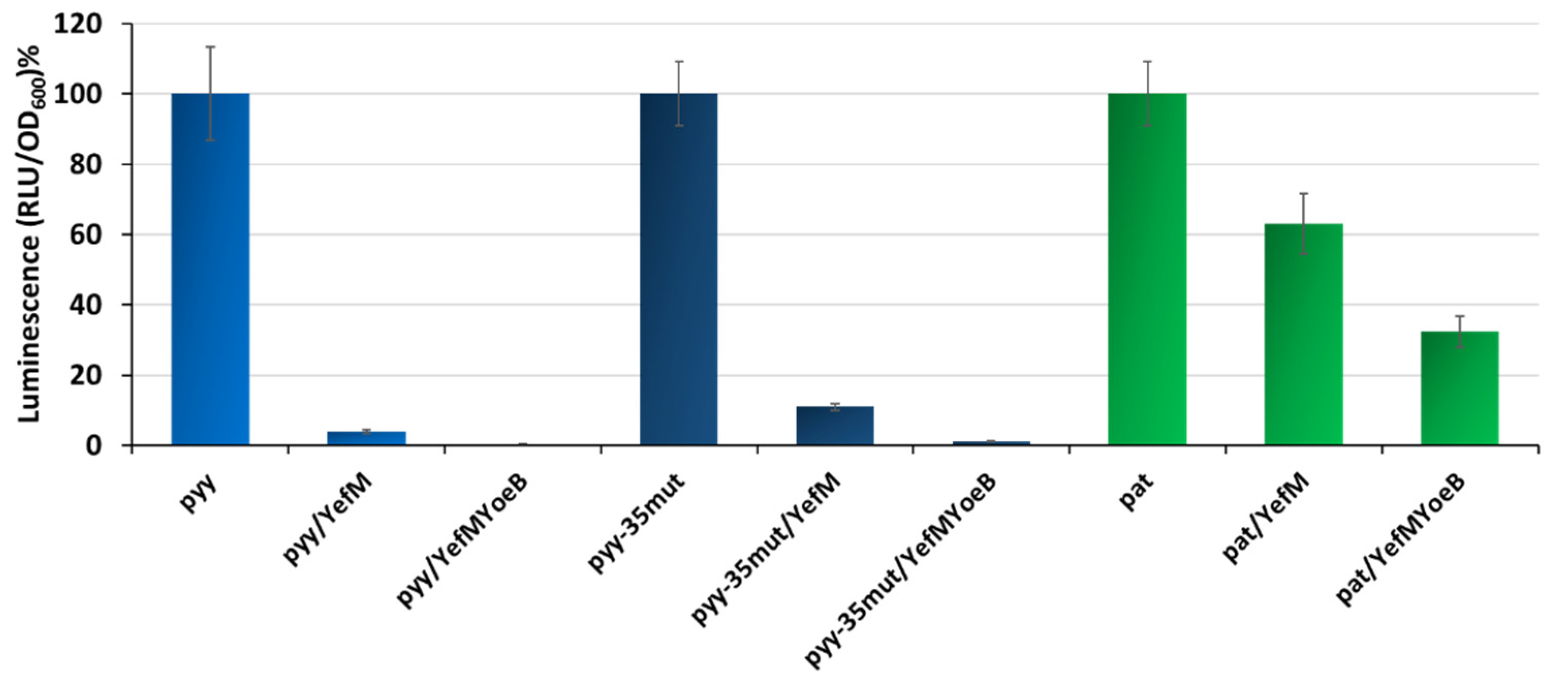
2.3. YefM-YoeB Complex Represses Transcription at the pat and pyy Promoters with Different Efficiency
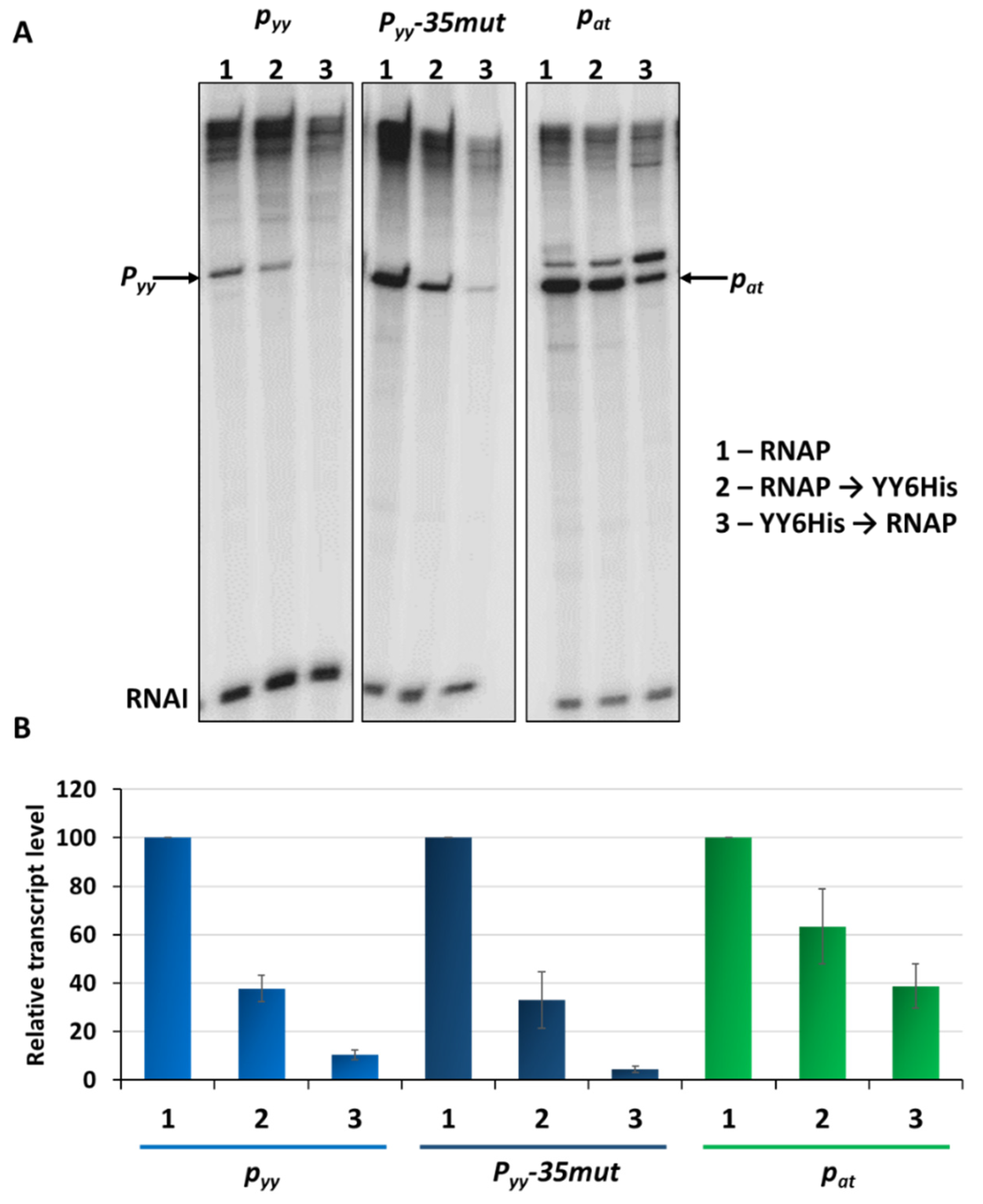
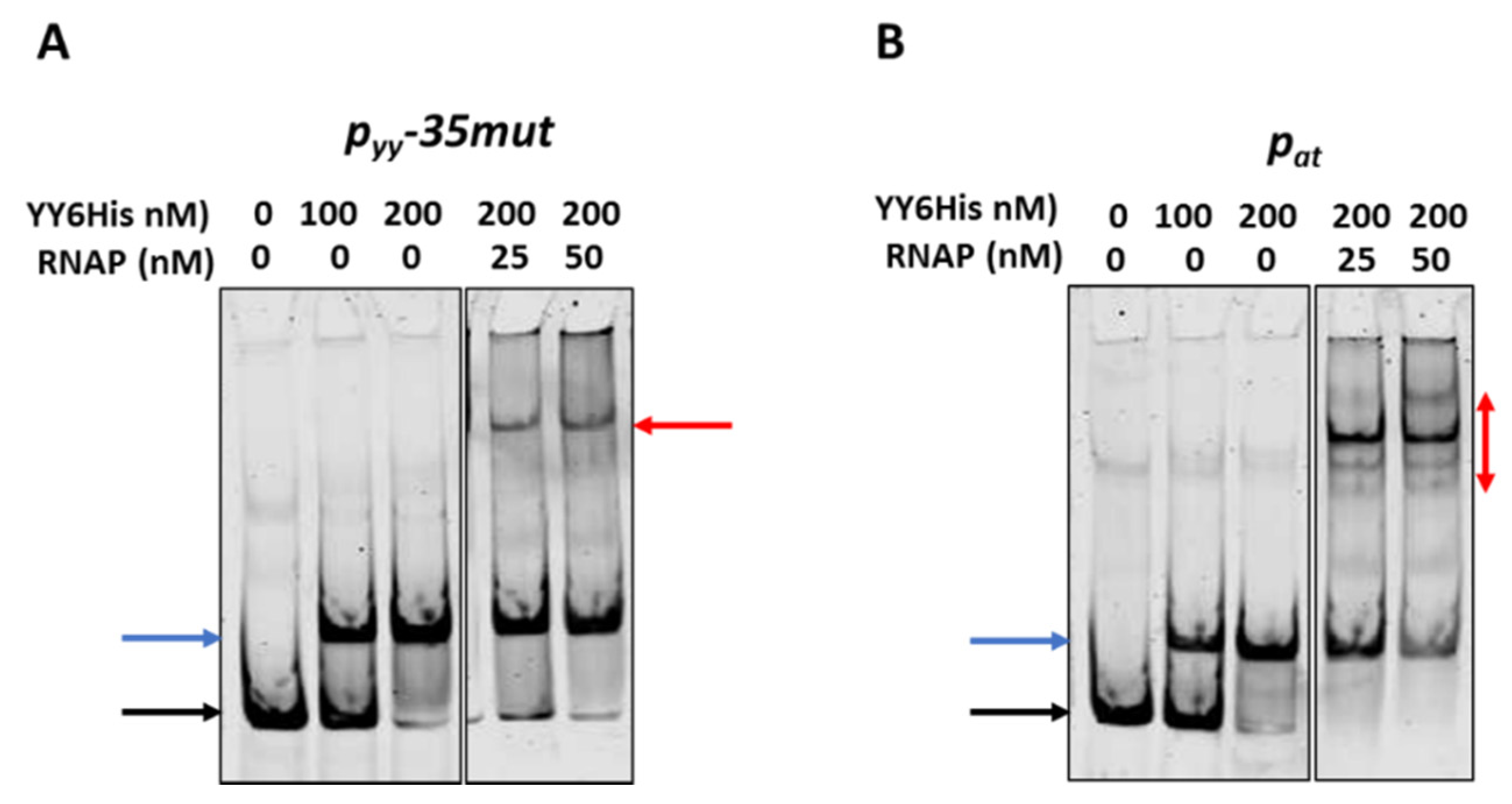
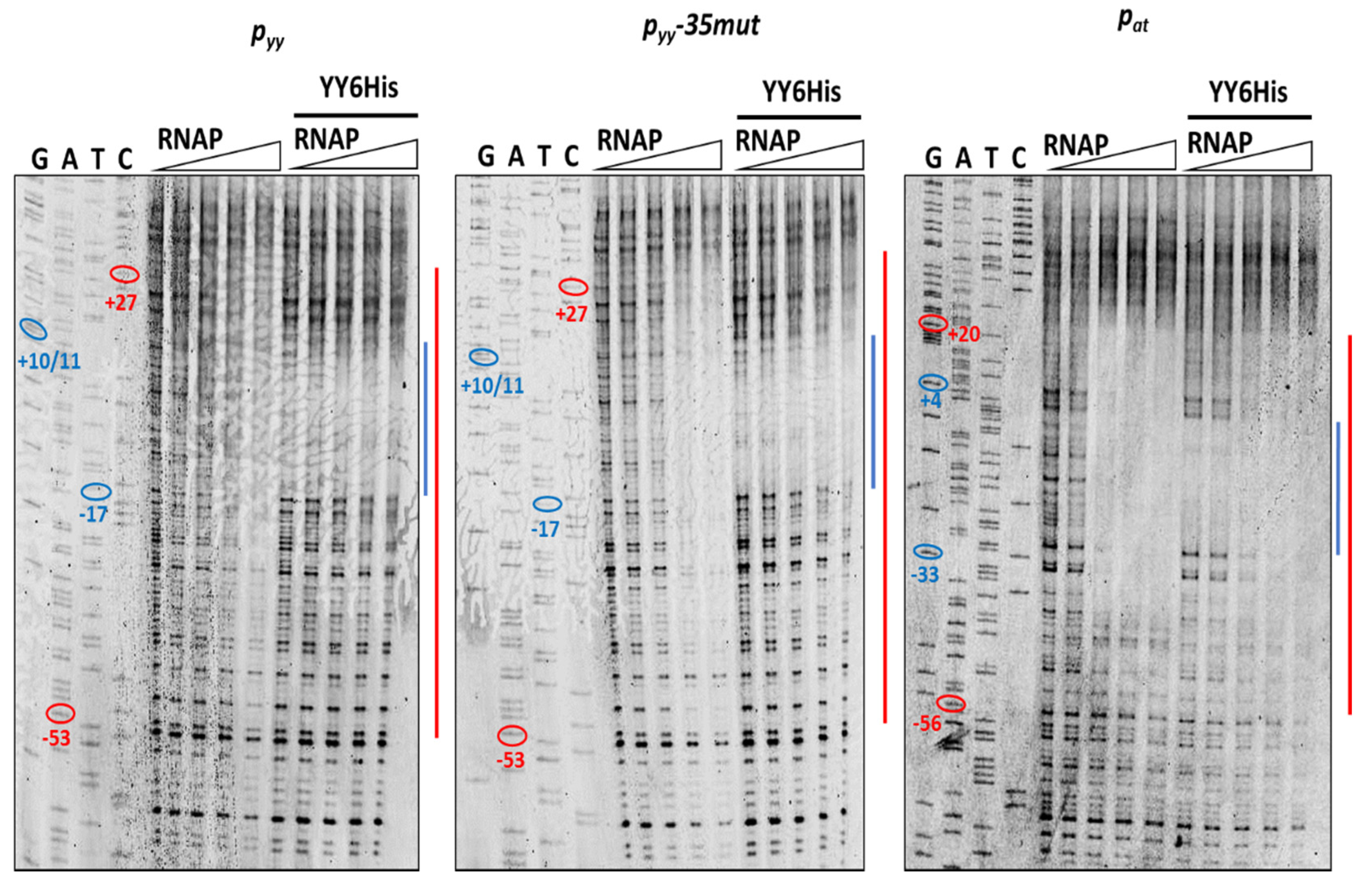
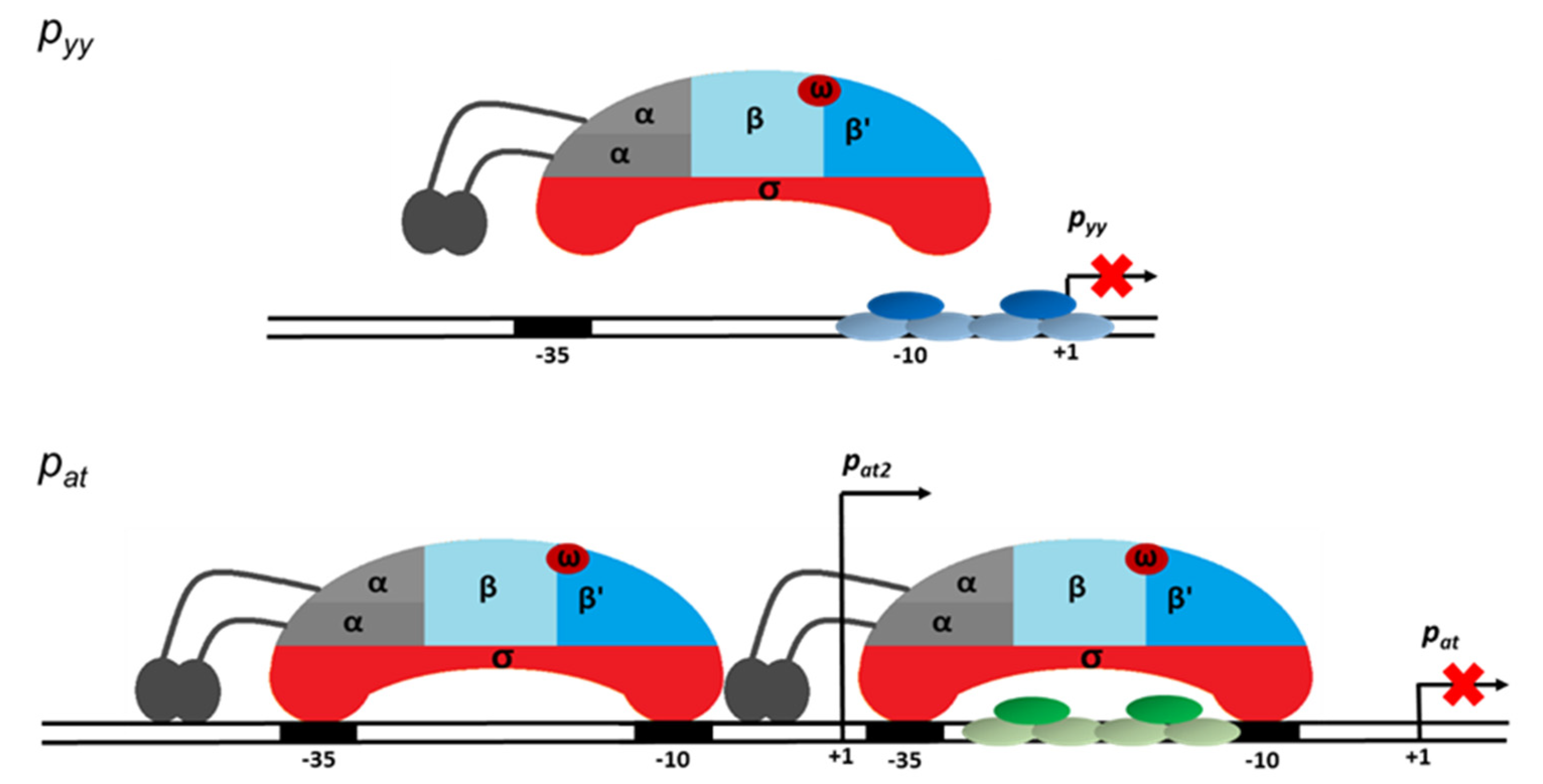
2.4. Different Repression Level of pyy and pat May Result from Repressor Acting on Different Steps in the Transcription Initiation Process
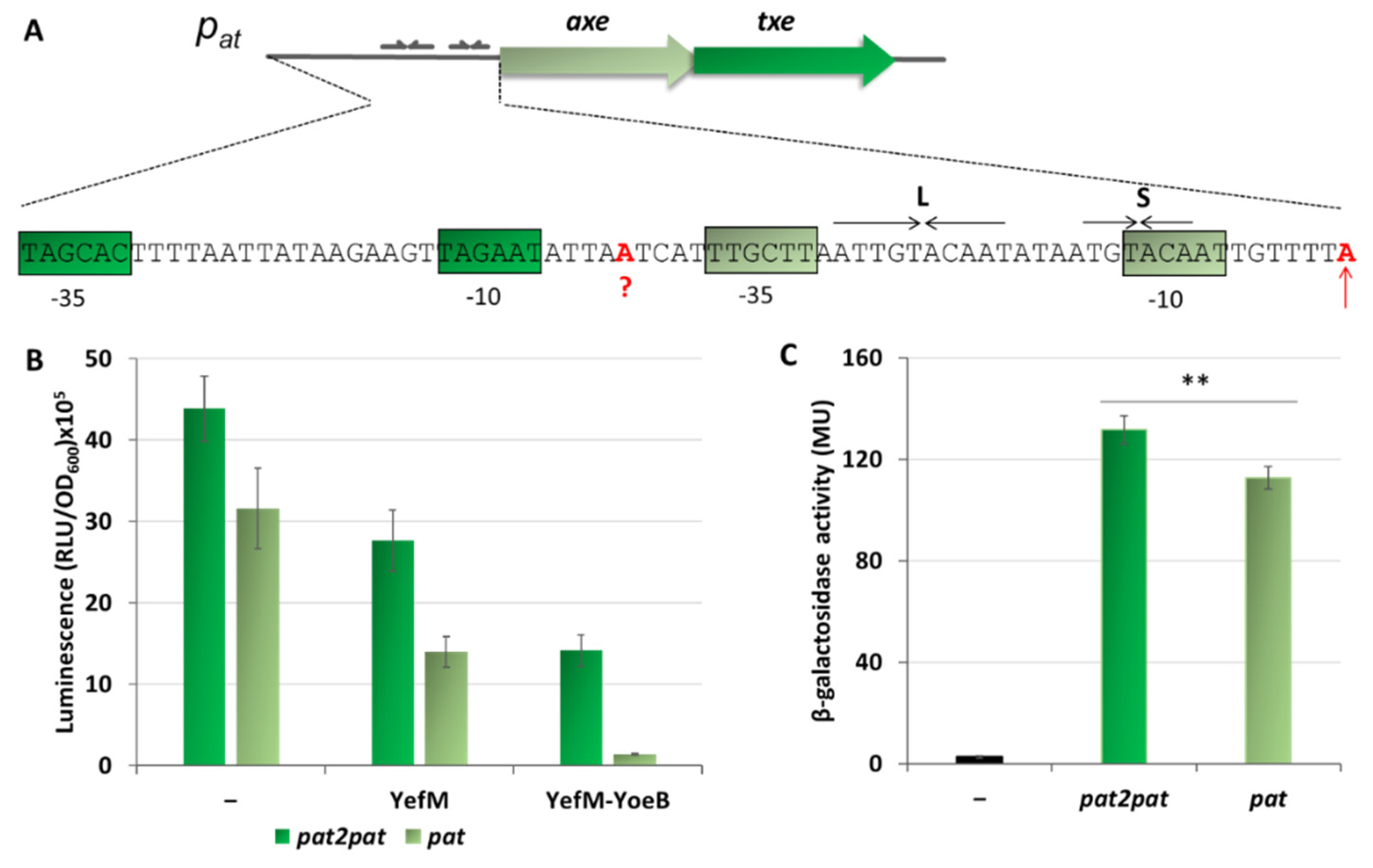
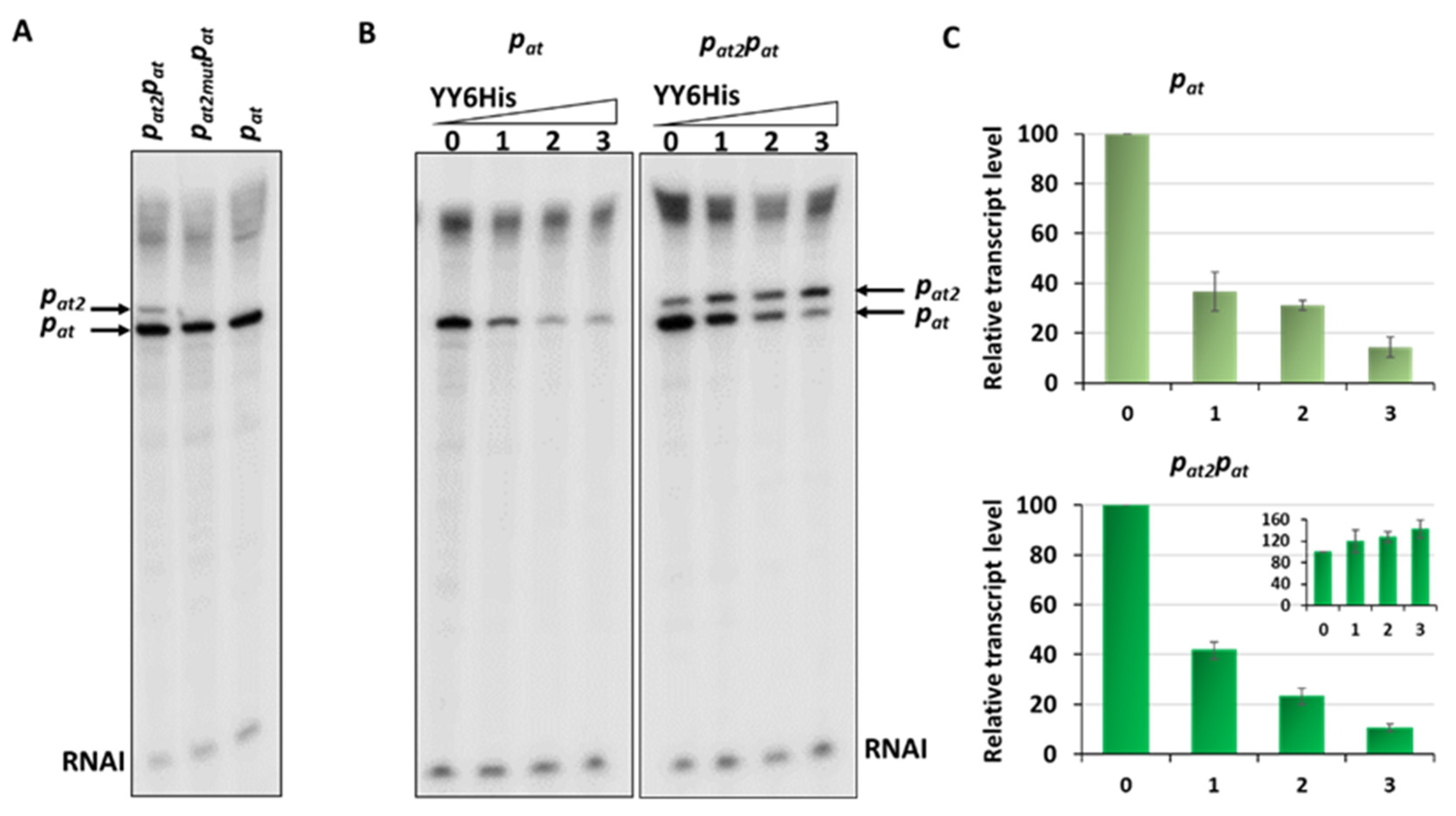
2.5. Additional Active Promoter that Contributes to the Overall Transcriptional Activity of the Axe-Txe Cassette Is Identified Just Upstream of pat
3. Discussion
4. Materials and Methods
4.1. Strains, Plasmids and Oligonucleotides
4.2. Crude Extract Preparation
4.3. YefM-YoeB Protein Complex Purification
4.4. RNA Polymerase Holoenzyme Purification
4.5. Promoter Fusion Studies and Bioluminescence Assays in Escherichia coli Cells
4.6. Electrophoretic Mobility Shift Assays (EMSA)
4.7. In vitro Transcription Analysis
4.8. DNaseI Footprinting Assays
4.9. Promoter Fusion Studies and β-Galactosidase Assays in Enterococcal cells
4.10. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Murakami, K.S.; Darst, S.A. Bacterial RNA polymerases: The wholo story. Curr. Opin. Struct. Biol. 2003, 13, 31–39. [Google Scholar] [CrossRef]
- Murakami, K.S.; Masuda, S.; Darst, S.A. Structural Basis of Transcription Initiation: RNA Polymerase Holoenzyme at 4 A Resolution. Science 2002, 296, 1280–1284. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.A.; Muzzin, O.; Chlenov, M.; Sun, J.L.; Olson, C.; Weinman, O.; Trester-Zedlitz, M.L.; Darst, S.A. Structure of the Bacterial RNA Polymerase Promoter Specificity σ Subunit. Mol. Cell 2002, 9, 527–539. [Google Scholar] [CrossRef]
- Murakami, K.S. X-ray Crystal Structure of Escherichia coli RNA Polymerase σ70 Holoenzyme. J. Biol. Chem. 2013, 288, 9126–9134. [Google Scholar] [CrossRef] [PubMed]
- Shultzaberger, R.K.; Chen, Z.; Lewis, K.A.; Schneider, T.D. Anatomy of Escherichia coli σ70 promoters. Nucleic Acids Res. 2007, 35, 771–788. [Google Scholar] [CrossRef] [PubMed]
