Rational Computational Design of Fourth-Generation EGFR Inhibitors to Combat Drug-Resistant Non-Small Cell Lung Cancer


Abstract
1. Introduction
2. Results and Discussion
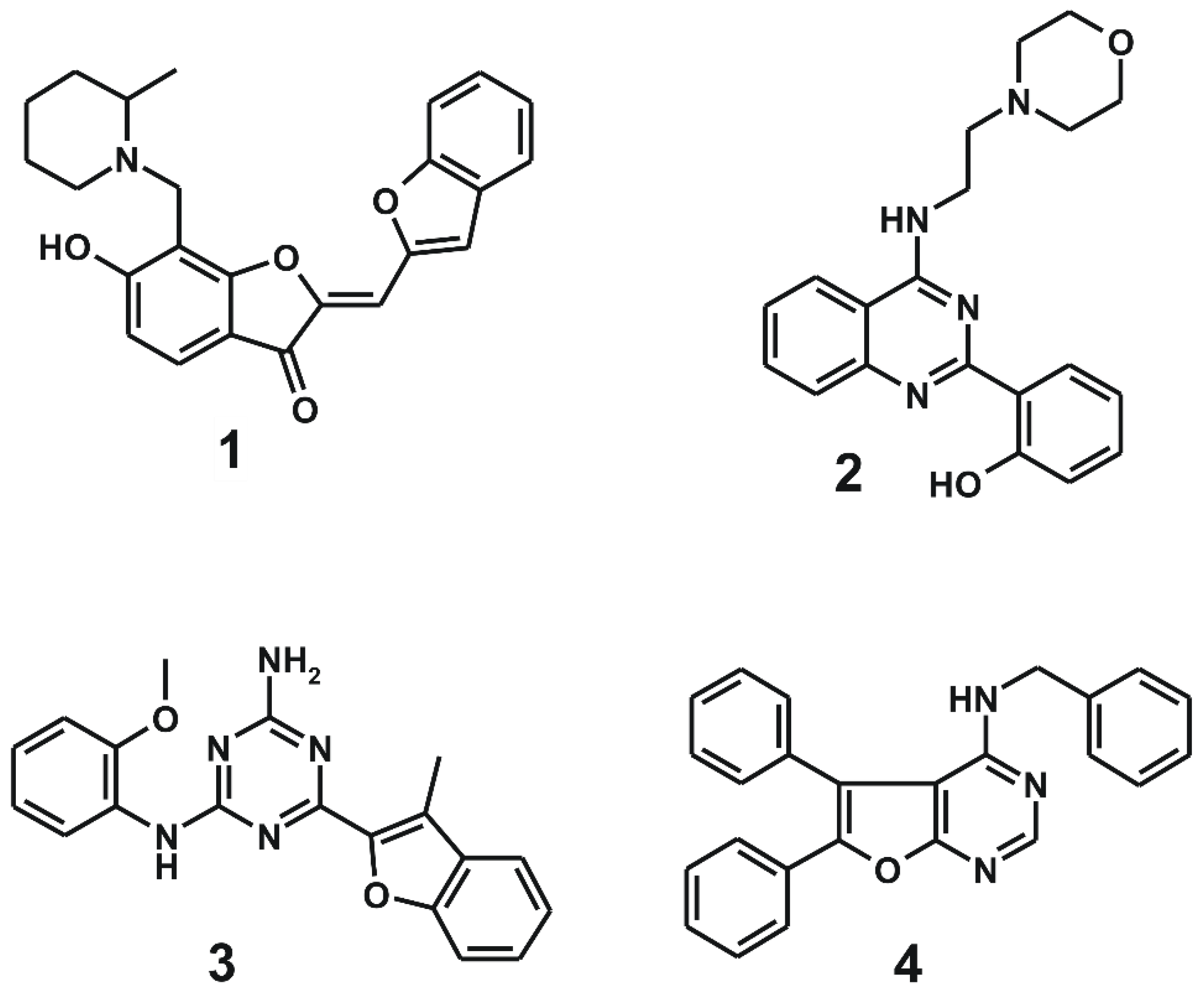
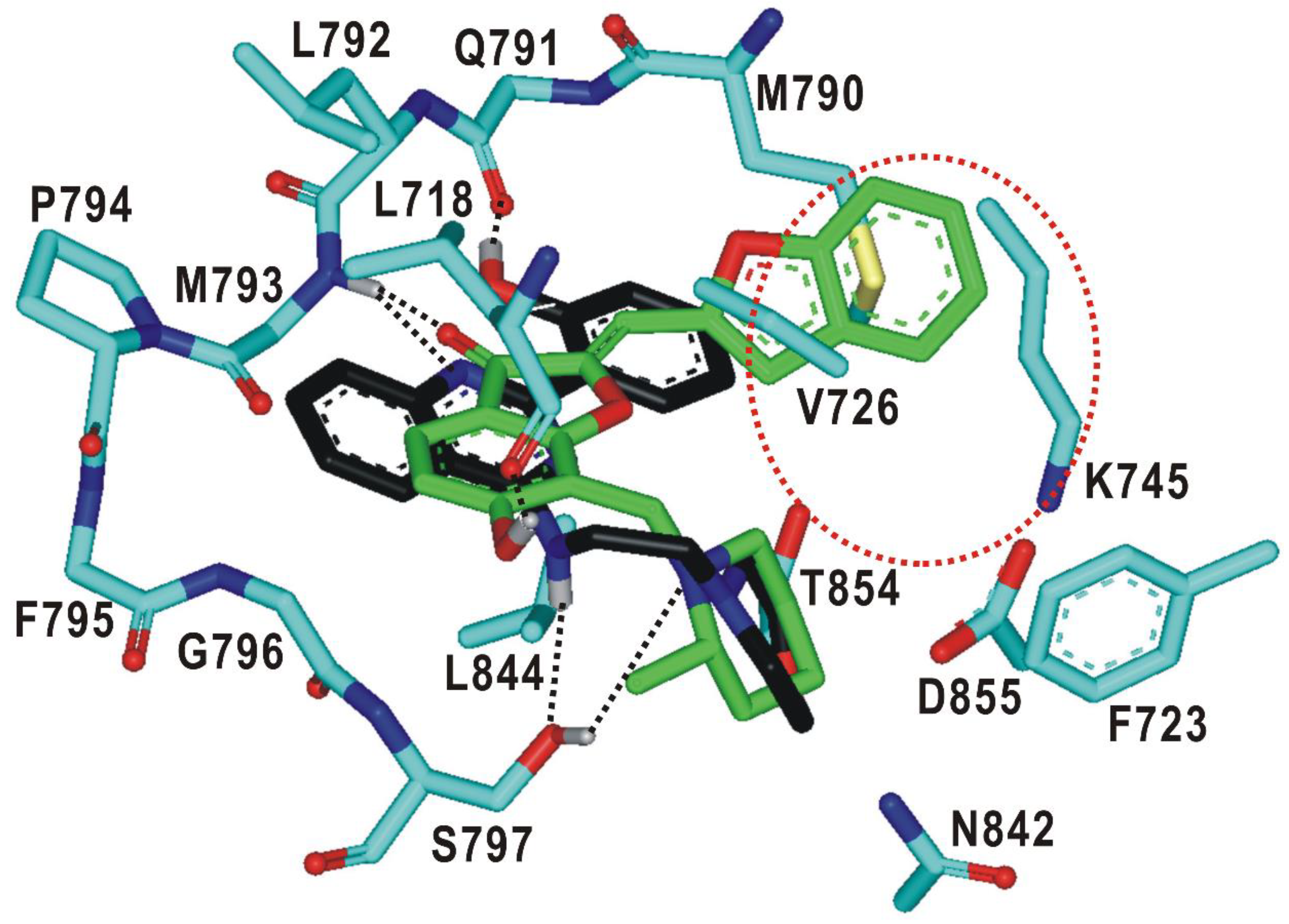
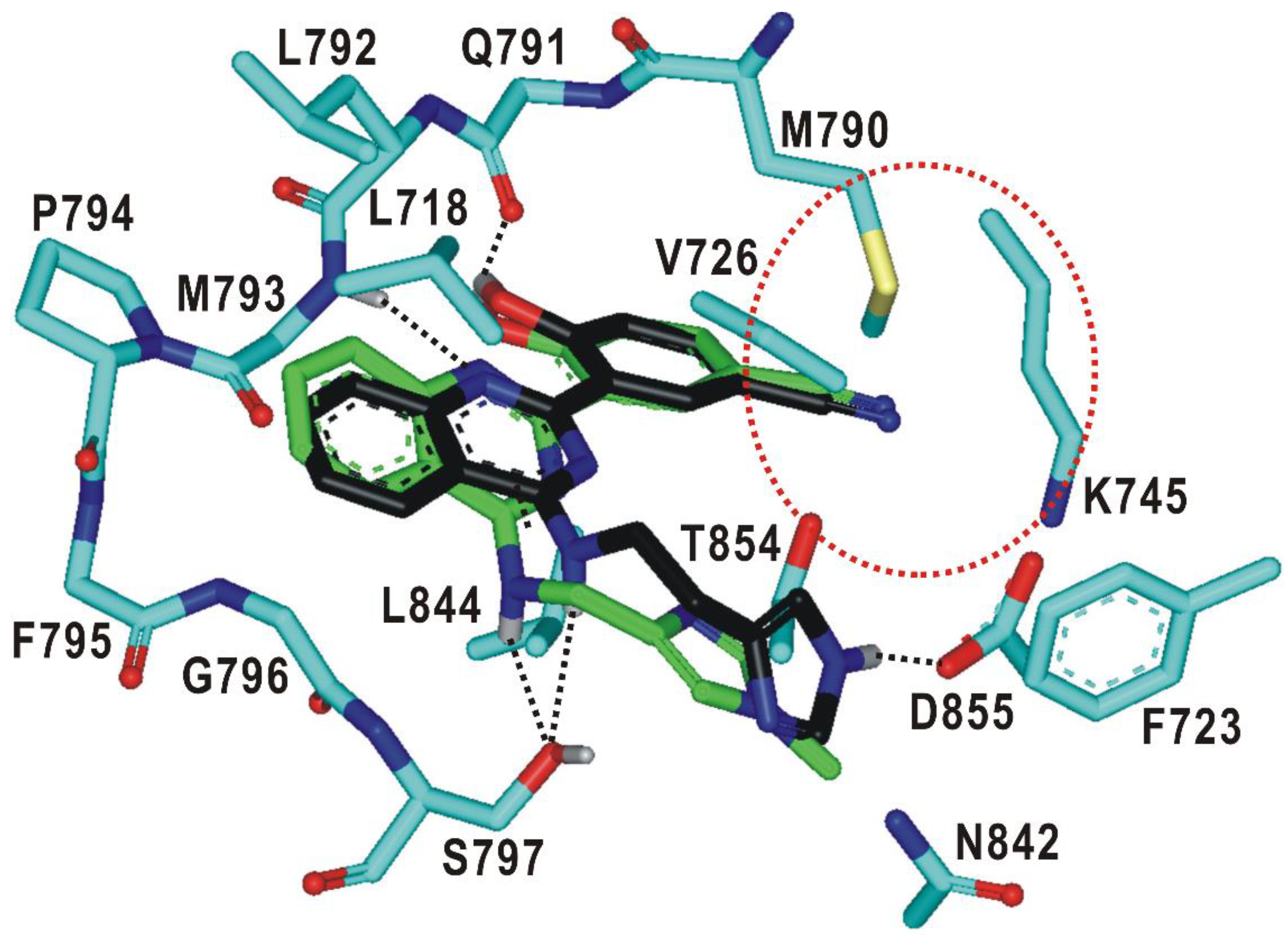
2.1. Structure-Based Virtual Screening of the Fourth-Generation EGFR Inhibitors
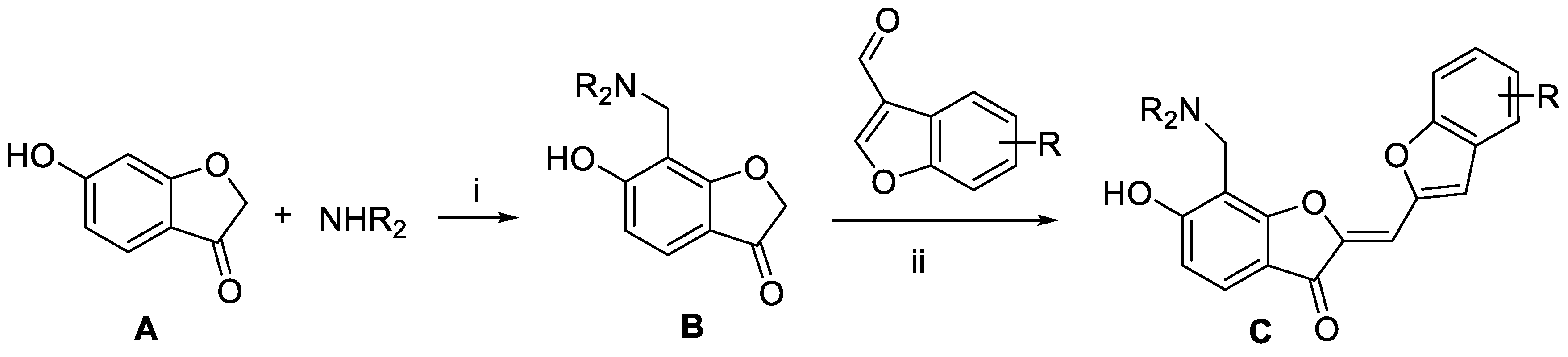
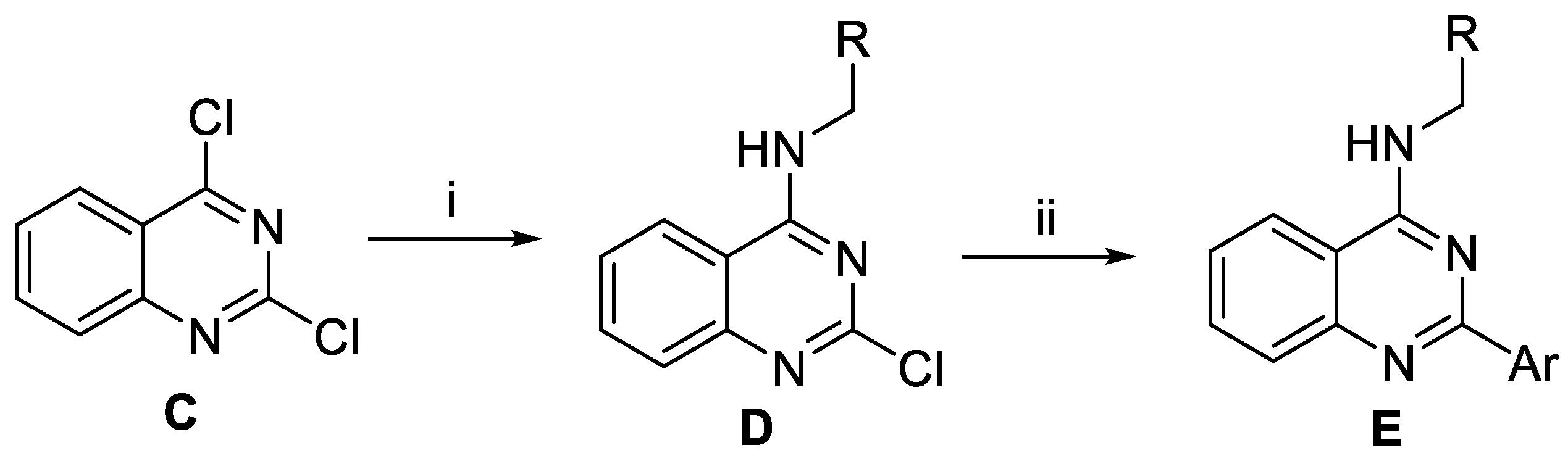
2.2. Synthesis of the Derivatives of 1 and 2 Generated from de novo Design
2.3. Biochemical Potencies of the Newly Synthesized Compounds
3. Materials and Methods
3.1. Structural Preparations of d746-750/T790M/C797S Mutant and Wild Type of EGFR
3.2. Two-Track Virtual Screening to Identify the Fourth-Generation EGFR Inhibitors
3.3. De novo Design
3.4. Chemical Synthesis
3.4.1. General Methods
3.4.2. Synthesis of Compound C
3.4.3. Representative Procedure for Modification of Coumaranone (Step 1)
3.4.4. Representative Procedure for Preparing Aurone Derivatives (Step 2)
3.4.5. Synthesis of Compound E
3.5. Enzyme Inhibition Assays
3.6. Cell Proliferation Inhibition Assay
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| EGFR | Epidermal growth factor receptor |
| NSCLC | non-small cell lung cancer |
| d746-750 | deletion of Glu746-Ala750 |
| PAINS | pan assay interference compounds |
| Gly loop | glycine-rich loop |
| DGF | Asp855-Phe856-Gly857 |
| PDB | protein data bank |
References
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non–small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.M.; Jeong, Y.; Hong, S. Strategies to overcome acquired resistances conferred by mutations in the kinase domain of EGFR. Future Med. Chem. 2016, 8, 853–878. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Heymach, J.V.; Lippman, S.M. Lung cancer. N. Engl. J. Med. 2008, 359, 1367–1380. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Bell, D.W.; Settleman, J.; Haber, D.A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. 2007, 7, 169–181. [Google Scholar] [CrossRef]
