Emerging Kinase Therapeutic Targets in Pancreatic Ductal Adenocarcinoma and Pancreatic Cancer Desmoplasia

 ,
, 
 ,
, 
Abstract
1. Introduction
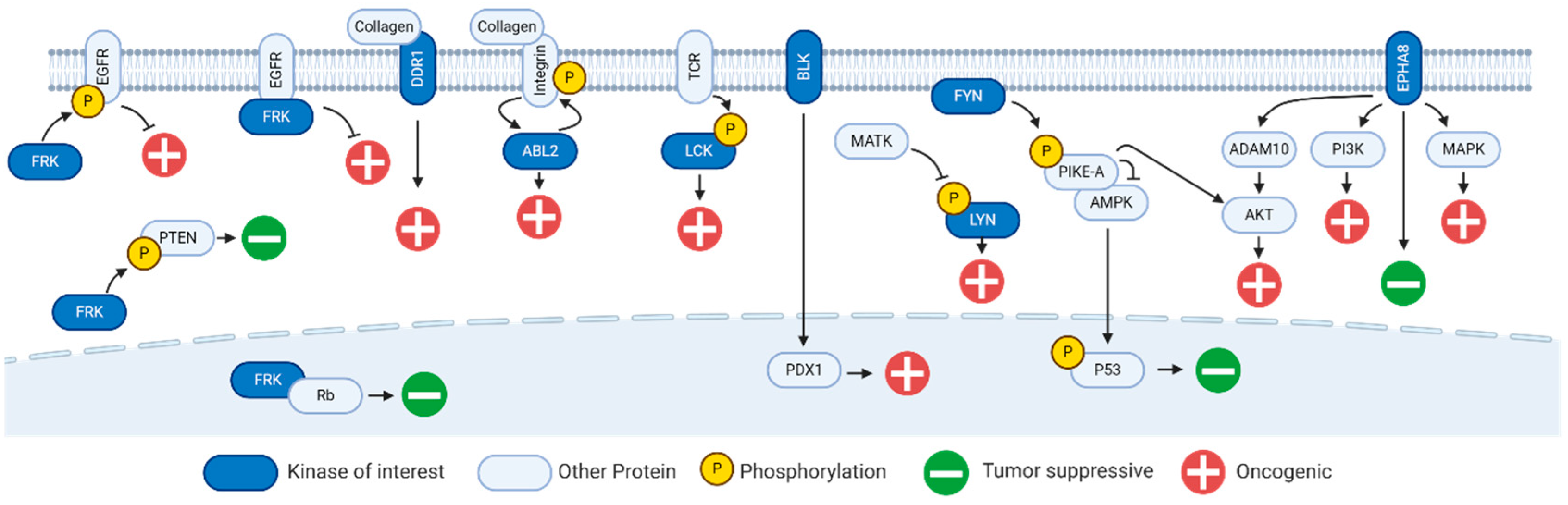
2. Fyn-Related Kinase (FRK)
3. B Lymphoid Kinase (BLK)
4. Hemopoietic Cell Kinase (HCK)
5. ABL Proto-Oncogene 2 Kinase (ABL2)
6. Discoidin Domain Receptor 1 Kinase (DDR1)
7. Lck/Yes-Related Novel Kinase (LYN)
8. Ephrin Receptor A8 Kinase (EPHA8)
9. FYN Proto-Oncogene Kinase (FYN)
10. Lymphocyte Cell-Specific Kinase (LCK)
11. Tec Protein Kinase (TEC)
12. Tyrosine Kinase Inhibitors
13. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABL2 | Abl proto-oncogene 2, nonreceptor tyrosine kinase |
| ADAM10 | A disintegrin and metalloprotease domain 10 |
| BLK | Blk proto-oncogene, Src family tyrosine kinase |
| BMX | Bmx nonreceptor tyrosine kinase |
| BTK | Bruton tyrosine kinase |
| DDR1 | Discoidin domain receptor tyrosine kinase 1 |
| EGFR | Epidermal growth factor receptor |
| EMT | Epithelial-to-mesenchymal transition |
| EPH | Ephrin receptor family kinases |
| EPHA8 | Eph receptor A8 |
| FRK | Fyn related Src family tyrosine kinase |
| FYN | FYN proto-oncogene, Src family tyrosine kinase |
| GLUT1 | Glucose transporter 1 |
| GLUT4 | Glucose transporter 4 |
| HCK | Hck proto-oncogene, Src family tyrosine kinase |
| IL2 | Interleukin-2 |
| INSR | Insulin receptor |
| IPF1 | Insulin promoter factor 1 |
| ITK | Il2 inducible T cell kinase |
| KRSA | Kinome reverse signature analyzer |
| LCK | Lck proto-oncogene, Src family tyrosine kinase |
| LYN | Lyn oroto-oncogene, Src family tyrosine kinase |
| MAPK | Mitogen-activated protein kinase |
| MATK | Megakaryocyte-associated tyrosine kinase |
| PDAC | Pancreatic ductal adenocarcinoma |
| PDCL | Patient-derived pancreatic ductal adenocarcinoma cell line |
| PDX1 | Pancreatic and duodenal homeobox 1 transcription factor |
| PI3K | Phosphatidylinositol 3-kinase |
| PTEN | Phosphatase and tensin homolog tumor suppressor protein |
| Rb | Retinoblastoma |
| RTK | Receptor tyrosine kinase |
| TCR | T-cell receptor |
| TEC | Tec protein tyrosine kinase |
| TP53 | Tumor protein 53 |
| TXK | Txk tyrosine kinase |
References
- Orth, M.; Metzger, P.; Gerum, S.; Mayerle, J.; Schneider, G.; Belka, C.; Schnurr, M.; Lauber, K. Pancreatic ductal adenocarcinoma: Biological hallmarks, current status, and future perspectives of combined modality treatment approaches. Radiat. Oncol. 2019, 14, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. The extracellular matrix: Not just pretty fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.; Cukierman, E. Stromal dynamic reciprocity in cancer: Intricacies of fibroblastic-ECM interactions. Curr. Opin. Cell Biol. 2016, 42, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Radhakrishnan, P. Tumor-stromal crosstalk in pancreatic cancer and tissue fibrosis. Mol. Cancer 2019, 18, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Melstrom, L.G.; Salazar, M.D.; Diamond, D.J. The pancreatic cancer microenvironment: A true double agent. J. Surg. Oncol. 2017, 116, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, T.M.N.W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef]
- Wormann, S.M.; Song, L.; Ai, J.; Diakopoulos, K.N.; Kurkowski, M.U.; Gorgulu, K.; Ruess, D.; Campbell, A.; Doglioni, C.; Jodrell, D.; et al. Loss of P53 Function Activates JAK2-STAT3 Signaling to Promote Pancreatic Tumor Growth, Stroma Modification, and Gemcitabine Resistance in Mice and Is Associated With Patient Survival. Gastroenterology 2016, 151, 180–193. [Google Scholar] [CrossRef]
- Sato, T.; Shibata, W.; Hikiba, Y.; Kaneta, Y.; Suzuki, N.; Ihara, S.; Ishii, Y.; Sue, S.; Kameta, E.; Sugimori, M.; et al. c-Jun N-terminal kinase in pancreatic tumor stroma augments tumor development in mice. Cancer Sci. 2017, 108, 2156–2165. [Google Scholar] [CrossRef]
- Zhang, D.; Li, L.; Jiang, H.; Li, Q.; Wang-Gillam, A.; Yu, J.; Head, R.; Liu, J.; Ruzinova, M.B.; Lim, K.-H. Tumor-Stroma IL1beta-IRAK4 Feedforward Circuitry Drives Tumor Fibrosis, Chemoresistance, and Poor Prognosis in Pancreatic Cancer. Cancer Res. 2018, 78, 1700–1712. [Google Scholar] [CrossRef]
- Lai, E.; Puzzoni, M.; Ziranu, P.; Pretta, A.; Impera, V.; Mariani, S.; Liscia, N.; Soro, P.; Musio, F.; Persano, M.; et al. New therapeutic targets in pancreatic cancer. Cancer Treat. Rev. 2019, 81, 101926. [Google Scholar] [CrossRef]
- Garber, K. Stromal Depletion Goes on Trial in Pancreatic Cancer. J. Natl. Cancer Inst. 2010, 102, 448–450. [Google Scholar] [CrossRef] [PubMed]
- Yabar, C.S.; Winter, J.M. Pancreatic Cancer: A Review. Gastroenterol. Clin. N. Am. 2016, 45, 429–445. [Google Scholar] [CrossRef] [PubMed]
- Creeden, J.F.; Alganem, K.; Imami, A.S.; Brunicardi, F.C.; Liu, S.-H.; Shukla, R.; Tomar, T.; Naji, F.; McCullumsmith, R.E. Kinome Array Profiling of Patient-Derived Pancreatic Ductal Adenocarcinoma Identifies Differentially Active Protein Tyrosine Kinases. Int. J. Mol. Sci. 2020, 21, 8679. [Google Scholar] [CrossRef] [PubMed]
- Goel, R.K.; Lukong, K.E. Understanding the cellular roles of Fyn-related kinase (FRK): Implications in cancer biology. Cancer Metastasis Rev. 2016, 35, 179–199. [Google Scholar] [CrossRef]
- Meyer, T.; Xu, L.; Chang, J.; Liu, E.T.; Craven, R.J.; Cance, W. Breast cancer cell line proliferation blocked by the Src-related Rak tyrosine kinase. Int. J. Cancer 2003, 104, 139–146. [Google Scholar] [CrossRef]
- Yim, E.-K.; Peng, G.; Dai, H.; Hu, R.; Li, K.; Lu, Y.; Mills, G.B.; Meric-Bernstam, F.; Hennessy, B.T.; Craven, R.J.; et al. Rak Functions as a Tumor Suppressor by Regulating PTEN Protein Stability and Function. Cancer Cell 2009, 15, 304–314. [Google Scholar] [CrossRef]
- Zhou, X.; Hua, L.; Zhang, W.; Zhu, M.; Shi, Q.; Li, F.; Zhang, L.; Song, C.; Yu, R. FRK controls migration and invasion of human glioma cells by regulating JNK/c-Jun signaling. J. Neuro-Oncol. 2012, 110, 9–19. [Google Scholar] [CrossRef]
- Hua, L.; Zhu, M.; Song, X.; Wang, J.; Fang, Z.; Zhang, C.; Shi, Q.; Zhan, W.; Wang, L.; Meng, Q.; et al. FRK suppresses the proliferation of human glioma cells by inhibiting cyclin D1 nuclear accumulation. J. Neuro-Oncol. 2014, 119, 49–58. [Google Scholar] [CrossRef]
- Shi, Q.; Song, X.; Wang, J.; Gu, J.; Zhang, W.; Hu, J.; Zhou, X.; Yu, R. FRK inhibits migration and invasion of human glioma cells by promoting N-cadherin/beta-catenin complex formation. J. Mol. Neurosci. 2015, 55, 32–41. [Google Scholar] [CrossRef]
- Chen, J.S.; Hung, W.S.; Chan, H.H.; Tsai, S.J.; Sun, H.S. In silico identification of oncogenic potential of fyn-related kinase in hepatocellular carcinoma. Bioinformatics 2013, 29, 420–427. [Google Scholar] [CrossRef]
- Pilati, C.; Letouzé, E.; Nault, J.-C.; Imbeaud, S.; Boulai, A.; Calderaro, J.; Poussin, K.; Franconi, A.; Couchy, G.; Morcrette, G.; et al. Genomic Profiling of Hepatocellular Adenomas Reveals Recurrent FRK-Activating Mutations and the Mechanisms of Malignant Transformation. Cancer Cell 2014, 25, 428–441. [Google Scholar] [CrossRef] [PubMed]
- Je, D.W.; O, Y.M.; Ji, Y.G.; Cho, Y.; Lee, D.H. The inhibition of SRC family kinase suppresses pancreatic cancer cell proliferation, migration, and invasion. Pancreas 2014, 43, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, Y.; Chai, L.; Bu, H.; Yang, Y.; Huang, H.; Ran, J.; Zhu, Y.; Li, L.; Chen, F.; et al. FRK plays an oncogenic role in non-small cell lung cancer by enhancing the stemness phenotype via induction of metabolic reprogramming. Int. J. Cancer 2019, 146, 208–222. [Google Scholar] [CrossRef] [PubMed]
- Anneren, C. Dual role of the tyrosine kinase GTK and the adaptor protein SHB in beta-cell growth: Enhanced beta-cell replication after 60% pancreatectomy and increased sensitivity to streptozotocin. J. Endocrinol. 2002, 172, 145–153. [Google Scholar] [CrossRef][Green Version]
- Docherty, H.M.; Hay, C.W.; Ferguson, L.A.; Barrow, J.; Durward, E.; Docherty, K. Relative contribution of PDX-1, MafA and E47/beta2 to the regulation of the human insulin promoter. Biochem. J. 2005, 389, 813–820. [Google Scholar] [CrossRef]
- Glick, E.; Leshkowitz, D.; Walker, M.D. Transcription Factor BETA2 Acts Cooperatively with E2A and PDX1 to Activate the Insulin Gene Promoter. J. Biol. Chem. 2000, 275, 2199–2204. [Google Scholar] [CrossRef]
- Annerén, C.; Welsh, M. Increased cytokine-induced cytotoxicity of pancreatic islet cells from transgenic mice expressing the Src-like tyrosine kinase GTK. Mol. Med. 2001, 7, 301–310. [Google Scholar] [CrossRef]
- Craven, R.J.; Cance, W.; Liu, E.T. The nuclear tyrosine kinase Rak associates with the retinoblastoma protein pRb. Cancer Res. 1995, 55, 3969–3972. [Google Scholar]
- Jin, L.; Craven, R.J. The Rak/Frk tyrosine kinase associates with and internalizes the epidermal growth factor receptor. Oncogene 2014, 33, 326–335. [Google Scholar] [CrossRef][Green Version]
- Tomas, A.; Futter, C.E.; Eden, E.R. EGF receptor trafficking: Consequences for signaling and cancer. Trends Cell Biol. 2014, 24, 26–34. [Google Scholar] [CrossRef]
- Keilhack, H.; Tenev, T.; Nyakatura, E.; Godovac-Zimmermann, J.; Nielsen, L.; Seedorf, K.; Böhmer, F.-D. Phosphotyrosine 1173 Mediates Binding of the Protein-tyrosine Phosphatase SHP-1 to the Epidermal Growth Factor Receptor and Attenuation of Receptor Signaling. J. Biol. Chem. 1998, 273, 24839–24846. [Google Scholar] [CrossRef] [PubMed]
- Emlet, D.R.; Moscatello, D.K.; Ludlow, L.B.; Wong, A.J. Subsets of Epidermal Growth Factor Receptors during Activation and Endocytosis. J. Biol. Chem. 1997, 272, 4079–4086. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.-M.; Chen, C.-T.; Chou, C.-K.; Kuo, H.-P.; Li, L.-Y.; Lin, C.-Y.; Lee, H.-J.; Wang, Y.-N.; Liu, M.; Liao, H.-W.; et al. Crosstalk between Arg 1175 methylation and Tyr 1173 phosphorylation negatively modulates EGFR-mediated ERK activation. Nat. Cell Biol. 2011, 13, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef]