- Hook-Barnard, I.G.; Hinton, D.M. Transcription Initiation by Mix and Match Elements: Flexibility for Polymerase Binding to Bacterial Promoters. Gene Regul. Syst. Biol. 2007, 1, 275–293. [Google Scholar] [CrossRef]
- Ruff, E.F.; Record, M.T.; Artsimovitch, I. Initial Events in Bacterial Transcription Initiation. Biomolecules 2015, 5, 1035–1062. [Google Scholar] [CrossRef] [PubMed]
- Meng, C.A.; Fazal, F.M.; Block, S.M. Real-time observation of polymerase-promoter contact remodeling during transcription initiation. Nat. Commun. 2017, 8, 1178. [Google Scholar] [CrossRef]
- Glyde, R.; Ye, F.; Jovanovic, M.; Kotta-Loizou, I.; Buck, M.; Zhang, X. Structures of Bacterial RNA Polymerase Complexes Reveal the Mechanism of DNA Loading and Transcription Initiation. Mol. Cell 2018, 70, 1111–1120.e3. [Google Scholar] [CrossRef]
- Mazumder, A.; Kapanidis, A.N. Recent Advances in Understanding σ70-Dependent Transcription Initiation Mechanisms. J. Mol. Biol. 2019, 431, 3947–3959. [Google Scholar] [CrossRef]
- Saecker, R.M.; Record, M.T.; DeHaseth, P.L. Mechanism of Bacterial Transcription Initiation: RNA Polymerase—Promoter Binding, Isomerization to Initiation-Competent Open Complexes, and Initiation of RNA Synthesis. J. Mol. Biol. 2011, 412, 754–771. [Google Scholar] [CrossRef] [PubMed]
- Feklistov, A. RNA polymerase: In search of promoters. Ann. N. Y. Acad. Sci. 2013, 1293, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Hawley, D.K.; McClure, W.R. Compilation and analysis of Escherichia coli promoter DNA sequences. Nucleic Acids Res. 1983, 11, 2237–2255. [Google Scholar] [CrossRef] [PubMed]
- Huerta, A.M.; Collado-Vides, J. Sigma70 Promoters in Escherichia coli: Specific Transcription in Dense Regions of Overlapping Promoter-like Signals. J. Mol. Biol. 2003, 333, 261–278. [Google Scholar] [CrossRef]
- Huerta, A.M.; Francino, M.P.; Morett, E.; Collado-Vides, J. Selection for Unequal Densities of σ70 Promoter-Like Signals in Different Regions of Large Bacterial Genomes. PLoS Genet. 2006, 2, e185. [Google Scholar] [CrossRef]