- Kawahara, A.; Yamamoto, C.; Nakashima, K.; Azuma, K.; Hattori, S.; Kashihara, M.; Aizawa, H.; Basaki, Y.; Kuwano, M.; Kage, M.; et al. Molecular diagnosis of activating EGFR mutations in non–small cell lung cancer using mutation-specific antibodies for immunohistochemical analysis. Clin. Cancer Res. 2010, 16, 3163–3170. [Google Scholar] [CrossRef]
- Rosell, R.; Moran, T.; Queralt, C.; Porta, R.; Cardenal, F.; Camps, C.; Majem, M.; Lopez-Vivanco, G.; Isla, D.; Provencio, M.; et al. Screening for epidermal growth factor receptor mutations in lung cancer. N. Engl. J. Med. 2009, 361, 958–967. [Google Scholar] [CrossRef]
- Wang, S.; Wang, Z. EGFR mutations in patients with non-small cell lung cancer from mainland China and their relationships with clinicopathological features: A meta-analysis. Int. J. Clin. Exp. Med. 2014, 7, 1967–1978. [Google Scholar]
- Shepherd, F.A.; Pereira, J.R.; Ciuleanu, T.; Tan, E.H.; Hirsh, V.; Thongprasert, S.; Campos, D.; Maoleekoonpiroj, S.; Smylie, M.; Martins, R.; et al. Erlotinib in previously treated non–small-cell lung cancer. N. Engl. J. Med. 2005, 353, 123–132. [Google Scholar] [CrossRef]
- Park, J.H.; Liu, Y.; Lemmon, M.A. Radhakrishnan, Erlotinib binds both inactive and active conformations of the EGFR tyrosine kinase domain. Biochem. J. 2012, 448, 417–423. [Google Scholar] [CrossRef]
- Barker, A.J.; Gibson, K.H.; Grundy, W.; Godfrey, A.A.; Barlow, J.J.; Healy, M.P.; Woodburn, J.R.; Ashton, S.E.; Curry, B.J.; Scarlett, L.; et al. Studies leading to the identification of ZD1839 (IRESSA): An orally active, selective epidermal growth factor receptor tyrosine kinase inhibitor targeted to the treatment of cancer. Bioorg. Med. Chem. Lett. 2001, 11, 1911–1914. [Google Scholar] [CrossRef]