- Alganem, K.; Shukla, R.; Eby, H.; Abel, M.; Zhang, X.; McIntyre, W.B.; Lee, J.; Au-Yeung, C.; Asgariroozbehani, R.; Panda, R.; et al. Kaleidoscope: A New Bioinformatics Pipeline Web Application for In Silico Hypothesis Exploration of Omics Signatures. bioRxiv 2020. [Google Scholar] [CrossRef]
- Guo, Y.; Liu, Y.; Hu, N.; Yu, D.; Zhou, C.; Shi, G.; Zhang, B.; Wei, M.; Liu, J.; Luo, L.; et al. Discovery of Zanubrutinib (BGB-3111), a Novel, Potent, and Selective Covalent Inhibitor of Bruton’s Tyrosine Kinase. J. Med. Chem. 2019, 62, 7923–7940. [Google Scholar] [CrossRef]
- Kneidinger, M.; Schmidt, U.; Rix, U.; Gleixner, K.V.; Vales, A.; Baumgartner, C.; Lupinek, C.; Weghofer, M.; Bennett, K.L.; Herrmann, H.; et al. The effects of dasatinib on IgE receptor–dependent activation and histamine release in human basophils. Blood 2008, 111, 3097–3107. [Google Scholar] [CrossRef]
- Rolf, M.G.; Curwen, J.O.; Veldmanjones, M.H.; Eberlein, C.; Wang, J.; Harmer, A.; Hellawell, C.J.; Braddock, M. In vitro pharmacological profiling of R406 identifies molecular targets underlying the clinical effects of fostamatinib. Pharmacol. Res. Perspect. 2015, 3, e00175. [Google Scholar] [CrossRef]
- Krejsgaard, T.; Vetter-Kauczok, C.S.; Woetmann, A.; Kneitz, H.; Eriksen, K.W.; Lovato, P.; Zhang, Q.; Wasik, M.A.; Geisler, C.; Ralfkiaer, E.; et al. Ectopic expression of B-lymphoid kinase in cutaneous T-cell lymphoma. Blood 2009, 113, 5896–5904. [Google Scholar] [CrossRef]
- Ratner, L.; Rauch, D.; Abel, H.; Caruso, B.; Noy, A.; Barta, S.K.; Parekh, S.; Ramos, J.C.; Ambinder, R.; Phillips, A.; et al. Dose-adjusted EPOCH chemotherapy with bortezomib and raltegravir for human T-cell leukemia virus-associated adult T-cell leukemia lymphoma. Blood Cancer J. 2016, 6, e408. [Google Scholar] [CrossRef]
- Petersen, D.L.; Berthelsen, J.; Willerslev-Olsen, A.; Fredholm, S.; Dabelsteen, S.; Bonefeld, C.M.; Geisler, C.; Woetmann, A. A novel BLK-induced tumor model. Tumour Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Malek, S.N.; Dordai, D.I.; Reim, J.; Dintzis, H.; Desiderio, S. Malignant transformation of early lymphoid progenitors in mice expressing an activated Blk tyrosine kinase. Proc. Natl. Acad. Sci. USA 1998, 95, 7351–7356. [Google Scholar] [CrossRef] [PubMed]
- Petersen, D.L.; Krejsgaard, T.; Berthelsen, J.; Fredholm, S.; Willerslev-Olsen, A.; A Sibbesen, N.; Bonefeld, C.M.; Andersen, M.H.; Francavilla, C.; Olsen, J.V.; et al. B-lymphoid tyrosine kinase (Blk) is an oncogene and a potential target for therapy with dasatinib in cutaneous T-cell lymphoma (CTCL). Leukemia 2014, 28, 2109–2112. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lun, X.K.; Szklarczyk, D.; Gábor, A.; Dobberstein, N.; Zanotelli, V.R.T.; Saez-Rodriguez, J.; Von Mering, C.; Bodenmiller, B. Analysis of the Human Kinome and Phosphatome by Mass Cytometry Reveals Overexpression-Induced Effects on Cancer-Related Signaling. Mol. Cell 2019, 74, 1086–1102. [Google Scholar] [CrossRef]
- Kim, E.; Hurtz, C.; Koehrer, S.; Wang, Z.; Balasubramanian, S.; Chang, B.Y.; Müschen, M.; Davis, R.E.; Burger, J.A. Ibrutinib inhibits pre-BCR+ B-cell acute lymphoblastic leukemia progression by targeting BTK and BLK. Blood 2017, 129, 1155–1165. [Google Scholar] [CrossRef]
- Kim, J.E.; Kim, J.H.; Lee, Y.; Yang, H.; Heo, Y.S.; Bode, A.M.; Lee, K.W.; Dong, Z. Bakuchiol suppresses proliferation of skin cancer cells by directly targeting Hck, Blk, and p38 MAP kinase. Oncotarget 2016, 7, 14616–14627. [Google Scholar] [CrossRef]
- Montero, J.C.; Seoane, S.; Ocaña, A.; Pandiella, A. Inhibition of Src Family Kinases and Receptor Tyrosine Kinases by Dasatinib: Possible Combinations in Solid Tumors. Clin. Cancer Res. 2011, 17, 5546–5552. [Google Scholar] [CrossRef]
- Chen, R.; Chen, B.-A. The role of dasatinib in the management of chronic myeloid leukemia. Drug Des. Dev. Ther. 2015, 9, 773–779. [Google Scholar] [CrossRef]
- Fallacara, A.L.; Passannanti, R.; Mori, M.; Iovenitti, G.; Musumeci, F.; Greco, C.; Crespan, E.; Kissova, M.; Maga, G.; Tarantelli, C.; et al. Identification of a new family of pyrazolo[3,4-d]pyrimidine derivatives as multitarget Fyn-Blk-Lyn inhibitors active on B- and T-lymphoma cell lines. Eur. J. Med. Chem. 2019, 181, 111545. [Google Scholar] [CrossRef]
- Zhang, H.; Peng, C.; Hu, Y.; Li, H.; Sheng, Z.; Chen, Y.; Sullivan, C.; Cerny, J.; Hutchinson, L.; Higgings, A.; et al. The Blk pathway functions as a tumor suppressor in chronic myeloid leukemia stem cells. Nat. Genet. 2012, 44, 861–871. [Google Scholar] [CrossRef]
- Crivellaro, S.; Carrà, G.; Panuzzo, C.; Taulli, R.; Guerrasio, A.; Saglio, G.; Morotti, A. The non-genomic loss of function of tumor suppressors: An essential role in the pathogenesis of chronic myeloid leukemia chronic phase. BMC Cancer 2016, 16, 314. [Google Scholar] [CrossRef] [PubMed]
- Borowiec, M.; Liew, C.W.; Thompson, R.; Boonyasrisawat, W.; Hu, J.; Mlynarski, W.M.; El Khattabi, I.; Kim, S.H.; Marselli, L.; Rich, S.S.; et al. Mutations at the BLK locus linked to maturity onset diabetes of the young and beta-cell dysfunction. Proc. Natl. Acad. Sci. USA 2009, 106, 14460–14465. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Chen, M.; Wang, J.; Xia, H.H.; Zhu, S.; Liang, Y.; Gu, Q.; Qiao, L.; Dai, Y.; Zou, B.; et al. Pancreatic duodenal homeobox-1 (PDX1) functions as a tumor suppressor in gastric cancer. Carcinogenesis 2008, 29, 1327–1333. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Initiative, A.P.C.G.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Roy, N.; Takeuchi, K.K.; Ruggeri, J.M.; Bailey, P.; Chang, D.; Li, J.; Leonhardt, L.; Puri, S.; Hoffman, M.T.; Gao, S.; et al. PDX1 dynamically regulates pancreatic ductal adenocarcinoma initiation and maintenance. Genes Dev. 2016, 30, 2669–2683. [Google Scholar] [CrossRef]
- Jay, C.M.; Ruoff, C.; Kumar, P.; Maass, H.; Spanhel, B.; Miller, M.; Arrington, A.; Montalvo, N.; Gresham, V.; Rao, D.D.; et al. Assessment of intravenous pbi-shRNA PDX1 nanoparticle (OFHIRNA-PDX1) in yucatan swine. Cancer Gene Ther. 2013, 20, 683–689. [Google Scholar] [CrossRef][Green Version]
- Wu, J.X.; Liu, S.; Yu, J.; Zhou, G.; Rao, D.; Jay, C.M.; Kumar, P.; Sanchez, R.; Templeton, N.; Senzer, N.; et al. Vertically integrated translational studies of PDX1 as a therapeutic target for pancreatic cancer via a novel bifunctional RNAi platform. Cancer Gene Ther. 2014, 21, 48–53. [Google Scholar] [CrossRef]
- Yu, J.; Liu, S.-H.; Sanchez, R.; Nemunaitis, J.; Rozengurt, E.; Brunicardi, F.C. PDX1 associated therapy in translational medicine. Ann. Transl. Med. 2016, 4, 214. [Google Scholar] [CrossRef]
- Ballian, N.; Liu, S.-H.; Brunicardi, F.C. Transcription factor PDX-1 in human colorectal adenocarcinoma: A potential tumor marker? World J. Gastroenterol. 2008, 14, 5823–5826. [Google Scholar] [CrossRef]
- Duarte-Medrano, G.; Lopez-Mendez, I.; Ramirez-Luna, M.A.; Valdovinos-Andraca, F.; Cruz-Martinez, R.; Medina-Vera, I.; Perez-Monter, C.; Tellez-Avila, F.I. Analysis of circulating blood and tissue biopsy PDX1 and MSX2 gene expression in patients with pancreatic cancer: A case-control experimental study. Medicine 2019, 98, e15954. [Google Scholar] [CrossRef]
- Marzioni, M.; Germani, U.; Agostinelli, L.; Bedogni, G.; Saccomanno, S.; Marini, F.; Bellentani, S.; Barbera, C.; De Minicis, S.; Rychlicki, C.; et al. PDX-1 mRNA expression in endoscopic ultrasound-guided fine needle cytoaspirate: Perspectives in the diagnosis of pancreatic cancer. Dig. Liver Dis. 2015, 47, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.H.; Patel, S.; Gingras, M.C.; Nemunaitis, J.; Zhou, G.; Chen, C.; Li, M.; Fisher, W.; Gibbs, R.; Brunicardi, F.C. PDX-1: Demonstration of oncogenic properties in pancreatic cancer. Cancer 2011, 117, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.P.; Li, Z.J.; Magnusson, J.; Brunicardi, F.C. Tissue MicroArray analyses of pancreatic duodenal homeobox-1 in human cancers. World J. Surg. 2005, 29, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Alalem, M.; Ray, B.K. Insulin signaling network in cancer. Indian J. Biochem. Biophys. 2014, 51, 493–498. [Google Scholar] [PubMed]
- Home, P. Insulin therapy and cancer. Diabetes Care 2013, 36, S240–S244. [Google Scholar] [CrossRef]
- Djiogue, S.; Kamdje, A.H.N.; Vecchio, L.; Kipanyula, M.J.; Farahna, M.; Aldebasi, Y.; Etet, P.F. Insulin resistance and cancer: The role of insulin and IGFs. Endocr. Relat. Cancer 2012, 20, R1–R17. [Google Scholar] [CrossRef]
- Bose, S.; Le, A. Glucose Metabolism in Cancer. Adv. Exp. Med. Biol. 2018, 1063, 3–12. [Google Scholar]
- Orgel, E.; Mittelman, S.D. The Links Between Insulin Resistance, Diabetes, and Cancer. Curr. Diabetes Rep. 2013, 13, 213–222. [Google Scholar] [CrossRef]
- Vigneri, R.; Goldfine, I.D.; Frittitta, L. Insulin, insulin receptors, and cancer. J. Endocrinol. Investig. 2016, 39, 1365–1376. [Google Scholar] [CrossRef]
- Dai, L.; Qi, Y.; Chen, J.; Kaczorowski, D.; Di, W.; Wang, W.; Xia, P. Sphingosine kinase (SphK) 1 and SphK2 play equivalent roles in mediating insulin’s mitogenic action. Mol. Endocrinol. 2014, 28, 197–207. [Google Scholar] [CrossRef]
- Pollak, M. Insulin and insulin-like growth factor signalling in neoplasia. Nat. Rev. Cancer 2008, 8, 915–928. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M. The insulin and insulin-like growth factor receptor family in neoplasia: An update. Nat. Rev. Cancer 2012, 12, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Castillo, J.J.; Munshi, M.; Hunter, Z.; Xu, L.; Kofides, A.; Tsakmaklis, N.; Demos, M.G.; Guerrera, M.L.; Chan, G.G.; et al. Expression of the prosurvival kinase HCK requires PAX5 and mutated MYD88 signaling in MYD88-driven B-cell lymphomas. Blood Adv. 2020, 4, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, T.; Kiyokawa, N.; Sato, N.; Saito, M.; Fujimoto, J. Characteristic expression of Hck in human B-cell precursors. Exp. Hematol. 2000, 28, 55–64. [Google Scholar] [CrossRef]