- Goodrich, J.A.; McClure, W.R. Competing promoters in prokaryotic transcription. Trends Biochem. Sci. 1991, 16, 394–397. [Google Scholar] [CrossRef]
- Browning, D.F.; Busby, S.J.W. The regulation of bacterial transcription initiation. Nat. Rev. Genet. 2004, 2, 57–65. [Google Scholar] [CrossRef]
- Bintu, L.; Buchler, N.E.; Garcia, H.G.; Gerland, U.; Hwa, T.; Kondev, J.; Phillips, R. Transcriptional regulation by the numbers: Models. Curr. Opin. Genet. Dev. 2005, 15, 116–124. [Google Scholar] [CrossRef]
- Payankaulam, S.; Li, L.M.; Arnosti, D.N. Transcriptional Repression: Conserved and Evolved Features. Curr. Biol. 2010, 20, R764–R771. [Google Scholar] [CrossRef]
- Browning, D.F.; Butala, M.; Busby, S.J. Bacterial Transcription Factors: Regulation by Pick “N” Mix. J. Mol. Biol. 2019, 431, 4067–4077. [Google Scholar] [CrossRef]
- Collado-Vides, J.; Magasanik, B.; Gralla, J.D. Control site location and transcriptional regulation in Escherichia coli. Microbiol. Rev. 1991, 55, 371–394. [Google Scholar] [CrossRef] [PubMed]
- Segal, E.; Widom, J. From DNA sequence to transcriptional behaviour: A quantitative approach. Nat. Rev. Genet. 2009, 10, 443–456. [Google Scholar] [CrossRef] [PubMed]
- Garcia, H.G.; Sanchez, A.; Boedicker, J.Q.; Osborne, M.L.; Gelles, J.; Kondev, J.; Phillips, R. Operator Sequence Alters Gene Expression Independently of Transcription Factor Occupancy in Bacteria. Cell Rep. 2012, 2, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Kędzierska, B.; Hayes, F. Transcriptional Control of Toxin-Antitoxin Expression: Keeping Toxins under Wraps Until the Time is Right. In Stress and Environmental Regulation of Gene Expression and Adaptation in Bacteria; John Wiley & Sons: Hoboken, NJ, USA, 2016; Volume 1, pp. 463–472. [Google Scholar]
- Hayes, F.; Kędzierska, B. Regulating Toxin-Antitoxin Expression: Controlled Detonation of Intracellular Molecular Timebombs. Toxins 2014, 6, 337–358. [Google Scholar] [CrossRef]
