Neutrophils as a Novel Target of Modified Low-Density Lipoproteins and an Accelerator of Cardiovascular Diseases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Induction of NETs
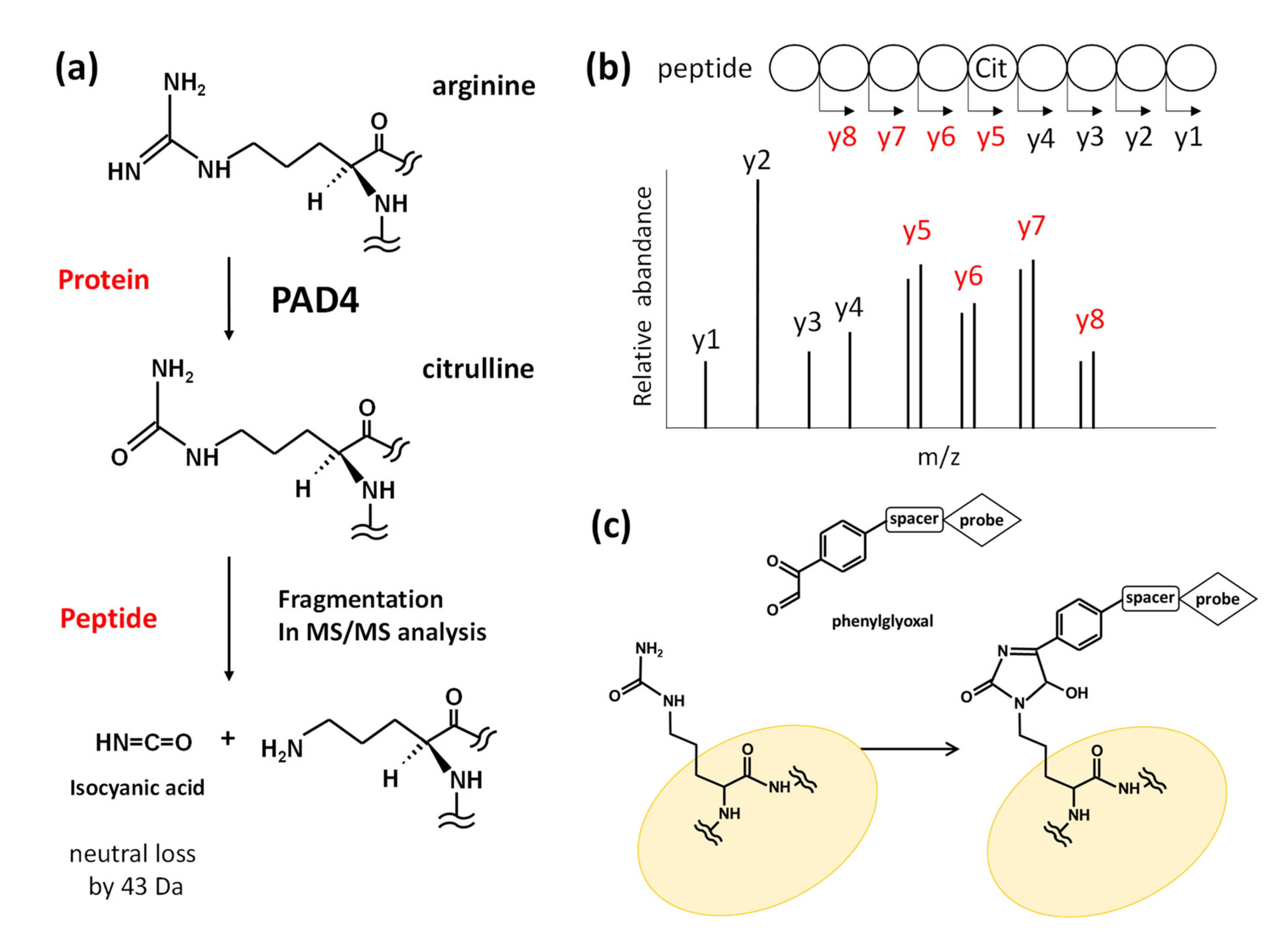
3. Detection and Analysis of Protein Citrullination
4. NET-Related Receptors
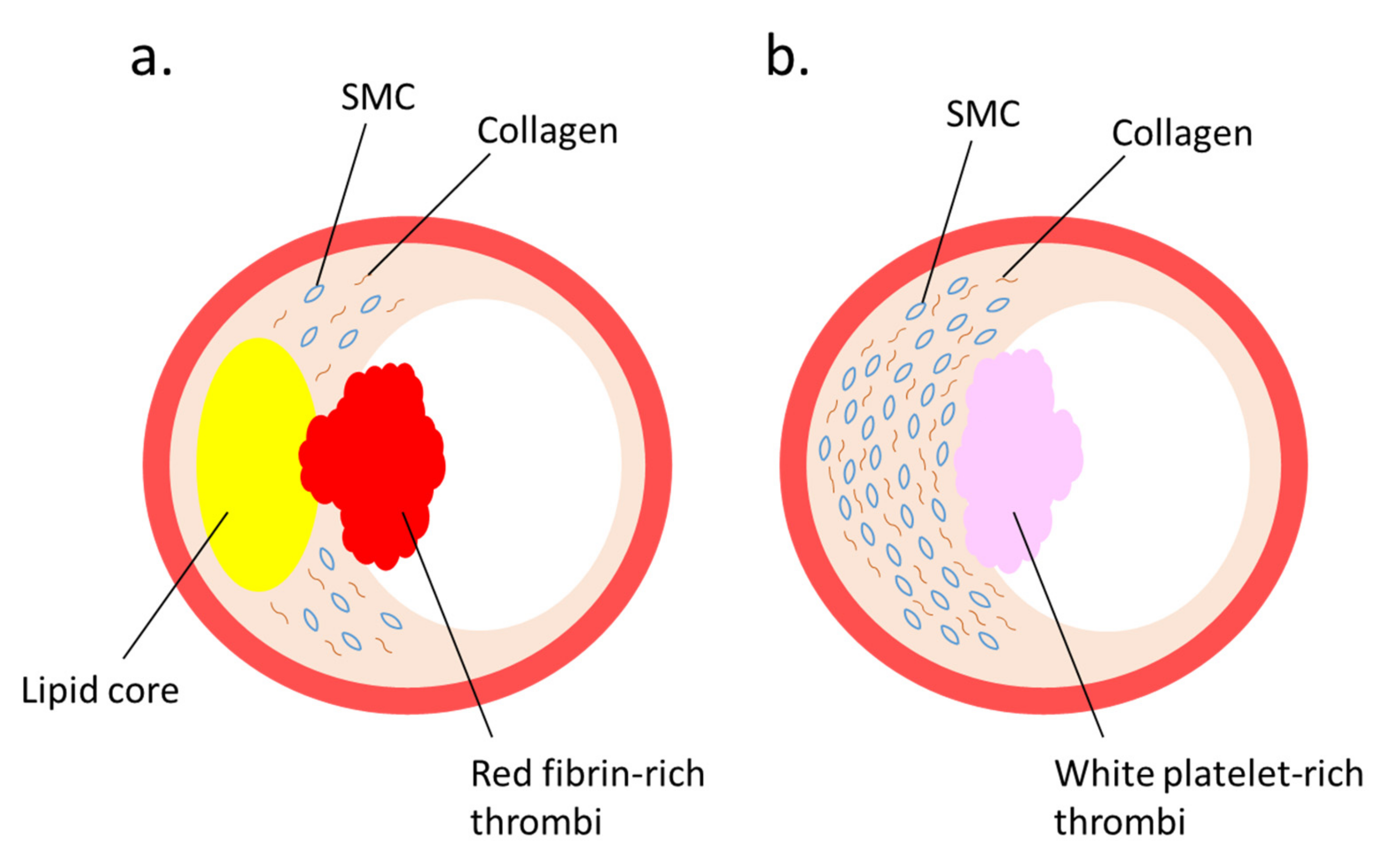
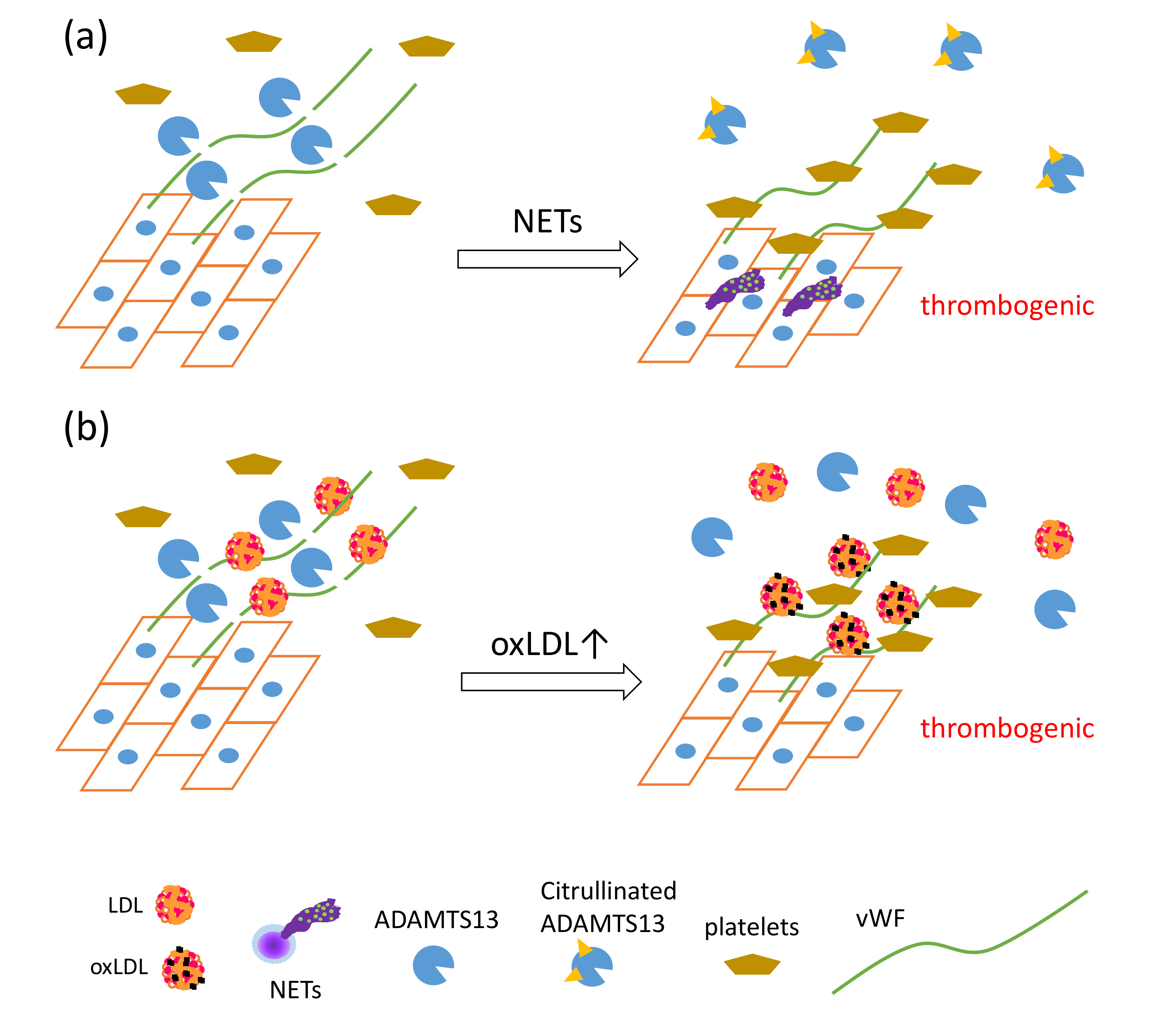
5. NETs and Cardiovascular Diseases
6. The Impact of Modified LDL and Its Components on Vascular Cells
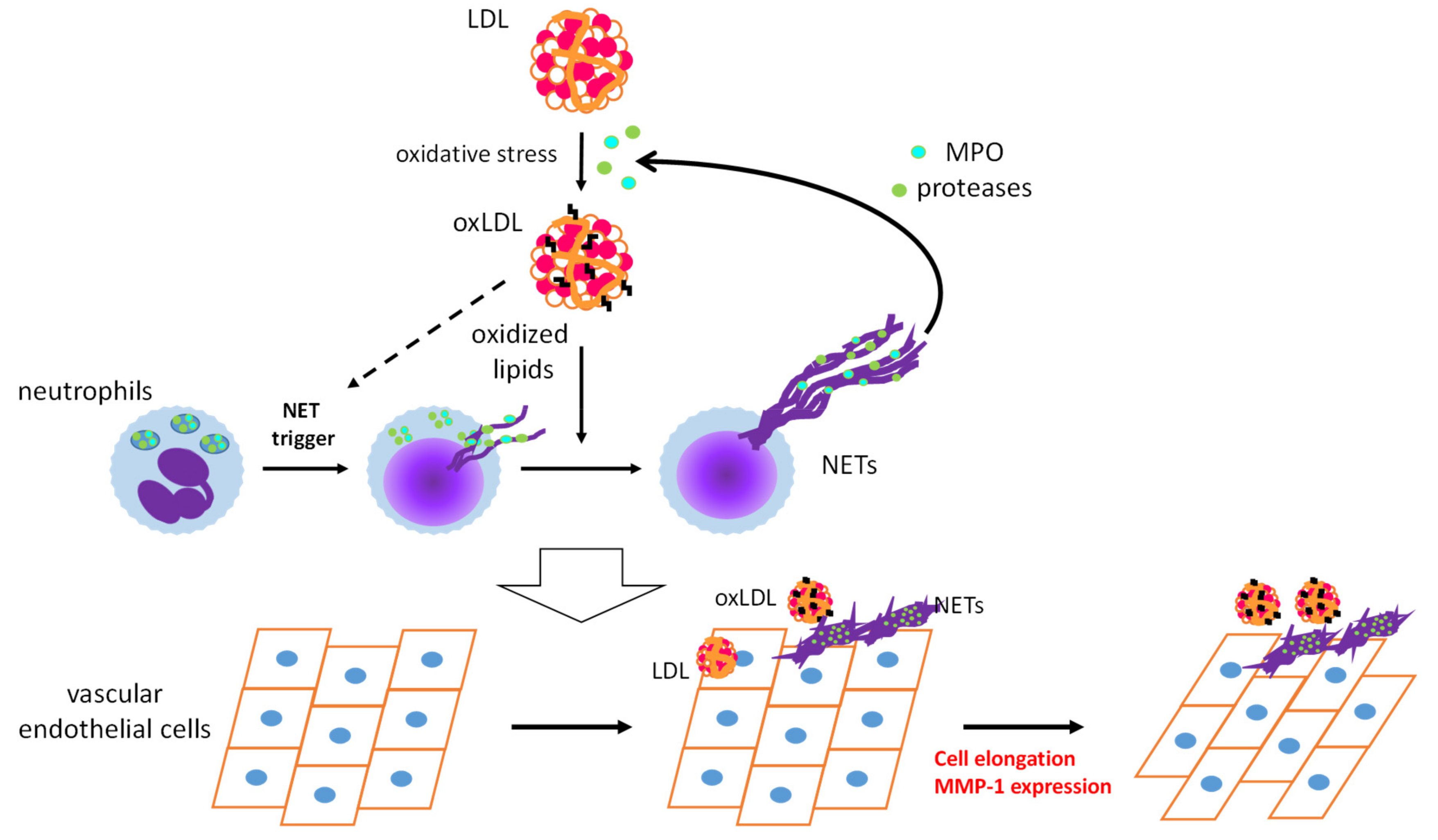
7. Modified LDL and NET Formation
8. Significance of OxLDL in Autoimmune Diseases
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NETs | neutrophil extracellular traps |