- Poh, A.R.; O’Donoghue, R.J.; Ernst, M. Hematopoietic cell kinase (HCK) as a therapeutic target in immune and cancer cells. Oncotarget 2015, 6, 15752–15771. [Google Scholar] [CrossRef]
- Poh, A.R.; Love, C.G.; Masson, F.; Preaudet, A.; Tsui, C.; Whitehead, L.; Monard, S.; Khakham, Y.; Burstroem, L.; Lessene, G.; et al. Inhibition of Hematopoietic Cell Kinase Activity Suppresses Myeloid Cell-Mediated Colon Cancer Progression. Cancer Cell 2017, 31, 563–575. [Google Scholar] [CrossRef]
- Roseweir, A.K.; Powell, A.; Horstman, S.L.; Inthagard, J.; Park, J.H.; McMillan, N.C.; Horgan, P.G.; Edwards, J. Src family kinases, HCK and FGR, associate with local inflammation and tumour progression in colorectal cancer. Cell. Signal. 2019, 56, 15–22. [Google Scholar] [CrossRef]
- Loukopoulos, P.; Shibata, T.; Katoh, H.; Kokubu, A.; Sakamoto, M.; Yamazaki, K.; Kosuge, T.; Kanai, Y.; Hosoda, F.; Imoto, I.; et al. Genome-wide array-based comparative genomic hybridization analysis of pancreatic adenocarcinoma: Identification of genetic indicators that predict patient outcome. Cancer Sci. 2007, 98, 392–400. [Google Scholar] [CrossRef]
- Dorman, H.R.; Close, D.; Wingert, B.M.; Camacho, C.J.; Johnston, P.A.; Smithgall, T.E. Discovery of Non-peptide Small Molecule Allosteric Modulators of the Src-family Kinase, Hck. Front. Chem. 2019, 7, 822. [Google Scholar] [CrossRef]
- Liu, X.F.; Xiang, L.; FitzGerald, D.J.; Pastan, I. Antitumor effects of immunotoxins are enhanced by lowering HCK or treatment with SRC kinase inhibitors. Mol. Cancer Ther. 2014, 13, 82–89. [Google Scholar] [CrossRef]
- Wei, C.; Margulies, I.; Menon, M.C.; Zhang, W.; Fu, J.; Kidd, B.; Keung, K.L.; Woytovich, C.; Greene, I.; Xiao, W.; et al. Genomic Analysis of Kidney Allograft Injury Identifies Hematopoietic Cell Kinase as a Key Driver of Renal Fibrosis. J. Am. Soc. Nephrol. 2016, 28, 1385–1393. [Google Scholar] [CrossRef] [PubMed]
- Smolinska, M.J.; Page, T.H.; Urbaniak, A.M.; Mutch, B.E.; Horwood, N.J. Hck Tyrosine Kinase Regulates TLR4-Induced TNF and IL-6 Production via AP-1. J. Immunol. 2011, 187, 6043–6051. [Google Scholar] [CrossRef] [PubMed]
- Ernst, M.; Inglese, M.; Scholz, G.M.; Harder, K.W.; Clay, F.J.; Bozinovski, S.; Waring, P.; Darwiche, R.; Kay, T.; Sly, P.; et al. Constitutive Activation of the Src Family Kinase Hck Results in Spontaneous Pulmonary Inflammation and an Enhanced Innate Immune Response. J. Exp. Med. 2002, 196, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.G.; Li, Y.; Payne-Turner, D.; Harvey, R.C.; Yang, Y.-L.; Pei, D.; McCastlain, K.; Ding, L.; Lu, C.; Song, G.; et al. Targetable Kinase-Activating Lesions in Ph-like Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2014, 371, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.G.; Yang, Y.-L.; Payne-Turner, D.; Lin, W.; Files, J.K.; Dickerson, K.; Gu, Z.; Taunton, J.; Janke, L.J.; Chen, T.; et al. Oncogenic role and therapeutic targeting of ABL-class and JAK-STAT activating kinase alterations in Ph-like ALL. Blood Adv. 2017, 1, 1657–1671. [Google Scholar]
- Decool, G.; Domenech, C.; Grardel, N.; Plesa, A.; Raczkiewicz, I.; Ducourneau, B.; Ruminy, P.; Pages, M.P.; Girard, S.; Fenwarth, L.; et al. Efficacy of Tyrosine Kinase Inhibitor Therapy in a Chemotherapy-refractory B-cell Precursor Acute Lymphoblastic Leukemia With ZC3HAV1-ABL2 Fusion. Hemasphere 2019, 3, e193. [Google Scholar] [CrossRef]
- Crnogorac-Jurcevic, T.; Efthimiou, E.; Nielsen, T.; Loader, J.; Terris, B.; Stamp, G.; Baron, A.; Scarpa, A.; Lemoine, N.R. Expression profiling of microdissected pancreatic adenocarcinomas. Oncogene 2002, 21, 4587–4594. [Google Scholar] [CrossRef]
- Gu, J.J.; Rouse, C.; Xu, X.; Wang, J.; Onaitis, M.W.; Pendergast, A.M. Inactivation of ABL kinases suppresses non-small cell lung cancer metastasis. JCI Insight 2016, 1, e89647. [Google Scholar] [CrossRef][Green Version]
- Sos, M.L.; Michel, K.; Zander, T.; Weiss, J.; Frommolt, P.; Peifer, M.; Li, D.; Ullrich, R.; Koker, M.; Fischer, F.; et al. Predicting drug susceptibility of non–small cell lung cancers based on genetic lesions. J. Clin. Investig. 2009, 119, 1727–1740. [Google Scholar] [CrossRef]
- Xing, Q.T.; Qu, C.M.; Wang, G. Overexpression of Abl2 predicts poor prognosis in hepatocellular carcinomas and is associated with cancer cell migration and invasion. OncoTargets Ther. 2014, 7, 881–885. [Google Scholar] [CrossRef]
- Chen, L.; Zhu, M.; Yu, S.; Hai, L.; Zhang, L.; Zhang, C.; Zhao, P.; Zhou, H.; Wang, S.; Yang, X. Arg kinase mediates CXCL12/CXCR4-induced invadopodia formation and invasion of glioma cells. Exp. Cell Res. 2020, 389, 111893. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Shao, C.; Zhu, L.; Jiang, S.; Li, G.; Zhang, W.; Lin, Y.; Ni, Y.; Cao, H.; Shao, S.H. High Expression of ABL2 Suppresses Apoptosis in Gastric Cancer. Dig. Dis. Sci. 2018, 63, 2294–2300. [Google Scholar] [CrossRef] [PubMed]
- Kazi, J.U.; Rupar, K.; Marhäll, A.; Moharram, S.A.; Khanum, F.; Shah, K.; Gazi, M.; Nagaraj, S.R.M.; Sun, J.; Chougule, R.A.; et al. ABL2 suppresses FLT3-ITD-induced cell proliferation through negative regulation of AKT signaling. Oncotarget 2017, 8, 12194–12202. [Google Scholar] [CrossRef] [PubMed]
- Qiang, X.F.; Zhang, Z.W.; Liu, Q.; Sun, N.; Pan, L.L.; Shen, J.; Li, T.; Yun, C.; Li, H.; Shi, L.H. miR-20a promotes prostate cancer invasion and migration through targeting ABL2. J. Cell Biochem. 2014, 115, 1269–1276. [Google Scholar] [CrossRef]
- Kain, K.H.; Klemke, R.L. Inhibition of Cell Migration by Abl Family Tyrosine Kinases through Uncoupling of Crk-CAS Complexes. J. Biol. Chem. 2001, 276, 16185–16192. [Google Scholar] [CrossRef]
- Peacock, J.G.; Miller, A.L.; Bradley, W.D.; Rodriguez, O.C.; Webb, D.J.; Koleske, A.J. The Abl-related Gene Tyrosine Kinase Acts through p190RhoGAP to Inhibit Actomyosin Contractility and Regulate Focal Adhesion Dynamics upon Adhesion to Fibronectin. Mol. Biol. Cell 2007, 18, 3860–3872. [Google Scholar] [CrossRef]
- Gil-Henn, H.; Patsialou, A.; Wang, Y.; Warren, M.S.; Condeelis, J.S.; Koleske, A.J. Arg/Abl2 promotes invasion and attenuates proliferation of breast cancer in vivo. Oncogene 2013, 32, 2622–2630. [Google Scholar] [CrossRef]
- Hamada, S.; Masamune, A. Elucidating the link between collagen and pancreatic cancer: What’s next? Expert Rev. Gastroenterol. Hepatol. 2018, 12, 315–317. [Google Scholar] [CrossRef]
- Whatcott, C.J.; Diep, C.H.; Jiang, P.; Watanabe, A.; LoBello, J.; Sima, C.; Hostetter, G.; Shepard, H.M.; Von Hoff, D.D.; Han, H. Desmoplasia in Primary Tumors and Metastatic Lesions of Pancreatic Cancer. Clin. Cancer Res. 2015, 21, 3561–3568. [Google Scholar] [CrossRef]
- Zeltz, C.; Orgel, J.; Gullberg, D. Molecular composition and function of integrin-based collagen glues-introducing COLINBRIs. Biochim. Biophys. Acta 2014, 1840, 2533–2548. [Google Scholar] [CrossRef]
- Apte, M.V.; Park, S.; Phillips, P.A.; Santucci, N.; Goldstein, D.; Kumar, R.K.; Ramm, G.A.; Buchler, M.; Friess, H.; McCarroll, J.A.; et al. Desmoplastic Reaction in Pancreatic Cancer: Role of Pancreatic Stellate Cells. Pancreas 2004, 29, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Simpson, M.A.; Bradley, W.D.; Harburger, D.; Parsons, M.; Calderwood, D.A.; Koleske, A.J. Direct interactions with the integrin beta1 cytoplasmic tail activate the Abl2/Arg kinase. J. Biol. Chem. 2015, 290, 8360–8372. [Google Scholar] [CrossRef] [PubMed]
- Pandol, S.; Edderkaoui, M.; Gukovsky, I.; Lugea, A.; Gukovskaya, A. Desmoplasia of pancreatic ductal adenocarcinoma. Clin. Gastroenterol. Hepatol. 2009, 7, S44–S47. [Google Scholar] [CrossRef] [PubMed]
- Cannon, A.; Thompson, C.; Hall, B.R.; Jain, M.; Kumar, S.; Batra, S.K. Desmoplasia in pancreatic ductal adenocarcinoma: Insight into pathological function and therapeutic potential. Genes Cancer 2018, 9, 78–86. [Google Scholar] [CrossRef]
- Schnittert, J.; Bansal, R.; Mardhian, D.F.; Van Baarlen, J.; Östman, A.; Prakash, J. Integrin α11 in pancreatic stellate cells regulates tumor stroma interaction in pancreatic cancer. FASEB J. 2019, 33, 6609–6621. [Google Scholar] [CrossRef]
- Yang, D.; Shi, J.; Fu, H.; Wei, Z.; Xu, J.; Hu, Z.; Zhang, Y.; Yan, R.; Cai, Q. Integrinbeta1 modulates tumour resistance to gemcitabine and serves as an independent prognostic factor in pancreatic adenocarcinomas. Tumour Biol. 2016, 37, 12315–12327. [Google Scholar] [CrossRef]
- Yang, D.; Tang, Y.; Fu, H.; Xu, J.; Hu, Z.; Zhang, Y.; Cai, Q. Integrin beta1 promotes gemcitabine resistance in pancreatic cancer through Cdc42 activation of PI3K p110beta signaling. Biochem. Biophys. Res. Commun. 2018, 505, 215–221. [Google Scholar] [CrossRef]
- Beaty, B.T.; Sharma, V.P.; Bravo-Cordero, J.J.; Simpson, M.A.; Eddy, R.J.; Koleske, A.J.; Condeelis, J. beta1 integrin regulates Arg to promote invadopodial maturation and matrix degradation. Mol. Biol. Cell 2013, 24, 1661–1675. [Google Scholar] [CrossRef]
- Torsello, B.; De Marco, S.; Bombelli, S.; Chisci, E.; Cassina, V.; Corti, R.; Bernasconi, D.; Giovannoni, R.; Bianchi, C.; Perego, R.A. The 1ALCTL and 1BLCTL isoforms of Arg/Abl2 induce fibroblast activation and extra cellular matrix remodelling differently. Biol. Open 2019, 8, bio038554. [Google Scholar] [CrossRef]
- Lapetina, S.; Mader, C.C.; Machida, K.; Mayer, B.J.; Koleske, A.J. Arg interacts with cortactin to promote adhesion-dependent cell edge protrusion. J. Cell Biol. 2009, 185, 503–519. [Google Scholar] [CrossRef]