- Echan, W.T.; Espinosa, M.; Yeo, C.C. Keeping the Wolves at Bay: Antitoxins of Prokaryotic Type II Toxin-Antitoxin Systems. Front. Mol. Biosci. 2016, 3, 9. [Google Scholar] [CrossRef]
- Harms, A.; Brodersen, D.E.; Mitarai, N.; Gerdes, K. Toxins, Targets, and Triggers: An Overview of Toxin-Antitoxin Biology. Mol. Cell 2018, 70, 768–784. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Park, J.-H.; Inouye, M. Toxin-Antitoxin Systems in Bacteria and Archaea. Annu. Rev. Genet. 2011, 45, 61–79. [Google Scholar] [CrossRef]
- Hayes, F.; Van Melderen, L. Toxins-antitoxins: Diversity, evolution and function. Crit. Rev. Biochem. Mol. Biol. 2011, 46, 386–408. [Google Scholar] [CrossRef]
- Jurėnas, D.; Van Melderen, L. The Variety in the Common Theme of Translation Inhibition by Type II Toxin-Antitoxin Systems. Front. Genet. 2020, 11, 1–19. [Google Scholar] [CrossRef]
- Lee, K.-Y.; Lee, B.-J. Structure, Biology, and Therapeutic Application of Toxin-Antitoxin Systems in Pathogenic Bacteria. Toxins 2016, 8, 305. [Google Scholar] [CrossRef]
- Page, R.; Peti, W. Toxin-antitoxin systems in bacterial growth arrest and persistence. Nat. Chem. Biol. 2016, 12, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Pino, A.; Balasubramanian, S.; Wyns, L.; Gazit, E.; De Greve, H.; Magnuson, R.D.; Charlier, D.; Van Nuland, N.A.; Loris, R. Allostery and Intrinsic Disorder Mediate Transcription Regulation by Conditional Cooperativity. Cell 2010, 142, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Overgaard, M.; Borch, J.; Jørgensen, M.G.; Gerdes, K. Messenger RNA interferase RelE controlsrelBEtranscription by conditional cooperativity. Mol. Microbiol. 2008, 69, 841–857. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, N. Autoregulation of bacterial gene expression: Lessons from the MazEF toxin-antitoxin system. Curr. Genet. 2018, 65, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Cataudella, I.; Trusina, A.; Sneppen, K.; Gerdes, K.; Mitarai, N. Conditional cooperativity in toxin-antitoxin regulation prevents random toxin activation and promotes fast translational recovery. Nucleic Acids Res. 2012, 40, 6424–6434. [Google Scholar] [CrossRef]