| DAMPs | damage-associated molecular patterns |
| oxLDL | oxidized low-density lipoprotein |
| CVD | cardiovascular disease |
| MPO | myeloperoxidase |
| ROS | reactive oxygen species |
| NE | neutrophil elastase |
| PMNs | polymorphonuclear leukocytes |
| PMA | phorbol myristate acetate |
| PAD4 | peptidylarginine deiminase 4 |
| MS | mass spectrometry |
| PG | phenylglyoxal |
| ADAMTS13 | a disintegrin-like and metalloproteinase with thrombospondin type 1 motif 13 |
| vWF | von Willebrand factor |
| ECs | endothelial cells |
| TLRs | toll-like receptors |
| RAGE | receptor for advanced glycation end products |
| HMGB1 | high-mobility group box 1 |
| SLE | systemic lupus erythematosus |
| SMCs | smooth muscle cells |
| HDL | high-density lipoprotein |
| EndMT | endothelial-to-mesenchymal transition |
| PLs | phospholipids |
| Lp-PLA2 | lipoprotein-associated phospholipase A2 |
| E-LDL | enzymatically-modified LDL |
| LDL(-) | electronegative LDL |
| MMP | matrix metalloproteinase |
| LOX-1 | lectin-like oxLDL receptor 1 |
| ICAM-1 | intercellular adhesion molecule-1 |
| TGs | triglycerides |
| AMI | acute myocardial infarction |
| oxPC | oxidized phosphatidylcholine |
| sdLDL | small dense LDL |
| S1P | sphingosine-1-phosphate |
| LDN | low-density neutrophil |
| GPI | glycoprotein I |
References
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Castanheira, F.V.S.; Kubes, P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood 2019, 133, 2178–2185. [Google Scholar] [CrossRef]