- Paez, J.G.; Janne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef]
- Morgillo, F.; Della Corte, C.M.; Fasano, M.; Ciardiello, F. Mechanisms of resistance to EGFR-targeted drugs: Lung cancer. ESMO Open 2016, 1, e000060. [Google Scholar] [CrossRef]
- Eck, M.J.; Yun, C.-H. Structural and mechanistic underpinnings of the differential drug sensitivity of EGFR mutations in non-small cell lung cancer. Biochim. Biophys. Acta 2010, 1804, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef] [PubMed]
- Stewart, E.L.; Tan, S.Z.; Liu, G.; Tsao, M.-S. Known and putative mechanisms of resistance to EGFR targeted therapies in NSCLC patients with EGFR mutations–a review. Transl. Lung Cancer Res. 2015, 4, 67–81. [Google Scholar] [PubMed]
- Godin-Heymann, N.; Ulkus, L.; Brannigan, B.W.; McDermott, U.; Lamb, J.; Maheswaran, S.; Settleman, J.; Haber, D.A. The T790M “gatekeeper” mutation in EGFR mediates resistance to low concentrations of an irreversible EGFR inhibitor. Mol. Cancer Ther. 2008, 7, 874–879. [Google Scholar] [CrossRef]
- Wang, S.; Cang, S.; Liu, D. Third-generation inhibitors targeting EGFR T790M mutation in advanced non-small cell lung cancer. J. Hematol. Oncol. 2016, 9, 34. [Google Scholar] [CrossRef]
- Walter, A.O.; Sjin, R.T.T.; Haringsma, H.J.; Ohashi, K.; Sun, J.; Lee, K.; Dubrovskiy, A.; Labenski, M.; Zhu, Z.; Wang, Z.; et al. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov. 2013, 1404–1415. [Google Scholar] [CrossRef]
- Zhou, W.; Ercan, D.; Chen, L.; Yun, C.-H.; Li, D.; Capelletti, M.; Cortot, A.B.; Chirieac, L.; Iacob, R.E.; Padera, R.; et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 2009, 462, 1070–1074. [Google Scholar] [CrossRef]
- Wang, S.; Tsui, S.T.; Liu, C.; Song, Y.; Liu, D. EGFR C797S mutation mediates resistance to third-generation inhibitors in T790M-positive non-small cell lung cancer. J. Hematol. Oncol. 2016, 9, 59. [Google Scholar] [CrossRef]