- Miller, A.L.; Wang, Y.; Mooseker, M.S.; Koleske, A.J. The Abl-related gene (Arg) requires its F-actin—microtubule cross-linking activity to regulate lamellipodial dynamics during fibroblast adhesion. J. Cell Biol. 2004, 165, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Montenegro, R.; Howarth, A.; Ceroni, A.; Fedele, V.; Farran, B.; Mesquita, F.P.; Frejno, M.; Berger, B.-T.; Heinzlmeir, S.; Sailem, H.Z.; et al. Identification of molecular targets for the targeted treatment of gastric cancer using dasatinib. Oncotarget 2020, 11, 535–549. [Google Scholar] [CrossRef] [PubMed]
- Hantschel, O.; Rix, U.; Schmidt, U.; Burckstummer, T.; Kneidinger, M.; Schutze, G.; Colinge, J.; Bennett, K.L.; Ellmeier, W.; Valent, P.; et al. The Btk tyrosine kinase is a major target of the Bcr-Abl inhibitor dasatinib. Proc. Natl. Acad. Sci. USA 2007, 104, 13283–13288. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Wei, J.; Su, G.H.; Lin, J. Dasatinib can enhance paclitaxel and gemcitabine inhibitory activity in human pancreatic cancer cells. Cancer Biol. Ther. 2019, 20, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Huo, Y.; Yang, M.-W.; Liu, W.; Yang, J.; Fu, X.; Liu, D.-J.; Li, J.; Zhang, J.; Hua, R.; Sun, Y. High expression of DDR1 is associated with the poor prognosis in Chinese patients with pancreatic ductal adenocarcinoma. J. Exp. Clin. Cancer Res. 2015, 34, 88. [Google Scholar] [CrossRef] [PubMed]
- Moll, S.; Desmouliere, A.; Moeller, M.J.; Pache, J.C.; Badi, L.; Arcadu, F.; Richter, H.; Satz, A.; Uhles, S.; Cavalli, A.; et al. DDR1 role in fibrosis and its pharmacological targeting. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118474. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.; Zhang, Y.; He, S.J.; Li, M.M.; Cai, X.L.; Wang, H.; Xu, L.M.; Cao, J. TM4SF1 Promotes Metastasis of Pancreatic Cancer via Regulating the Expression of DDR1. Sci. Rep. 2017, 7, 45895. [Google Scholar] [CrossRef]
- Aguilera, K.Y.; Huang, H.; Du, W.; Hagopian, M.M.; Wang, Z.; Hinz, S.; Hwang, T.H.; Wang, H.; Fleming, J.B.; Castrillon, D.H.; et al. Inhibition of Discoidin Domain Receptor 1 Reduces Collagen-mediated Tumorigenicity in Pancreatic Ductal Adenocarcinoma. Mol. Cancer Ther. 2017, 16, 2473–2485. [Google Scholar] [CrossRef]
- Gao, M.; Duan, L.; Luo, J.; Zhang, L.; Lu, X.; Zhang, Y.; Zhang, Z.; Tu, Z.; Xu, Y.; Ren, X.; et al. Discovery and optimization of 3-(2-(Pyrazolo[1,5-a]pyrimidin-6-yl)ethynyl)benzamides as novel selective and orally bioavailable discoidin domain receptor 1 (DDR1) inhibitors. J. Med. Chem. 2013, 56, 3281–3295. [Google Scholar] [CrossRef]
- Wang, Z.; Bian, H.; Bartual, S.G.; Du, W.; Luo, J.; Zhao, H.; Zhang, S.; Mo, C.; Zhou, Y.; Xu, Y.; et al. Structure-Based Design of Tetrahydroisoquinoline-7-carboxamides as Selective Discoidin Domain Receptor 1 (DDR1) Inhibitors. J. Med. Chem. 2016, 59, 5911–5916. [Google Scholar] [CrossRef]
- Richter, H.; Satz, A.L.; Bedoucha, M.; Buettelmann, B.; Petersen, A.C.; Harmeier, A.; Hermosilla, R.; Hochstrasser, R.; Burger, D.; Gsell, B.; et al. DNA-Encoded Library-Derived DDR1 Inhibitor Prevents Fibrosis and Renal Function Loss in a Genetic Mouse Model of Alport Syndrome. ACS Chem. Biol. 2018, 14, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, Y.; Bartual, S.G.; Luo, J.; Xu, T.; Du, W.; Xun, Q.; Tu, Z.; Brekken, R.A.; Ren, X.; et al. Tetrahydroisoquinoline-7-carboxamide Derivatives as New Selective Discoidin Domain Receptor 1 (DDR1) Inhibitors. ACS Med. Chem. Lett. 2017, 8, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Zhang, M.; Wen, Z.; Wang, B.; Zhang, L.; Ou, Y.; Tang, X.; Yu, X.; Jiang, Q. Inhibition of EP300 and DDR1 synergistically alleviates pulmonary fibrosis in vitro and in vivo. Biomed. Pharmacother. 2018, 106, 1727–1733. [Google Scholar] [CrossRef] [PubMed]
- Olivares, O.; Vasseur, S. Metabolic rewiring of pancreatic ductal adenocarcinoma: New routes to follow within the maze. Int. J. Cancer 2015, 138, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Cameron, M.E.; Yakovenko, A.; Trevino, J.G. Glucose and Lactate Transport in Pancreatic Cancer: Glycolytic Metabolism Revisited. J. Oncol. 2018, 2018, 6214838. [Google Scholar] [CrossRef]
- Ding, X.Z.; Fehsenfeld, D.M.; Murphy, L.O.; Permert, J.; Adrian, T.E. Physiological concentrations of insulin augment pancreatic cancer cell proliferation and glucose utilization by activating MAP kinase, PI3 kinase and enhancing GLUT-1 expression. Pancreas 2000, 21, 310–320. [Google Scholar] [CrossRef]
- Kumari, S.; Khan, S.; Gupta, S.C.; Kashyap, V.K.; Yallapu, M.M.; Chauhan, S.C.; Jaggi, M. MUC13 contributes to rewiring of glucose metabolism in pancreatic cancer. Oncogenesis 2018, 7, 1–12. [Google Scholar] [CrossRef]
- Muller, G.; Schulz, A.; Wied, S.; Frick, W. Regulation of lipid raft proteins by glimepiride- and insulin-induced glycosylphosphatidylinositol-specific phospholipase C in rat adipocytes. Biochem. Pharmacol. 2005, 69, 761–780. [Google Scholar] [CrossRef]
- Thirone, A.C.; Huang, C.; Klip, A. Tissue-specific roles of IRS proteins in insulin signaling and glucose transport. Trends Endocrinol. Metab. 2006, 17, 72–78. [Google Scholar] [CrossRef]
- Hribal, M.L.; Federici, M.; Porzio, O.; Lauro, D.; Borboni, P.; Accili, D.; Lauro, R.; Sesti, G. The Gly-->Arg972 amino acid polymorphism in insulin receptor substrate-1 affects glucose metabolism in skeletal muscle cells. J. Clin. Endocrinol. Metab. 2000, 85, 2004–2013. [Google Scholar] [CrossRef]
- Sakurai, Y.; Kubota, N.; Takamoto, I.; Obata, A.; Iwamoto, M.; Hayashi, T.; Aihara, M.; Kubota, T.; Nishihara, H.; Kadowaki, T. Role of insulin receptor substrates in the progression of hepatocellular carcinoma. Sci. Rep. 2017, 7, 5387. [Google Scholar] [CrossRef] [PubMed]
- Ochman, A.R.; Lipinski, C.A.; Handler, J.A.; Reaume, A.G.; Saporito, M.S. The Lyn kinase activator MLR-1023 is a novel insulin receptor potentiator that elicits a rapid-onset and durable improvement in glucose homeostasis in animal models of type 2 diabetes. J. Pharmacol. Exp. Ther. 2012, 342, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Reaume, A.G. High throughput in vivo phenotypic screening for drug repurposing: Discovery of MLR-1023 a novel insulin sensitizer and novel Lyn kinase activator with clinical proof of concept. Bioorg. Med. Chem. 2020, 28, 115425. [Google Scholar] [CrossRef] [PubMed]
- Saporito, M.S.; Ochman, A.R.; Lipinski, C.A.; Handler, J.A.; Reaume, A.G. MLR-1023 is a potent and selective allosteric activator of Lyn kinase in vitro that improves glucose tolerance in vivo. J. Pharmacol. Exp. Ther. 2012, 342, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Su, R.; Zhang, J. Oncogenic role of LYN in human gastric cancer via the Wnt/beta-catenin and AKT/mTOR pathways. Exp. Ther. Med. 2020, 20, 646–654. [Google Scholar] [PubMed]
- Liu, S.; Hao, X.; Ouyang, X.; Dong, X.; Yang, Y.; Yu, T.; Hu, J.; Hu, L. Tyrosine kinase LYN is an oncotarget in human cervical cancer: A quantitative proteomic based study. Oncotarget 2016, 7, 75468–75481. [Google Scholar] [CrossRef]
- Goldenberg-Furmanov, M.; Stein, I.; Pikarsky, E.; Rubin, H.; Kasem, S.; Wygoda, M.; Weinstein, I.; Reuveni, H.; Ben-Sasson, S.A. Lyn is a target gene for prostate cancer: Sequence-based inhibition induces regression of human tumor xenografts. Cancer Res. 2004, 64, 1058–1066. [Google Scholar] [CrossRef]
- Guan, H.; Zhou, Z.; Gallick, G.E.; Jia, S.F.; Morales, J.; Sood, A.K.; Corey, S.J.; Kleinerman, E.S. Targeting Lyn inhibits tumor growth and metastasis in Ewing’s sarcoma. Mol. Cancer Ther. 2008, 7, 1807–1816. [Google Scholar] [CrossRef]
- Roseweir, A.K.; Qayyum, T.; Lim, Z.; Hammond, R.; MacDonald, A.I.; Fraser, S.; Oades, G.M.; Aitchison, M.; Jones, R.J.; Edwards, J. Nuclear expression of Lyn, a Src family kinase member, is associated with poor prognosis in renal cancer patients. BMC Cancer 2016, 16, 1–10. [Google Scholar] [CrossRef]
- Mao, L.; Deng, W.W.; Yu, G.T.; Bu, L.L.; Liu, J.F.; Ma, S.R.; Wu, L.; Kulkarni, A.B.; Zhang, W.F.; Sun, Z.J. Inhibition of SRC family kinases reduces myeloid-derived suppressor cells in head and neck cancer. Int. J. Cancer. 2017, 140, 1173–1185. [Google Scholar] [CrossRef]
- Kim, Y.J.; Hong, S.; Sung, M.; Park, M.J.; Jung, K.; Noh, K.-W.; Oh, D.-Y.; Lee, M.-S.; Oh, E.; Shin, Y.K.; et al. LYN expression predicts the response to dasatinib in a subpopulation of lung adenocarcinoma patients. Oncotarget 2016, 7, 82876–82888. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Choi, Y.L.; Bocanegra, M.; Kwon, M.J.; Shin, Y.K.; Nam, S.J.; Yang, J.H.; Kao, J.; Godwin, A.K.; Pollack, J.R. LYN is a mediator of epithelial-mesenchymal transition and a target of dasatinib in breast cancer. Cancer Res. 2010, 70, 2296–2306. [Google Scholar] [CrossRef] [PubMed]
- Stettner, M.R.; Wang, W.; Nabors, L.B.; Bharara, S.; Flynn, D.C.; Grammer, J.R.; Gillespie, G.Y.; Gladson, C.L. Lyn kinase activity is the predominant cellular SRC kinase activity in glioblastoma tumor cells. Cancer Res. 2005, 65, 5535–5543. [Google Scholar] [CrossRef] [PubMed]
- Dolivo, D.M.; Larson, S.A.; Dominko, T. Tryptophan metabolites kynurenine and serotonin regulate fibroblast activation and fibrosis. Cell Mol. Life Sci. 2018, 75, 3663–3681. [Google Scholar] [CrossRef]
- Tawfik, M.K.; Makary, S. 5-HT7 receptor antagonism (SB-269970) attenuates bleomycin-induced pulmonary fibrosis in rats via downregulating oxidative burden and inflammatory cascades and ameliorating collagen deposition: Comparison to terguride. Eur. J. Pharmacol. 2017, 814, 114–123. [Google Scholar] [CrossRef]
- Lofdahl, A.; Rydell-Tormanen, K.; Muller, C.; Martina Holst, C.; Thiman, L.; Ekstrom, G.; Wenglen, C.; Larsson-Callerfelt, A.K.; Westergren-Thorsson, G. 5-HT2B receptor antagonists attenuate myofibroblast differentiation and subsequent fibrotic responses in vitro and in vivo. Physiol. Rep. 2016, 4, e12873. [Google Scholar] [CrossRef]