- Kędzierska, B.; Lian, L.-Y.; Hayes, F. Toxin-antitoxin regulation: Bimodal interaction of YefM-YoeB with paired DNA palindromes exerts transcriptional autorepression. Nucleic Acids Res. 2006, 35, 325–339. [Google Scholar] [CrossRef]
- Bailey, S.E.S.; Hayes, F. Influence of Operator Site Geometry on Transcriptional Control by the YefM-YoeB Toxin-Antitoxin Complex. J. Bacteriol. 2008, 191, 762–772. [Google Scholar] [CrossRef]
- Chan, W.T.; Nieto, C.; Harikrishna, J.A.; Khoo, S.K.; Othman, R.Y.; Espinosa, M.; Yeo, C.C. Genetic Regulation of the yefM-yoeB Toxin-Antitoxin Locus of Streptococcus pneumoniae. J. Bacteriol. 2011, 193, 4612–4625. [Google Scholar] [CrossRef]
- Krügel, H.; Klimina, K.M.; Mrotzek, G.; Tretyakov, A.; Schöfl, G.; Saluz, H.P.; Brantl, S.; Poluektova, E.U.; Danilenko, V.N. Expression of the toxin-antitoxin genes yefMLrh, yoeBLrh in human Lactobacillus rhamnosus isolates. J. Basic Microbiol. 2015, 55, 982–991. [Google Scholar] [CrossRef]
- Boss, L.; Labudda, Ł.; Węgrzyn, G.; Hayes, F.; Kędzierska, B. The Axe-Txe Complex of Enterococcus faecium Presents a Multilayered Mode of Toxin-Antitoxin Gene Expression Regulation. PLoS ONE 2013, 8, e73569. [Google Scholar] [CrossRef]
- Grady, R.; Hayes, F. Axe-Txe, a broad-spectrum proteic toxin-antitoxin system specified by a multidrug-resistant, clinical isolate of Enterococcus faecium. Mol. Microbiol. 2003, 47, 1419–1432. [Google Scholar] [CrossRef] [PubMed]
- Kamada, K.; Hanaoka, F. Conformational Change in the Catalytic Site of the Ribonuclease YoeB Toxin by YefM Antitoxin. Mol. Cell 2005, 19, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Połom, D.; Boss, L.; Węgrzyn, G.; Hayes, F.; Kędzierska, B.; Kȩdzierska, B. Amino acid residues crucial for specificity of toxin-antitoxin interactions in the homologous Axe-Txe and YefM-YoeB complexes. FEBS J. 2013, 280, 5906–5918. [Google Scholar] [CrossRef] [PubMed]
- Nieto, C.; Cherny, I.; Khoo, S.K.; De Lacoba, M.G.; Chan, W.T.; Yeo, C.C.; Gazit, E.; Espinosa, M. The yefM-yoeB Toxin-Antitoxin Systems of Escherichia coli and Streptococcus pneumoniae: Functional and Structural Correlation. J. Bacteriol. 2007, 189, 1266–1278. [Google Scholar] [CrossRef]