- Ayeni, D.; Politi, K.; Goldberg, S.B. Emerging agents and new mutations in EGFR-mutant lung cancer. Clin. Cancer Res. 2015, 21, 3818–3820. [Google Scholar] [CrossRef] [PubMed]
- Costa, D.B.; Kobayashi, S.S. Whacking a molecule: Clinical activity and mechanisms of resistance to third generation EGFR inhibitors in EGFR mutated lung cancers with EGFR-T790M. Transl. Lung Cancer Res. 2015, 4, 809–815. [Google Scholar] [PubMed]
- Ercan, D.; Choi, H.G.; Yun, C.-H.; Capelletti, M.; Xie, T.; Eck, M.J.; Gray, N.S.; Jänne, P.A. EGFR mutations and resistance to irreversible pyrimidine-based EGFR inhibitors. Clin. Cancer Res. 2015, 21, 3913–3923. [Google Scholar] [CrossRef]
- Jia, Y.; Yun, C.-H.; Park, E.; Ercan, D.; Manuia, M.; Juarez, J.; Xu, C.; Rhee, K.; Chen, T.; Zhang, H.; et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature 2016, 534, 129–132. [Google Scholar] [CrossRef]
- Engel, J.; Becker, C.; Lategahn, J.; Keul, M.; Ketzer, J.; Muhlenberg, T.; Kollipara, L.; Schultz-Fademrecht, C.; Zahedi, R.P.; Bauer, S.; et al. Insight into the inhibition of drug-resistant mutants of the receptor tyrosine kinase EGFR. Angew. Chem. Int. Ed. 2016, 55, 10909–10912. [Google Scholar] [CrossRef]
- Gunther, M.; Juchum, M.; Kelter, G.; Fiebig, H.; Laufer, S. Lung cancer: EGFR inhibitors with low nanomolar activity against a therapy-resistant L858R/T790M/C797S mutant. Angew. Chem. Int. Ed. 2016, 55, 10890–10894. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Jung, H.-Y.; Mah, S.; Hong, S. Discovery of EGF receptor inhibitors that are selective for the d746-750/T790M/C797S mutant. Angew. Chem. Int. Ed. 2017, 56, 7634–7638. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhang, T.; Zhu, S.J.; Sun, M.; Tong, L.; Lai, M.; Zhang, R.; Xu, W.; Wu, R.; Ding, J.; et al. Structure-Based design of 5-methylpyrimidopyridone derivatives as new wild-type sparing inhibitors of the epidermal growth factor receptor triple mutant (EGFRL858R/T790M/C797S). J. Med. Chem. 2019, 62, 7302–7308. [Google Scholar] [CrossRef] [PubMed]