- Elaidy, S.M.; Essawy, S.S. The antifibrotic effects of alveolar macrophages 5-HT2C receptors blockade on bleomycin-induced pulmonary fibrosis in rats. Pharmacol. Rep. 2016, 68, 1244–1253. [Google Scholar] [CrossRef]
- Ruddell, R.G.; Oakley, F.; Hussain, Z.; Yeung, I.; Bryan-Lluka, L.J.; Ramm, G.A.; Mann, D.A. A role for serotonin (5-HT) in hepatic stellate cell function and liver fibrosis. Am. J. Pathol. 2006, 169, 861–876. [Google Scholar] [CrossRef]
- Erikci, A.; Ucar, G.; Yabanoglu-Ciftci, S. Role of serotonin in the regulation of renal proximal tubular epithelial cells. Ren. Fail. 2016, 38, 1141–1150. [Google Scholar] [CrossRef]
- Hamasaki, Y.; Doi, K.; Maeda-Mamiya, R.; Ogasawara, E.; Katagiri, D.; Tanaka, T.; Yamamoto, T.; Sugaya, T.; Nangaku, M.; Noiri, E. A 5-hydroxytryptamine receptor antagonist, sarpogrelate, reduces renal tubulointerstitial fibrosis by suppressing PAI-1. Am. J. Physiol. Renal Physiol. 2013, 305, F1796–F1803. [Google Scholar] [CrossRef]
- Soll, C.; Jang, J.H.; Riener, M.O.; Moritz, W.; Wild, P.J.; Graf, R.; Clavien, P.A. Serotonin promotes tumor growth in human hepatocellular cancer. Hepatology 2010, 51, 1244–1254. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.H.; Li, J.; Dong, F.Y.; Yang, J.Y.; Liu, D.J.; Yang, X.M.; Wang, Y.H.; Yang, M.W.; Fu, X.L.; Zhang, X.X.; et al. Increased Serotonin Signaling Contributes to the Warburg Effect in Pancreatic Tumor Cells Under Metabolic Stress and Promotes Growth of Pancreatic Tumors in Mice. Gastroenterology 2017, 153, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Zrihan-Licht, S.; Deng, B.; Yarden, Y.; McShan, G.; Keydar, I.; Avraham, H.K. Csk Homologous Kinase, a Novel Signaling Molecule, Directly Associates with the Activated ErbB-2 Receptor in Breast Cancer Cells and Inhibits Their Proliferation. J. Biol. Chem. 1998, 273, 4065–4072. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Zagozdzon, R.; Avraham, R.; Avraham, H.K. CHK negatively regulates Lyn kinase and suppresses pancreatic cancer cell invasion. Int. J. Oncol. 2006, 29, 1453–1458. [Google Scholar] [CrossRef] [PubMed]
- Britton, D.; Zen, Y.; Quaglia, A.; Selzer, S.; Mitra, V.; Lobetaner, C.; Jung, S.; Bohm, G.; Schmid, P.; Prefot, P.; et al. Quantification of pancreatic cancer proteome and phosphorylome: Indicates molecular events likely contributing to cancer and activity of drug targets. PLoS ONE 2014, 9, e90948. [Google Scholar] [CrossRef]
- Chen, Z.-Y.; Cai, L.; Bie, P.; Wang, S.-G.; Jiang, Y.; Dong, J.-H.; Li, X.-W. Roles of Fyn in pancreatic cancer metastasis. J. Gastroenterol. Hepatol. 2010, 25, 293–301. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Cai, L.; Zhu, J.; Chen, M.; Chen, J.; Li, Z.H.; Liu, X.D.; Wang, S.G.; Bie, P.; Jiang, P.; et al. Fyn requires HnRNPA2B1 and Sam68 to synergistically regulate apoptosis in pancreatic cancer. Carcinogenesis 2011, 32, 1419–1426. [Google Scholar] [CrossRef]
- Dong, W.; Sun, S.J.; Qin, J.J.; Liu, G.M. Fyn stimulates the progression of pancreatic cancer via Fyn-GluN2b-AKT axis. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 109–121. [Google Scholar]
- Batlle, E.; Wilkinson, D.G. Molecular mechanisms of cell segregation and boundary formation in development and tumorigenesis. Cold Spring Harb. Perspect. Biol. 2012, 4, a008227. [Google Scholar] [CrossRef]
- Klein, R. Eph/ephrin signalling during development. Development 2012, 139, 4105–4109. [Google Scholar] [CrossRef]
- Pitulescu, M.E.; Adams, R.H. Eph/ephrin molecules—A hub for signaling and endocytosis. Genes Dev. 2010, 24, 2480–2492. [Google Scholar] [CrossRef] [PubMed]
- Huusko, P.; Ponciano-Jackson, D.; Wolf, M.; Kiefer, J.; O Azorsa, D.; Tuzmen, S.; Weaver, D.; Robbins, C.; Moses, T.; Allinen, M.; et al. Nonsense-mediated decay microarray analysis identifies mutations of EPHB2 in human prostate cancer. Nat. Genet. 2004, 36, 979–983. [Google Scholar] [CrossRef] [PubMed]
- Sulman, E.P.; Tang, X.X.; Allen, C.; Biegel, J.A.; Pleasure, D.E.; Brodeur, G.M.; Ikegaki, N. ECK, a human EPH-related gene, maps to 1p36.1, a common region of alteration in human cancers. Genomics 1997, 40, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Bardelli, A.; Parsons, D.W.; Silliman, N.; Ptak, J.; Szabo, S.; Saha, S.; Markowitz, S.; Willson, J.K.; Parmigiani, G.; Kinzler, K.W.; et al. Mutational analysis of the tyrosine kinome in colorectal cancers. Science 2003, 300, 949. [Google Scholar] [CrossRef] [PubMed]
- Noblitt, L.W.; Bangari, D.S.; Shukla, S.; Knapp, D.W.; Mohammed, S.; Kinch, M.S.; Mittal, S.K. Decreased tumorigenic potential of EphA2-overexpressing breast cancer cells following treatment with adenoviral vectors that express EphrinA1. Cancer Gene Ther. 2004, 11, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Pasquale, E.B. Eph receptor signalling casts a wide net on cell behaviour. Nat. Rev. Mol. Cell Biol. 2005, 6, 462–475. [Google Scholar] [CrossRef]
- Liu, C.; Huang, H.; Wang, C.; Kong, Y.; Zhang, H. Involvement of ephrin receptor A4 in pancreatic cancer cell motility and invasion. Oncol. Lett. 2014, 7, 2165–2169. [Google Scholar] [CrossRef][Green Version]
- Lu, Z.; Zhang, Y.; Li, Z.; Yu, S.; Zhao, G.; Li, M.; Wang, Z.; Wang, Q.; Yang, Y. Overexpression of the B-type Eph and ephrin genes correlates with progression and pain in human pancreatic cancer. Oncol. Lett. 2012, 3, 1207–1212. [Google Scholar] [CrossRef]
- Rudno-Rudzinska, J.; Kielan, W.; Frejlich, E.; Kotulski, K.; Hap, W.; Kurnol, K.; Dzierzek, P.; Zawadzki, M.; Halon, A. A review on Eph/ephrin, angiogenesis and lymphangiogenesis in gastric, colorectal and pancreatic cancers. Chin. J. Cancer Res. 2017, 29, 303–312. [Google Scholar] [CrossRef]
- Giaginis, C.; Tsourouflis, G.; Zizi-Serbetzoglou, A.; Kouraklis, G.; Chatzopoulou, E.; Dimakopoulou, K.; Theocharis, S.E. Clinical significance of ephrin (eph)- A1, -A2, -a4, -a5 and -a7 receptors in pancreatic ductal adenocarcinoma. Pathol. Oncol. Res. 2010, 16, 267–276. [Google Scholar] [CrossRef]
- Lisabeth, E.M.; Falivelli, G.; Pasquale, E.B. Eph receptor signaling and ephrins. Cold Spring Harb. Perspect. Biol. 2013, 5, a009159. [Google Scholar] [CrossRef] [PubMed]
- Tuzi, N.L.; Gullick, W.J. Eph, the largest known family of putative growth factor receptors. Br. J. Cancer 1994, 69, 417–421. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Katoh, M.; Katoh, M. Comparative integromics on Eph family. Int. J. Oncol. 2006, 28, 1243–1247. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Basturk, O.; Tan, M.; Bhanot, U.; Allen, P.; Adsay, V.; Scott, S.N.; Shah, R.; Berger, M.F.; Askan, G.; Dikoglu, E.; et al. The oncocytic subtype is genetically distinct from other pancreatic intraductal papillary mucinous neoplasm subtypes. Mod. Pathol. 2016, 29, 1058–1069. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Park, S. The EphA8 receptor regulates integrin activity through p110gamma phosphatidylinositol-3 kinase in a tyrosine kinase activity-independent manner. Mol. Cell. Biol. 2001, 21, 4579–4597. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Topalovski, M.; Brekken, R.A. Matrix control of pancreatic cancer: New insights into fibronectin signaling. Cancer Lett. 2016, 381, 252–258. [Google Scholar] [CrossRef]
- Pankov, R.; Yamada, K.M. Fibronectin at a glance. J. Cell Sci. 2002, 115, 3861–3863. [Google Scholar] [CrossRef]
- Gu, C.; Shim, S.; Shin, J.; Kim, J.; Park, J.; Han, K.; Park, S. The EphA8 receptor induces sustained MAP kinase activation to promote neurite outgrowth in neuronal cells. Oncogene 2005, 24, 4243–4256. [Google Scholar] [CrossRef]
- Yan, Y.; Wang, Q.; Yan, X.L.; Zhang, Y.; Li, W.; Tang, F.; Li, X.; Yang, P. miR-10a controls glioma migration and invasion through regulating epithelial-mesenchymal transition via EphA8. FEBS Lett. 2015, 589, 756–765. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, N.; Li, P.; Wu, H.; Wang, Q.; Gao, X.; Wang, X.; Huang, J. EphA8 acts as an oncogene and contributes to poor prognosis in gastric cancer via regulation of ADAM10. J. Cell Physiol. 2019, 234, 20408–20419. [Google Scholar] [CrossRef]
- Liu, L.; Wang, X.; Ge, W. EphA8 is a Prognostic Factor for Oral Tongue Squamous Cell Carcinoma. Med. Sci. Monit. 2018, 24, 7213–7222. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, Q.; Zhao, L.; Jiang, L.; Qi, A.; Wei, Q.; Song, X.; Wang, L.; Zhang, L.; Zhao, Y.; et al. A Combined four-mRNA Signature Associated with Lymphatic Metastasis for Prognosis of Colorectal Cancer. J. Cancer 2020, 11, 2139–2149. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xu, Y.; Jin, Q.; Wang, W.; Zhang, S.; Wang, X.; Zhang, Y.; Xu, X.; Huang, J. EphA8 is a prognostic marker for epithelial ovarian cancer. Oncotarget 2016, 7, 20801–20809. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Park, S. Phosphorylation at Tyr-838 in the kinase domain of EphA8 modulates Fyn binding to the Tyr-615 site by enhancing tyrosine kinase activity. Oncogene 1999, 18, 5413–5422. [Google Scholar] [CrossRef]
- Lin, L.; Shi, C.; Sun, Z.; Le, N.T.; Abe, J.I.; Hu, K. The Ser/Thr kinase p90RSK promotes kidney fibrosis by modulating fibroblast-epithelial crosstalk. J. Biol. Chem. 2019, 294, 9901–9910. [Google Scholar] [CrossRef]
- Abe, J.; Berk, B. Fyn-Dependent Activation of p90 Ribosomal S6 Kinase (RSK) by H2O2: New Redox Sensitive Pathway. In Circulation; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 1998; p. 220. [Google Scholar]
- Calautti, E.; Grossi, M.; Mammucari, C.; Aoyama, Y.; Pirro, M.; Ono, Y.; Li, J.; Dotto, G.P. Fyn tyrosine kinase is a downstream mediator of Rho/PRK2 function in keratinocyte cell–cell adhesion. J. Cell Biol. 2002, 156, 137–148. [Google Scholar] [CrossRef]
- Gujral, T.S.; Chan, M.; Peshkin, L.; Sorger, P.K.; Kirschner, M.W.; MacBeath, G. A noncanonical Frizzled2 pathway regulates epithelial-mesenchymal transition and metastasis. Cell 2014, 159, 844–856. [Google Scholar] [CrossRef]