- Christensen, S.K.; Maenhaut-Michel, G.; Mine, N.; Gottesman, S.; Gerdes, K.; Van Melderen, L. Overproduction of the Lon protease triggers inhibition of translation in Escherichia coli: Involvement of the yefM-yoeB toxin-antitoxin system. Mol. Microbiol. 2004, 51, 1705–1717. [Google Scholar] [CrossRef]
- De Bast, M.S.; Mine, N.; Van Melderen, L. Chromosomal Toxin-Antitoxin Systems May Act as Antiaddiction Modules. J. Bacteriol. 2008, 190, 4603–4609. [Google Scholar] [CrossRef]
- Weiss, A.; Shaw, L.N. Small things considered: The small accessory subunits of RNA polymerase in Gram-positive bacteria. FEMS Microbiol. Rev. 2015, 39, 541–554. [Google Scholar] [CrossRef]
- Escolar, L.; Pérez-Martín, J.; De Lorenzo, V. Coordinated Repression In vitro of the DivergentfepA-fes Promoters of Escherichia coli by the Iron Uptake Regulation (Fur) Protein. J. Bacteriol. 1998, 180, 2579–2582. [Google Scholar] [CrossRef]
- Hawley, D.K.; Johnson, A.D.; McClure, W.R. Functional and physical characterization of transcription initiation complexes in the bacteriophage λ O(R) region. J. Biol. Chem. 1985, 260, 8618–8626. [Google Scholar]
- Monsalve, M.; Calles, B.; Mencía, M.; Rojo, F.; Salas, M. Binding of phage Φ29 protein p4 to the early A2c promoter: Recruitment of a repressor by the RNA polymerase. J. Mol. Biol. 1998, 283, 559–569. [Google Scholar] [CrossRef]
- Choy, H.E.; Hanger, R.R.; Aki, T.; Mahoney, M.; Murakami, K.; Ishihama, A.; Adhya, S. Repression and activation of promoter-bound RNA polymerase activity by gal repressor. J. Mol. Biol. 1997, 272, 293–300. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Williams, D.R.; Motallebi-veshareh, M.; Thomas, C.M. Multifunctional repressor KorB can block transcription by preventing isomerization of RNA polymerase-promoter complexes. Nucleic Acids Res. 1993, 21, 1141–1148. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Greene, E.A.; Spiegelman, G.B. The Spo0A Protein of Bacillus subtilis Inhibits Transcription of the abrB Gene without Preventing Binding of the Polymerase to the Promoter. J. Biol. Chem. 1996, 271, 11455–11461. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.L.; Sauer, R.T. Dual regulation of open-complex formation and promoter clearance by Arc explains a novel repressor to activator switch. Proc. Natl. Acad. Sci. USA 1996, 93, 8868–8872. [Google Scholar] [CrossRef] [PubMed]
- Ansari, A.Z.; Bradner, J.E.; O’Halloran, T.V. DNA-bend modulation in a repressor-to-activator switching mechanism. Nat. Cell Biol. 1995, 374, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Grainger, D.C.; Goldberg, M.D.; Lee, D.J.; Busby, S.J.W. Selective repression by Fis and H-NS at the Escherichia coli dpspromoter. Mol. Microbiol. 2008, 68, 1366–1377. [Google Scholar] [CrossRef] [PubMed]
- Sztiller-Sikorska, M.; Heyduk, E.; Heyduk, T. Promoter spacer DNA plays an active role in integrating the functional consequences of RNA polymerase contacts with −10 and −35 promoter elements. Biophys. Chem. 2011, 159, 73–81. [Google Scholar] [CrossRef][Green Version]
- Aizenman, E.; Engelberg-Kulka, H.; Glaser, G. An Escherichia coli chromosomal “addiction module” regulated by guanosine [corrected] 3’,5’-bispyrophosphate: A model for programmed bacterial cell death. Proc. Natl. Acad. Sci. USA 1996, 93, 6059–6063. [Google Scholar] [CrossRef]
- Marianovsky, I.; Aizenman, E.; Engelberg-Kulka, H.; Glaser, G. The Regulation of the Escherichia coli mazEF Promoter Involves an Unusual Alternating Palindrome. J. Biol. Chem. 2000, 276, 5975–5984. [Google Scholar] [CrossRef]
- Turnbull, K.J.; Gerdes, K. HicA toxin of Escherichia coli derepresses hic AB transcription to selectively produce HicB antitoxin. Mol. Microbiol. 2017, 104, 781–792. [Google Scholar] [CrossRef]
- Poyart, C. A broad-host-range mobilizable shuttle vector for the construction of transcriptional fusions to β-galactosidase in Gram-positive bacteria. FEMS Microbiol. Lett. 1997, 156, 193–198. [Google Scholar] [CrossRef]