- Janne, P.A.; Von Pawel, J.; Cohen, R.B.; Crino, L.; Butts, C.A.; Olson, S.S.; Eiseman, I.A.; Chiappori, A.A.; Yeap, B.Y.; Lenehan, P.F.; et al. Multicenter, randomized, phase II trial of CI-1033, an irreversible pan-ERBB inhibitor, for previously treated advanced non–small-cell lung cancer. J. Clin. Oncol. 2007, 25, 3936–3944. [Google Scholar] [CrossRef]
- Ayati, A.; Moghimi, S.; Salarinejad, S.; Safavi, M.; Pouramiri, B.; Foroumadi, A. A review on progression of epidermal growth factor receptor (EGFR) inhibitors as an efficient approach in cancer targeted therapy. Bioorg. Chem. 2020, 99, 103811. [Google Scholar] [CrossRef]
- Shoichet, B.K.; Leach, A.R.; Kuntz, I.D. Ligand solvation in molecular docking. Proteins 1999, 34, 4–16. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef]
- Yan, Y.; Wang, W.; Sun, Z.; Zhang, J.Z.H.; Ji, C. Protein-ligand empirical interaction components for virtual screening. J. Chem. Inf. Model. 2017, 57, 1793–1806. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Su, M.; Han, L.; Liu, J.; Yang, Q.; Li, Y.; Wang, R. Forging the basis for developing protein-ligand interaction scoring functions. Acc. Chem. Res. 2017, 50, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliver. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15–Ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef]
- Muley, L.; Baum, B.; Smolinski, M.; Freindorf, M.; Heine, A.; Klebe, G.; Hangauer, D.G. Enhancement of hydrophobic interactions and hydrogen bond strength by cooperativity: Synthesis, modeling, and molecular dynamics simulations of a congeneric series of thrombin Inhibitors. J. Med. Chem. 2010, 53, 2126–2135. [Google Scholar] [CrossRef]