- Cary, L.A.; Chang, J.F.; Guan, J.L. Stimulation of cell migration by overexpression of focal adhesion kinase and its association with Src and Fyn. J. Cell Sci. 1996, 109, 1787–1794. [Google Scholar]
- Lewis-Tuffin, L.J.; Feathers, R.; Hari, P.; Durand, N.; Li, Z.; Rodriguez, F.J.; Bakken, K.; Carlson, B.; Schroeder, M.; Sarkaria, J.N.; et al. Src family kinases differentially influence glioma growth and motility. Mol. Oncol. 2015, 9, 1783–1798. [Google Scholar] [CrossRef]
- Jensen, A.R.; David, S.Y.; Liao, C.; Dai, J.; Keller, E.T.; Al-Ahmadie, H.; Dakin-Hache, K.; Usatyuk, P.; Sievert, M.F.; Paner, G.P.; et al. Fyn is downstream of the HGF/MET signaling axis and affects cellular shape and tropism in PC3 cells. Clin. Cancer Res. 2011, 17, 3112–3122. [Google Scholar] [CrossRef]
- Posadas, E.M.; Al-Ahmadie, H.; Robinson, V.L.; Jagadeeswaran, R.; Otto, K.; Kasza, K.E.; Tretiakov, M.; Siddiqui, J.; Pienta, K.J.; Stadler, W.M.; et al. FYN is overexpressed in human prostate cancer. BJU Int. 2009, 103, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, Y.; Hu, Y.; Dang, D.; Regezi, J.; Schmidt, B.L.; Atakilit, A.; Chen, B.; Ellis, D.; Ramos, D.M. Alphavbeta6-Fyn signaling promotes oral cancer progression. J. Biol. Chem. 2003, 278, 41646–41653. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, R.; Kubo, S.; Takeda, H.; Ohta, T.; Tabata, C.; Ogawa, H.; Nakano, T.; Fujimori, Y. Deficiency of Fyn protein is prerequisite for apoptosis induced by Src family kinase inhibitors in human mesothelioma cells. Carcinogenesis 2012, 33, 969–975. [Google Scholar] [CrossRef] [PubMed]
- Elias, D.; Ditzel, H.J. Fyn is an important molecule in cancer pathogenesis and drug resistance. Pharmacol. Res. 2015, 100, 250–254. [Google Scholar] [CrossRef]
- Yin, L.; Wang, Y.; Guo, X.; Xu, C.; Yu, G. Comparison of gene expression in liver regeneration and hepatocellular carcinoma formation. Cancer Manag. Res. 2018, 10, 5691–5708. [Google Scholar] [CrossRef]
- Elias, D.; Vever, H.; Laenkholm, A.V.; Gjerstorff, M.F.; Yde, C.W.; Lykkesfeldt, A.E.; Ditzel, H.J. Gene expression profiling identifies FYN as an important molecule in tamoxifen resistance and a predictor of early recurrence in patients treated with endocrine therapy. Oncogene 2015, 34, 1919–1927. [Google Scholar] [CrossRef]
- Singh, M.M.; Howard, A.; Irwin, M.E.; Gao, Y.; Lu, X.; Multani, A.; Chandra, J. Expression and activity of Fyn mediate proliferation and blastic features of chronic myelogenous leukemia. PLoS ONE 2012, 7, e51611. [Google Scholar] [CrossRef]
- Grosso, S.; Puissant, A.; Dufies, M.; Colosetti, P.; Jacquel, A.; Lebrigand, K.; Barbry, P.; Deckert, M.; Cassuto, J.P.; Mari, B.; et al. Gene expression profiling of imatinib and PD166326-resistant CML cell lines identifies Fyn as a gene associated with resistance to BCR-ABL inhibitors. Mol. Cancer Ther. 2009, 8, 1924–1933. [Google Scholar] [CrossRef]
- Zhang, X.; Huang, Z.; Guo, Y.; Xiao, T.; Tang, L.; Zhao, S.; Wu, L.; Su, J.; Zeng, W.; Huang, H.; et al. The phosphorylation of CD147 by Fyn plays a critical role for melanoma cells growth and metastasis. Oncogene 2020, 39, 4183–4197. [Google Scholar] [CrossRef]
- Liu, D.; Gao, M.; Wu, K.; Zhu, D.; Yang, Y.; Zhao, S. LINC00152 facilitates tumorigenesis in esophageal squamous cell carcinoma via miR-153-3p/FYN axis. Biomed. Pharmacother. 2019, 112, 108654. [Google Scholar] [CrossRef]
- Wang, Y.; Lin, R.; Ling, H.; Ke, Y.; Zeng, Y.; Xiong, Y.; Zhou, Q.; Zhou, F.; Zhou, Y. Dual inhibition of CDK4 and FYN leads to selective cell death in KRAS-mutant colorectal cancer. Signal Transduct. Target. Ther. 2019, 4, 52. [Google Scholar] [CrossRef] [PubMed]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435. [Google Scholar] [CrossRef] [PubMed]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Qi, Q.; Chan, C.B.; Zhou, W.; Chen, J.; Luo, H.R.; Appin, C.; Brat, D.J.; Ye, K. Fyn-phosphorylated PIKE-A binds and inhibits AMPK signaling, blocking its tumor suppressive activity. Cell Death Differ. 2016, 23, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef]
- Ahn, J.Y.; Rong, R.; Kroll, T.G.; Van Meir, E.G.; Snyder, S.H.; Ye, K. PIKE (phosphatidylinositol 3-kinase enhancer)-A GTPase stimulates Akt activity and mediates cellular invasion. J. Biol. Chem. 2004, 279, 16441–16451. [Google Scholar] [CrossRef]
- Whiteman, E.L.; Cho, H.; Birnbaum, M.J. Role of Akt/protein kinase B in metabolism. Trends Endocrinol. Metab. 2002, 13, 444–451. [Google Scholar] [CrossRef]
- Sudhagar, S.; Sathya, S.; Gokulapriya, G.; Lakshmi, B.S. AKT-p53 axis protect cancer cells from autophagic cell death during nutrition deprivation. Biochem. Biophys. Res. Commun. 2016, 471, 396–401. [Google Scholar] [CrossRef]
- Stirnweiss, A.; Hartig, R.; Gieseler, S.; Lindquist, J.A.; Reichardt, P.; Philipsen, L.; Simeoni, L.; Poltorak, M.; Merten, C.; Zuschratter, W.; et al. T cell activation results in conformational changes in the Src family kinase Lck to induce its activation. Sci. Signal. 2013, 6, ra13. [Google Scholar] [CrossRef]
- Philipsen, L.; Reddycherla, A.V.; Hartig, R.; Gumz, J.; Kastle, M.; Kritikos, A.; Poltorak, M.P.; Prokazov, Y.; Turbin, E.; Weber, A.; et al. De novo phosphorylation and conformational opening of the tyrosine kinase Lck act in concert to initiate T cell receptor signaling. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef]
- Bommhardt, U.H.; Schraven, B.; Simeoni, L. Beyond TCR Signaling: Emerging Functions of Lck in Cancer and Immunotherapy. Int. J. Mol. Sci. 2019, 20, 3500. [Google Scholar] [CrossRef] [PubMed]
- Koster, A.; Landgraf, S.; Leipold, A.; Sachse, R.; Gebhart, E.; Tulusan, A.H.; Ronay, G.; Schmidt, C.; Dingermann, T. Expression of oncogenes in human breast cancer specimens. Anticancer Res. 1991, 11, 193–201. [Google Scholar] [PubMed]
- Rupniewska, E.; Roy, R.; Mauri, F.A.; Liu, X.; Kaliszczak, M.; Bellezza, G.; Cagini, L.; Barbareschi, M.; Ferrero, S.; Tommasi, A.M.; et al. Targeting autophagy sensitises lung cancer cells to Src family kinase inhibitors. Oncotarget 2018, 9, 27346–27362. [Google Scholar] [CrossRef] [PubMed]
- Sugihara, T.; Werneburg, N.W.; Hernandez, M.C.; Yang, L.; Kabashima, A.; Hirsova, P.; Yohanathan, L.; Sosa, C.; Truty, M.J.; Vasmatzis, G.; et al. YAP Tyrosine Phosphorylation and Nuclear Localization in Cholangiocarcinoma Cells Are Regulated by LCK and Independent of LATS Activity. Mol. Cancer Res. 2018, 16, 1556–1567. [Google Scholar] [CrossRef]
- Zepecki, J.P.; Snyder, K.M.; Moreno, M.M.; Fajardo, E.; Fiser, A.; Ness, J.; Sarkar, A.; Toms, S.A.; Tapinos, N. Regulation of human glioma cell migration, tumor growth, and stemness gene expression using a Lck targeted inhibitor. Oncogene 2019, 38, 1734–1750. [Google Scholar] [CrossRef]
- Clarke, C.N.; Lee, M.S.; Wei, W.; Manyam, G.; Jiang, Z.-Q.; Lu, Y.; Morris, J.; Broom, B.; Menter, D.; Vilar-Sanchez, E.; et al. Proteomic Features of Colorectal Cancer Identify Tumor Subtypes Independent of Oncogenic Mutations and Independently Predict Relapse-Free Survival. Ann. Surg. Oncol. 2017, 24, 4051–4058. [Google Scholar] [CrossRef]
- Janikowska, G.; Janikowski, T.; Pyka-Pajak, A.; Mazurek, U.; Janikowski, M.; Gonciarz, M.; Lorenc, Z. Potential biomarkers for the early diagnosis of colorectal adenocarcinoma—Transcriptomic analysis of four clinical stages. Cancer Biomark. 2018, 22, 89–99. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Marech, I.; Patruno, R.; Zizzo, N.; Gadaleta, C.; Introna, M.; Zito, A.F.; Gadaleta, C.D.; Ranieri, G. Masitinib (AB1010), from canine tumor model to human clinical development: Where we are? Crit. Rev. Oncol. Hematol. 2014, 91, 98–111. [Google Scholar] [CrossRef]
- Humbert, M.; Casteran, N.; Letard, S.; Hanssens, K.; Iovanna, J.; Finetti, P.; Bertucci, F.; Bader, T.; Mansfield, C.D.; Moussy, A.; et al. Masitinib combined with standard gemcitabine chemotherapy: In vitro and in vivo studies in human pancreatic tumour cell lines and ectopic mouse model. PLoS ONE 2010, 5, e9430. [Google Scholar] [CrossRef]
- Capurso, G.; Lattimore, S.; Crnogorac-Jurcevic, T.; Panzuto, F.; Milione, M.; Bhakta, V.; Campanini, N.; Swift, S.M.; Bordi, C.; Delle Fave, G.; et al. Gene expression profiles of progressive pancreatic endocrine tumours and their liver metastases reveal potential novel markers and therapeutic targets. Endocr. Relat. Cancer 2006, 13, 541–558. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Hu, C.; Wang, A.; Weisberg, E.L.; Chen, Y.; Yun, C.H.; Wang, W.; Liu, Y.; Liu, X.; Tian, B.; et al. Discovery of a BTK/MNK dual inhibitor for lymphoma and leukemia. Leukemia 2016, 30, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Bagheri-Yarmand, R.; Mandal, M.; Taludker, A.H.; Wang, R.A.; Vadlamudi, R.K.; Kung, H.J.; Kumar, R. Etk/Bmx tyrosine kinase activates Pak1 and regulates tumorigenicity of breast cancer cells. J. Biol. Chem. 2001, 276, 29403–29409. [Google Scholar] [CrossRef] [PubMed]
- Dai, B.; Kim, O.; Xie, Y.; Guo, Z.; Xu, K.; Wang, B.; Kong, X.; Melamed, J.; Chen, H.; Bieberich, C.J.; et al. Tyrosine Kinase Etk/BMX Is Up-regulated in Human Prostate Cancer and Its Overexpression Induces Prostate Intraepithelial Neoplasia in Mouse. Cancer Res. 2006, 66, 8058–8064. [Google Scholar] [CrossRef]
- Dai, B.; Chen, H.; Guo, S.; Yang, X.; Linn, D.E.; Sun, F.; Li, W.; Guo, Z.; Xu, K.; Kim, O.; et al. Compensatory Upregulation of Tyrosine Kinase Etk/BMX in Response to Androgen Deprivation Promotes Castration-Resistant Growth of Prostate Cancer Cells. Cancer Res. 2010, 70, 5587–5596. [Google Scholar] [CrossRef]
- Chen, C.; Wang, G.; Zhang, Z.M.; Xu, W.; Li, Q.; Hu, Q.; Wang, D.; Li, Z.P.; Yang, Z.X.; Suo, J.Y.; et al. The expression of Tec and the level of its phosphorylation in primary hepatic carcinomas. Zhonghua Gan Zang Bing Za Zhi 2007, 15, 910–913. [Google Scholar]