- Burgess, R.R.; Jendrisak, J.J. Procedure for the rapid, large-scale purification of Escherichia coli DNA-dependent RNA polymerase involving polymin P precipitation and DNA-cellulose chromatography. Biochemistry 1975, 14, 4634–4638. [Google Scholar] [CrossRef] [PubMed]
- Hager, D.A.; Jin, D.J.; Burgess, R.R. Use of mono Q high resolution ion exchange chromatography to obtain highly pure and active Escherichia coli RNA polymerase. Biochemistry 1990, 29, 7890–7894. [Google Scholar] [CrossRef] [PubMed]
- Guzman, L.M.; Belin, D.; Carson, M.J.; Beckwith, J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 1995, 177, 4121–4130. [Google Scholar] [CrossRef] [PubMed]
- Elliott, T.; Geiduschek, E. Defining a bacteriophage T4 late promoter: Absence of a “−35” region. Cell 1984, 36, 211–219. [Google Scholar] [CrossRef]
- Miller, J. Experiments in Molecular Genetics; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 1972; ISBN 9780879691066. [Google Scholar]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kędzierska, B.; Potrykus, K.; Szalewska-Pałasz, A.; Wodzikowska, B. Insights into Transcriptional Repression of the Homologous Toxin-Antitoxin Cassettes yefM-yoeB and axe-txe. Int. J. Mol. Sci. 2020, 21, 9062. https://doi.org/10.3390/ijms21239062
Kędzierska B, Potrykus K, Szalewska-Pałasz A, Wodzikowska B. Insights into Transcriptional Repression of the Homologous Toxin-Antitoxin Cassettes yefM-yoeB and axe-txe. International Journal of Molecular Sciences. 2020; 21(23):9062. https://doi.org/10.3390/ijms21239062
Chicago/Turabian StyleKędzierska, Barbara, Katarzyna Potrykus, Agnieszka Szalewska-Pałasz, and Beata Wodzikowska. 2020. "Insights into Transcriptional Repression of the Homologous Toxin-Antitoxin Cassettes yefM-yoeB and axe-txe" International Journal of Molecular Sciences 21, no. 23: 9062. https://doi.org/10.3390/ijms21239062
APA StyleKędzierska, B., Potrykus, K., Szalewska-Pałasz, A., & Wodzikowska, B. (2020). Insights into Transcriptional Repression of the Homologous Toxin-Antitoxin Cassettes yefM-yoeB and axe-txe. International Journal of Molecular Sciences, 21(23), 9062. https://doi.org/10.3390/ijms21239062