- Peng, Y.-H.; Ueng, S.-H.; Tseng, C.-T.; Hung, M.-S.; Song, J.-S.; Wu, J.-S.; Liao, F.-Y.; Fan, Y.-S.; Wu, M.-H.; Hsiao, W.-C.; et al. Important hydrogen bond networks in indoleamine 2,3-dioxygenase 1 (IDO1) inhibitor design revealed by crystal structures of imidazoleisoindole derivatives with IDO1. J. Med. Chem. 2016, 59, 282–293. [Google Scholar] [CrossRef]
- Gajiwala, K.S.; Feng, J.; Ferre, R.; Ryan, K.; Brodsky, O.; Weinrich, S.; Kath, J.C.; Stewart, A. Insights into the aberrant activity of mutant EGFR kinase domain and drug recognition. Structure 2013, 21, 209–219. [Google Scholar] [CrossRef]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Gureasko, J.; Shen, K.; Cole, P.A.; Kuriyan, J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 2006, 125, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, J.; Rudolph, C.; Sadowski, J. Automatic generation of 3D-atomic coordinates for organic molecules. Tetrahedron Comput. Methodol. 1990, 3, 537–547. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Park, H.; Lee, J.; Lee, S. Critical assessment of the automated AutoDock as a new docking tool for virtual screening. Proteins 2006, 65, 549–554. [Google Scholar] [CrossRef]
- Park, H.; Shin, Y.; Choe, H.; Hong, S. Computational design and discovery of nanomolar inhibitors of IκB kinase β. J. Am. Chem. Soc. 2015, 137, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Mah, S.; Hong, S.; Park, H. Discovery of low micromolar dual inhibitors for wild type and L1196M mutant of anaplastic lymphoma kinase through structure-based virtual screening. J. Chem. Inf. Model. 2016, 56, 802–810. [Google Scholar] [CrossRef]
- Park, H.; Shin, Y.; Kim, J.; Hong, S. Application of fragment-based de novo design to the discovery of selective picomolar inhibitors of glycogen synthase kinase-3 beta. J. Med. Chem. 2016, 59, 9018–9034. [Google Scholar] [CrossRef]