- Guryanova, O.A.; Wu, Q.; Cheng, L.; Lathia, J.D.; Huang, Z.; Yang, J.; MacSwords, J.; Eyler, C.E.; McLendon, R.E.; Heddleston, J.M.; et al. Nonreceptor Tyrosine Kinase BMX Maintains Self-Renewal and Tumorigenic Potential of Glioblastoma Stem Cells by Activating STAT3. Cancer Cell 2011, 19, 498–511. [Google Scholar] [CrossRef]
- Potter, D.S.; Galvin, M.; Brown, S.; Lallo, A.; Hodgkinson, C.L.; Blackhall, F.; Morrow, C.J.; Dive, C. Inhibition of PI3K/BMX Cell Survival Pathway Sensitizes to BH3 Mimetics in SCLC. Mol. Cancer Ther. 2016, 15, 1248–1260. [Google Scholar] [CrossRef]
- Potter, D.S.; Kelly, P.; Denneny, O.; Juvin, V.; Stephens, L.R.; Dive, C.; Morrow, C.J. BMX acts downstream of PI3K to promote colorectal cancer cell survival and pathway inhibition sensitizes to the BH3 mimetic ABT-737. Neoplasia 2014, 16, 147–157. [Google Scholar] [CrossRef]
- Paavonen, K.; Ekman, N.; Wirzenius, M.; Rajantie, I.; Poutanen, M.; Alitalo, K. Bmx Tyrosine Kinase Transgene Induces Skin Hyperplasia, Inflammatory Angiogenesis, and Accelerated Wound Healing. Mol. Biol. Cell 2004, 15, 4226–4233. [Google Scholar] [CrossRef]
- Singh, S.P.; Dammeijer, F.; Hendriks, R.W. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol. Cancer 2018, 17, 57. [Google Scholar] [CrossRef] [PubMed]
- Dubovsky, J.A.; Beckwith, K.A.; Natarajan, G.; Woyach, J.A.; Jaglowski, S.; Zhong, Y.; Hessler, J.D.; Liu, T.-M.; Chang, B.Y.; Larkin, K.M.; et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood 2013, 122, 2539–2549. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, K.; Sommers, C.L.; Decker, D.C.; Mitchell, E.O.; Korthauer, U.; Sperling, A.I.; Kozak, C.A.; Love, P.E.; Bluestone, J.A. Ribp, a Novel Rlk/Txk- and Itk-Binding Adaptor Protein That Regulates T Cell Activation. J. Exp. Med. 1999, 190, 1657–1668. [Google Scholar] [CrossRef]
- Chamorro, M.; Czar, M.J.; Debnath, J.; Cheng, G.; Lenardo, M.J.; Varmus, H.E.; Schwartzberg, P.L. Requirements for activation and RAFT localization of the T-lymphocyte kinase Rlk/Txk. BMC Immunol. 2001, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.; Guerette, B.; Guntermann, C.; Rudd, C.E. Resting Lymphocyte Kinase (Rlk/Txk) Targets Lymphoid Adaptor SLP-76 in the Cooperative Activation of Interleukin-2 Transcription in T-cells. J. Biol. Chem. 2000, 275, 3835–3840. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, E.M.; Debnath, J.; Yap, G.; McVicar, D.; Liao, X.C.; Littman, D.R.; Sher, A.; Varmus, H.E.; Lenardo, M.J.; Schwartzberg, P.L. Requirement for Tec Kinases Rlk and Itk in T Cell Receptor Signaling and Immunity. Science 1999, 284, 638–641. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Dong, S.; Strattan, E.; Ren, L.; Butchar, J.P.; Thornton, K.; Mishra, A.; Porcu, P.; Bradshaw, J.M.; Bisconte, A.; et al. Targeting Interleukin-2-inducible T-cell Kinase (ITK) and Resting Lymphocyte Kinase (RLK) Using a Novel Covalent Inhibitor PRN694. J. Biol. Chem. 2015, 290, 5960–5978. [Google Scholar] [CrossRef]
- Felices, M.; Falk, M.; Kosaka, Y.; Berg, L.J. Tec Kinases in T Cell and Mast Cell Signaling. In Advances in Immunology; Academic Press: Cambridge, MA, USA, 2007; Volume 93, pp. 145–184. [Google Scholar]
- Chen, R.; Kim, O.; Li, M.; Xiong, X.; Guan, J.-L.; Kung, H.-J.; Chen, H.; Shimizu, Y.; Qiu, Y. Regulation of the PH-domain-containing tyrosine kinase Etk by focal adhesion kinase through the FERM domain. Nat. Cell Biol. 2001, 3, 439–444. [Google Scholar] [CrossRef]
- Meng, Y.; Sha, S.; Yang, J.; Ren, H. Effects of Tec Tyrosine Kinase Inhibition on the Inflammatory Response of Severe Acute Pancreatitis-Associated Acute Lung Injury in Mice. Dig. Dis. Sci. 2019, 64, 2167–2176. [Google Scholar] [CrossRef]
- Whitcomb, D.C.; Frulloni, L.; Garg, P.; Greer, J.B.; Schneider, A.; Yadav, D.; Shimosegawa, T. Chronic pancreatitis: An international draft consensus proposal for a new mechanistic definition. Pancreatology 2016, 16, 218–224. [Google Scholar] [CrossRef]
- Raimondi, S.; Lowenfels, A.B.; Morselli-Labate, A.M.; Maisonneuve, P.; Pezzilli, R. Pancreatic cancer in chronic pancreatitis; aetiology, incidence, and early detection. Best Pr. Res. Clin. Gastroenterol. 2010, 24, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Aslan, M.; Shahbazi, R.; Ulubayram, K.; Ozpolat, B. Targeted Therapies for Pancreatic Cancer and Hurdles Ahead. Anticancer Res. 2018, 38, 6591–6606. [Google Scholar] [CrossRef] [PubMed]
- Valencia-Sama, I.; Ladumor, Y.; Kee, L.; Adderley, T.; Christopher, G.; Robinson, C.M.; Kano, Y.; Ohh, M.; Irwin, M.S. NRAS Status Determines Sensitivity to SHP2 Inhibitor Combination Therapies Targeting the RAS-MAPK Pathway in Neuroblastoma. Cancer Res. 2020, 80, 3413–3423. [Google Scholar] [CrossRef] [PubMed]
- Beyens, M.; Vandamme, T.; Peeters, M.; Van Camp, G.; De Beeck, K.O. Resistance to targeted treatment of gastroenteropancreatic neuroendocrine tumors. Endocr. Relat. Cancer 2019, 26, R109–R130. [Google Scholar] [CrossRef]
- Hu, H.; Zhang, Q.; Chen, W.; Wu, T.; Liu, S.; Li, X.; Luo, B.; Zhang, T.; Yan, G.; Lu, H.; et al. MicroRNA-301a promotes pancreatic cancer invasion and metastasis through the JAK/STAT3 signaling pathway by targeting SOCS5. Carcinogenesis 2020, 41, 502–514. [Google Scholar] [CrossRef]
- Hosein, A.N.; Brekken, R.A.; Maitra, A. Pancreatic cancer stroma: An update on therapeutic targeting strategies. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 487–505. [Google Scholar] [CrossRef]
- Huang, H.; Wright, S.; Zhang, J.; Brekken, R.A. Getting a grip on adhesion: Cadherin switching and collagen signaling. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118472. [Google Scholar] [CrossRef]
- Du, W.; Huang, H.; Sorrelle, N.; Brekken, R.A. Sitravatinib potentiates immune checkpoint blockade in refractory cancer models. JCI Insight 2018, 3, e124184. [Google Scholar] [CrossRef]
- Zhu, D.; Huang, H.; Pinkas, D.M.; Luo, J.; Ganguly, D.; Fox, A.E.; Arner, E.; Xiang, Q.; Tu, Z.C.; Bullock, A.N.; et al. 2-Amino-2,3-dihydro-1H-indene-5-carboxamide-Based Discoidin Domain Receptor 1 (DDR1) Inhibitors: Design, Synthesis, and in Vivo Antipancreatic Cancer Efficacy. J. Med. Chem. 2019, 62, 7431–7444. [Google Scholar] [CrossRef]
- Jing, Z.F.; Bi, J.B.; Li, Z.L.; Liu, X.K.; Li, J.; Zhu, Y.Y.; Zhang, X.T.; Zhang, Z.; Li, Z.H.; Kong, C.Z. miR-19 promotes the proliferation of clear cell renal cell carcinoma by targeting the FRK-PTEN axis. OncoTargets Ther. 2019, 12, 2713–2727. [Google Scholar] [CrossRef]
- Wang, Z.; Ying, C.; Zhang, A.; Xu, H.; Jiang, Y.; Lou, M. HCK promotes glioblastoma progression by TGFbeta signaling. Biosci. Rep. 2020, 40, BSR20200975. [Google Scholar] [CrossRef] [PubMed]
- Poh, A.R.; Dwyer, A.R.; Eissmann, M.F.; Chand, A.L.; Baloyan, D.; Boon, L.; Murrey, M.W.; Whitehead, L.; O’Brien, M.; Lowell, C.A.; et al. Inhibition of the SRC Kinase HCK Impairs STAT3-Dependent Gastric Tumor Growth in Mice. Cancer Immunol. Res. 2020, 8, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhou, Z.; Wei, Z.; Wu, J.; Ouyang, J.; Huang, W.; He, Y.; Zhang, C. FYN promotes gastric cancer metastasis by activating STAT3-mediated epithelial-mesenchymal transition. Transl. Oncol. 2020, 13, 100841. [Google Scholar] [CrossRef] [PubMed]
- Okada, J.; Yamada, E.; Saito, T.; Yokoo, H.; Osaki, A.; Shimoda, Y.; Ozawa, A.; Nakajima, Y.; Pessin, J.E.; Okada, S.; et al. Dapagliflozin Inhibits Cell Adhesion to Collagen I and IV and Increases Ectodomain Proteolytic Cleavage of DDR1 by Increasing ADAM10 Activity. Molecules 2020, 25, 495. [Google Scholar] [CrossRef]
- Serda, M.; Malarz, K.; Mrozek-Wilczkiewicz, A.; Wojtyniak, M.; Musiol, R.; Curley, S.A. Glycofullerenes as non-receptor tyrosine kinase inhibitors—towards better nanotherapeutics for pancreatic cancer treatment. Sci. Rep. 2020, 10, 260. [Google Scholar] [CrossRef]
- Sun, X.; Gao, H.; Yang, Y.; He, M.; Wu, Y.; Song, Y.; Tong, Y.; Rao, Y. PROTACs: Great opportunities for academia and industry. Signal Transduct. Target. Ther. 2019, 4, 64. [Google Scholar] [CrossRef]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. [Google Scholar] [CrossRef]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef]
- Chatterjee, N.; Bivona, T.G. Polytherapy and Targeted Cancer Drug Resistance. Trends Cancer 2019, 5, 170–182. [Google Scholar] [CrossRef]
- Konieczkowski, D.J.; Johannessen, C.M.; Garraway, L.A. A Convergence-Based Framework for Cancer Drug Resistance. Cancer Cell 2018, 33, 801–815. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
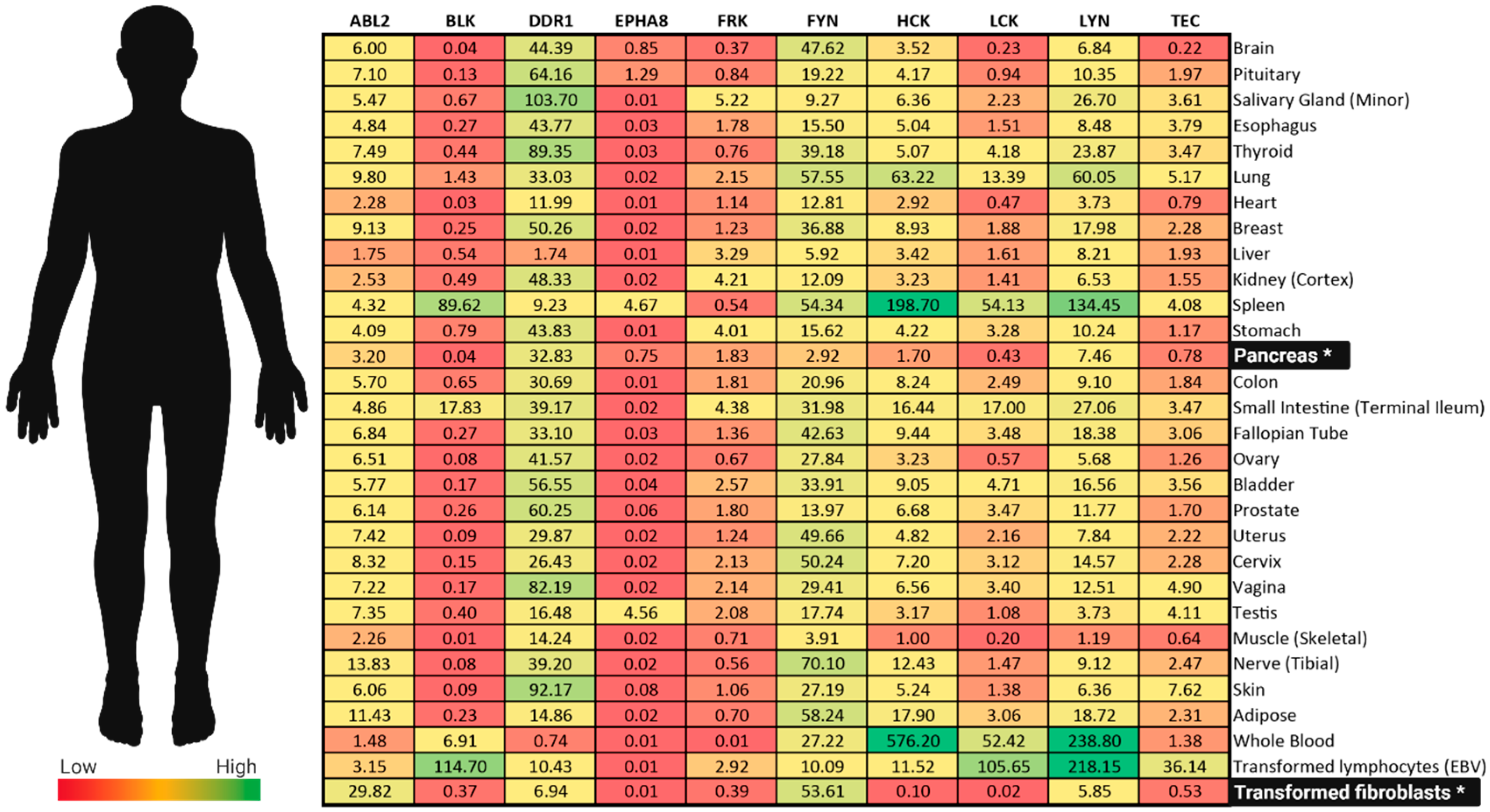
| Kinase | Study Samples | Implication | Citation |
|---|---|---|---|
| FRK | Pancreatic cancer cell lines: PANC-1 (pancreatic epithelioid carcinoma) MIA PaCa-2 (undifferentiated pancreatic carcinoma) Capan-1 (metastatic pancreatic adenocarcinoma obtained from liver) Capan-2 (pancreatic adenocarcinoma) HPAC (pancreatic adenocarcinoma). | FRK directly contributes to pancreatic cancer cell proliferation and migration in PANC-1, MIA Paca-2, Capan-1, and HPAC | [22] |
| HCK | Pancreatic ductal adenocarcinomas (PDACs) from 43 patients Well differentiated (n = 12) Moderately differentiated (n = 24) Poorly differentiated (n = 4) Lymph node metastasis Positive (n = 36) Negative (n = 6) | Gain of the HCK locus in PDAC patient biopsy predicts decreased patient survival | [78] |
| ABL2 | 22 adenocarcinoma samples 16 pancreatic cancer cell lines | In PDAC, ABL2 expression is often upregulated | [87] |
| DDR1 | 205 PDAC patient samples. T classification: T1 (n = 11); T2 (n = 31); T3 (n = 125); T4 (n = 38) n Classification: Absent (n = 136); Present (n =69) AJCC Stage: Stage I (n = 38); Stage II (n = 135); Stage III (n = 21); Stage IV (n = 14) Liver metastasis: Absent (n = 191); Present (n = 14) | DDR1 expression is associated with poor prognosis in PDAC | [115] |