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity a rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Mehler, E.L.; Solmajer, T. Electrostatic effects in proteins: Comparison of dielectric and charge models. Protein Eng. 1991, 4, 903–910. [Google Scholar] [CrossRef]
- Stouten, P.F.W.; Frömmel, C.; Nakamura, H.; Sander, C. An effective solvation term based on atomic occupancies for use in protein simulations. Mol. Simul. 1993, 10, 97–120. [Google Scholar] [CrossRef]
- Park, H. Extended solvent-contact model approach to SAMPL4 blind prediction challenge for hydration free energies. J. Comput. Aided Mol. Des. 2014, 28, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.-C.; Park, H. Accuracy enhancement in the estimation of molecular hydration free energies by implementing the intramolecular hydrogen bond effects. J. Cheminform. 2015, 7, 57. [Google Scholar] [CrossRef]
- Wang, R.; Gao, Y.; Lai, L. LigBuilder: A multi-purpose program for structure-based drug design. J. Mol. Model. 2000, 6, 498–516. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | IC50 (μM) | Selectivity Index (IC50WT/IC50mutant) | |
|---|---|---|---|
| Wild Type | d746-750/T790M/C797S | ||
| 1 | 72.7 ± 4.8 | 2.11 ± 0.56 | 34.5 |
| 2 | 37.1 ± 2.3 | 1.73 ± 0.38 | 21.4 |
| 3 | 35.5 ± 3.5 | 2.89 ± 0.91 | 12.3 |
| 4 | 0.00588 ± 0.00098 | 0.802 ± 0.055 | 0.00733 |

| X b | Y | EGFR | EGFRd746-750/T790M/C797S | Selectivity | |
|---|---|---|---|---|---|
| 1 |  | H | 72.7 ± 6.8 | 2.11 ± 0.31 | 34.5 |
| 5 |  | H | 68.7 ± 3.4 | 3.01 ± 0.73 | 22.8 |
| 6 |  | H | 65.7 ± 6.0 | 1.94 ± 0.35 | 33.9 |
| 7 |  | H | 78.7 ± 4.6 | 5.29 ± 1.80 | 14.8 |
| 8 |  | OCH3 | >100 | 20.1 ± 7.2 | >4.98 |
| 9 |  | H | 21.3 ± 6.3 | 1.10 ± 0.36 | 13.1 |
| 10 |  | OCH3 | 36.8 ± 3.9 | 1.63 ± 0.43 | 22.6 |
| 11 |  | OH | 1.04 ± 0.16 | 0.715 ± 0.242 | 1.45 |

| R1 b | R2 | EGFR | EGFRd746-750/T790M/C797S | Selectivity | |
|---|---|---|---|---|---|
| 2 |  | H | 37.1 ± 2.5 | 1.73 ± 0.43 | 21.4 |