| DDR1 | Pancreatic cancer cell lines: PANC-1 AsPC-1 (metastatic pancreatic adenocarcinoma obtained from ascites) | Upregulated DDR1 is associated with increased metastatic potential | [117] |
| DDR1 | Pancreatic cancer cell lines: AsPC-1 PANC-1 BxPC-3 (pancreatic adenocarcinoma) xenograft models | Activation of DDR1 contributes to tumorgenicity in PDAC. DDR1 inhibition reduces collagen-mediated tumorigenicity in PDAC | [118] |
| LYN | Pancreatic cancer cell lines: PANC-1 MIA PaCa-2 Capan-1 Capan-2 HPAC | LYN directly contributes to pancreatic cancer cell proliferation and migration in PANC-1, MIA Paca-2, Capan-1, Capan-2, and HPAC | [22] |
| FYN | 12 PDAC patient samples Stage IIA (n = 3) Stage IIB (n = 9) Lymph node metastasis: Yes (n = 9); No (n = 3) | Activator phosphorylation sites on FYN demonstrate a two-fold increase in tumor tissue compared to wild type pancreatic patient tissue | [155] |
| FYN | Pancreatic cancer cell lines: PANC-1 MIA PaCa-2 Capan-1 Capan-2 HPAC | FYN directly contributes to pancreatic cancer cell proliferation and migration in PANC-1, MIA Paca-2, Capan-1, Capan-2, and HPAC | [22] |
| FYN | 28 pancreatic cancer patient samples Staging: metastatic (n = 11); nonmetastatic (n = 17) TNM staging: T1 (n = 4); T2 (n = 9); T3 (n = 5); T4 (n = 10) Additional experiments were performed using BxPC3 cell lines and a nude mouse xenograft model | FYN detected in 24 (of 28) tumors Metastatic pancreatic cancer demonstrates increased FYN expression TNM staging did not correlate with FYN expression FYN inhibition reduces primary tumor weight and volume, as well as metastasis FYN inhibition decreases cell proliferation and increases apoptosis | [156] |
| FYN | Pancreatic cancer cell lines: BxPc3 AsPc1 PaCa2 28 pancreatic cancer patient samples TNM Staging: T1 (n = 4); T2 (n = 9); T3 (n = 5); T4 (n = 10) Staging: Nonmetastatic (n = 17); Metastatic (n = 11) Differentiation: High (n = 3); Middle (n = 16); Low (n = 9) | FYN activity is increased in metastatic pancreatic cancer tissue Mechanistic studies exploring the signaling axis of FYN in pancreatic cancer suggest significant coordination and regulation of apoptosis which promotes pancreatic cancer proliferation and metastasis | [157] |
| FYN | Pancreatic cell lines: HPDE6-C7 (immortalized human pancreatic duct epithelial cells) QGP1 (human pancreatic islet cell carcinoma) PANC-1 BxPC-3 SW1990 (metastatic pancreatic adenocarcinoma obtained from spleen) 30 pancreatic cancer patient samples TNM Staging: T1 (n = 4); T2 (n = 10); T3 (n = 6); T4 (n = 10) Staging: Nonmetastasis (n = 17); Metastasis (n = 13) Differentiation: High (n = 3); Middle (n = 15); Low (n = 12) | FYN mRNA expression is higher in tumor tissue compared to adjacent normal tissue FYN expression correlates with metastasis and staging | [158] |
| Kinase | Context | Implication | Citation |
|---|---|---|---|
| FRK | Breast cancer; brain cancer | Tumor suppressive functionality | [15,16,17,18,19] |
| FRK | Lung cancer; liver cancer | Oncogenic functionality | [20,21,23] |
| FRK | Transgenic mice | Increased expression increases beta cell mass, islet cell death, and beta cell proliferation after partial pancreatectomy | [24] |
| FRK | Cytokine treatment or antineoplastic therapy | Increased expression enhances beta cell death | [24,27] |
| FRK | Molecular | Direct binding to Rb proteins inside cellular nuclei alters Rb tumor suppressor activity | [28] |
| FRK | Molecular | Direct binding and phosphorylation of PTEN protects PTEN from degradation and maintains tumor suppression activity | [16] |
| FRK | Molecular | Direct binding of EGFR slows EGFR recycling and attenuates oncogenic effects | [29] |
| FRK | Molecular | Phosphorylation of EGFR downregulates tumorigenic EGFR pathways | [29,31,32,33,34] |
| BLK | Immune cells | Preferentially expressed in normal B cells and ectopically expressed in T cell malignancies | [39,40] |
| BLK | Lymphoma | Constitutive expression increases tumor development and malignant transformation | [41,42] |
| BLK | Melanoma cells | Amplifies drug resistance mechanisms in melanoma cells | [44] |
| BLK | Chronic myeloid leukemia | Tumor suppressor | [50,51] |
| BLK | Beta cells | Kinase activity enhances the synthesis and secretion of insulin by upregulating the transcription factor PDX1 | [52] |
| HCK | Leukemias; solid malignancies | Positive correlation between activity and cancer cell proliferation and survival | [75] |
| HCK | Colon cancer | Promotes tumor progression | [77] |
| HCK | Immunotoxin therapy | Anticancer effects of immunotoxin are augmented by HCK inhibition | [80] |
| HCK | Renal fibrosis | Overexpression activates fibrotic pathways; knockdown inhibits fibrotic pathways | [81] |
| HCK | Atherosclerosis; lung fibrosis | Implicated in inflammatory pathways | [82,83] |
| ABL2 | Lung cancer; hepatocellular carcinoma; glioma; gastric cancer | Oncogenic properties | [88,89,90,91,92] |
| ABL2 | Prostate cancer; breast cancer; other cancer models | Tumor suppressive properties | [93,94,95,96,97] |
| ABL2 | Fibroblast cells | Regulates proliferation and adhesion-dependent cell edge protrusions | [109,110,111] |
| DDR1 | Skin; kidney; lungs | Mediates fibrotic processes in the skin; plays a protective role in the kidney and lung Expression profiles are similar in the skin and kidney, but different in the kidney and lung | [116] |
| DDR1 | Fibrotic disease models | Inhibition reduces inflammation and fibrosis | [120,121,122,123] |
| LYN | Cervical cancer; prostate cancer; colon cancer; Ewing’s sarcoma | Upregulated | [135,136,137,138] |
| LYN | Renal cancer; head and neck squamous cell carcinoma; nonsmall cell lung cancer; breast cancer | Predicts poor prognosis | [139,140,141,142] |
| LYN | Glioblastoma | Kinase activity is elevated | [143] |
| EPHA8 | Neuroblastoma | Kinase-independent activation of MAPK to promote axonal projections | [178] |
| EPHA8 | Glioma | EPHA8-mediated inhibition of cell migration requires EPHA8 kinase activity | [179] |
| EPHA8 | Gastric cancer | Proliferation, migration, and invasion of gastric cancer cells are associated with EPHA8 kinase-mediated signaling involving ADAM10 and downstream AKT pathways | [180] |
| EPHA8 | Oral tongue squamous cell carcinoma; colorectal cancer; ovarian cancer | Expression associates with increased clinicopathological features or poor prognoses | [181,182,183] |
| FYN | Organ fibrosis | Regulates downstream serine-threonine kinase activity that modulates fibroblast–epithelial cell interactions and promotes organ fibrosis | [185,186] |
| FYN | Various experimental contexts | FYN signaling pathways regulate cell adhesion, drive epithelial-to-mesenchymal transition (EMT), and play a role in migration, cancer cell growth and motility; cancer progression; as well as antiapoptotic activity. | [187,188,189,190,191,192,193,194] |
| FYN | Hepatocellular carcinoma; oral cancer; mesothelioma; breast cancer; chronic myelogenous leukemia; prostate cancer; melanoma; brain cancer; esophageal squamous cell carcinoma | Varying degrees of evidence implicate FYN in the pathogenesis of these cancers | [190,191,192,193,194,195,196,197,198,199,200,201] |
| FYN | Colorectal cancer | Mechanistic studies suggest inhibition of FYN leads to greater cell death in KRAS mutant cells than in KRAS wild-type cells | [202] |
| LCK | T cells | Enzymatic activity is critical to TCR-induced downstream activation of T cells | [211] |
| LCK | Leukemia and immunotherapies | Implicated in several leukemias and immunotherapies | [212] |
| LCK | Breast cancer | Expressed in human breast cancer specimens | [213] |
| LCK | Lung cancer | Overexpressed and activated in lung cancer cell lines | [214] |
| LCK | Bile duct cancer | Upregulated in bile duct cancer cells and associates with early tumor recurrence | [215] |
| LCK | Glioma | Inhibition in human glioma cells decreases malignant progression | [216] |
| LCK | Colorectal cancer | Expression appears to be a positive prognostic marker; demonstrates potential as early diagnosis biomarker | [217,218] |
| LCK | Melanoma | Highly expressed in subsets of melanoma patients and associates with significantly improved survival | [219] |
| LCK | Pancreatic endocrine tumors | Overexpression | [222] |
| TEC | Liver cancer | Overexpression | [227] |
| TEC | Pancreatitis | Implicated in the inflammatory response associated with severe pancreatitis | [241] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Creeden, J.F.; Alganem, K.; Imami, A.S.; Henkel, N.D.; Brunicardi, F.C.; Liu, S.-H.; Shukla, R.; Tomar, T.; Naji, F.; McCullumsmith, R.E. Emerging Kinase Therapeutic Targets in Pancreatic Ductal Adenocarcinoma and Pancreatic Cancer Desmoplasia. Int. J. Mol. Sci. 2020, 21, 8823. https://doi.org/10.3390/ijms21228823
Creeden JF, Alganem K, Imami AS, Henkel ND, Brunicardi FC, Liu S-H, Shukla R, Tomar T, Naji F, McCullumsmith RE. Emerging Kinase Therapeutic Targets in Pancreatic Ductal Adenocarcinoma and Pancreatic Cancer Desmoplasia. International Journal of Molecular Sciences. 2020; 21(22):8823. https://doi.org/10.3390/ijms21228823
Chicago/Turabian StyleCreeden, Justin F., Khaled Alganem, Ali S. Imami, Nicholas D. Henkel, F. Charles Brunicardi, Shi-He Liu, Rammohan Shukla, Tushar Tomar, Faris Naji, and Robert E. McCullumsmith. 2020. "Emerging Kinase Therapeutic Targets in Pancreatic Ductal Adenocarcinoma and Pancreatic Cancer Desmoplasia" International Journal of Molecular Sciences 21, no. 22: 8823. https://doi.org/10.3390/ijms21228823
APA StyleCreeden, J. F., Alganem, K., Imami, A. S., Henkel, N. D., Brunicardi, F. C., Liu, S.-H., Shukla, R., Tomar, T., Naji, F., & McCullumsmith, R. E. (2020). Emerging Kinase Therapeutic Targets in Pancreatic Ductal Adenocarcinoma and Pancreatic Cancer Desmoplasia. International Journal of Molecular Sciences, 21(22), 8823. https://doi.org/10.3390/ijms21228823