| 12 |  | H | >50 | 0.516 ± 0.206 | >96.9 |
| 13 |  | CN | 8.45 ± 1.89 | 0.0265 ± 0.0084 | 319 |
| 14 |  | H | >50 | 1.12 ± 0.21 | >44.6 |
| 15 |  | CN | 1.04 ± 0.07 | 0.715 ± 0.092 | 1.45 |
| 16 |  | CN | >50 | 0.0721 ± 0024 | >693 |
| 17 |  | CN | >50 | 0.593 ± 0.076 | >84.3 |
| 18 |  | CN | >50 | 0.00747 ± 0.00039 | >6693 |
| 19 |  | CN | >50 | 0.00338 ± 0.00102 | >14793 |
| 20 |  | CN | >50 | 0.00484 ± 0.00063 | >10331 |
| 21 |  | CHO | 0.935 ± 0.173 | 0.00793 ± 0.00168 | 118 |
| Compound | IC50 (μM) | Selectivity | |
|---|---|---|---|
| Ba/F3 | Ba/F3d746-750/T790M/C797S | ||
| 18 | >5 | 0.78 ± 0.31 | >6.41 |
| 19 | 2.47 ± 0.89 | 0.74 ± 0.22 | 3.34 |
| Gefitinib | 3.72 ± 1.17 | 1.50 ± 0.36 | 2.48 |
| Brigatinib | 1.56 ± 0.45 | 0.067 ± 0.014 | 23.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, H.; Jung, H.-Y.; Kim, K.; Kim, M.; Hong, S. Rational Computational Design of Fourth-Generation EGFR Inhibitors to Combat Drug-Resistant Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2020, 21, 9323. https://doi.org/10.3390/ijms21239323
Park H, Jung H-Y, Kim K, Kim M, Hong S. Rational Computational Design of Fourth-Generation EGFR Inhibitors to Combat Drug-Resistant Non-Small Cell Lung Cancer. International Journal of Molecular Sciences. 2020; 21(23):9323. https://doi.org/10.3390/ijms21239323
Chicago/Turabian StylePark, Hwangseo, Hoi-Yun Jung, Kewon Kim, Myojeong Kim, and Sungwoo Hong. 2020. "Rational Computational Design of Fourth-Generation EGFR Inhibitors to Combat Drug-Resistant Non-Small Cell Lung Cancer" International Journal of Molecular Sciences 21, no. 23: 9323. https://doi.org/10.3390/ijms21239323
APA StylePark, H., Jung, H.-Y., Kim, K., Kim, M., & Hong, S. (2020). Rational Computational Design of Fourth-Generation EGFR Inhibitors to Combat Drug-Resistant Non-Small Cell Lung Cancer. International Journal of Molecular Sciences, 21(23), 9323. https://doi.org/10.3390/ijms21239323