- Sollberger, G.; Tilley, D.O.; Zychlinsky, A. Neutrophil extracellular traps: The biology of chromatin externalization. Dev. Cell 2018, 44, 542–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doring, Y.; Libby, P.; Soehnlein, O. Neutrophil extracellular traps participate in cardiovascular diseases: Recent experimental and clinical insights. Circ. Res. 2020, 126, 1228–1241. [Google Scholar] [CrossRef]
- Itabe, H.; Obama, T.; Kato, R. The dynamics of oxidized LDL during atherogenesis. J. Lipids 2011, 2011, 418313. [Google Scholar] [CrossRef] [Green Version]
- Binder, C.J.; Papac-Milicevic, N.; Witztum, J.L. Innate sensing of oxidation-specific epitopes in health and disease. Nat. Rev. Immunol. 2016, 16, 485–497. [Google Scholar] [CrossRef]
- Polacek, D.; Byrne, R.E.; Fless, G.M.; Scanu, A.M. In vitro proteolysis of human plasma low density lipoproteins by an elastase released from human blood polymorphonuclear cells. J. Biol. Chem. 1986, 261, 2057–2063. [Google Scholar] [PubMed]
- Polacek, D.; Byrne, R.E.; Scanu, A.M. Modification of low density lipoproteins by polymorphonuclear cell elastase leads to enhanced uptake by human monocyte-derived macrophages via the low density lipoprotein receptor pathway. J. Lipid Res. 1988, 29, 797–808. [Google Scholar]
- Manda-Handzlik, A.; Bystrzycka, W.; Wachowska, M.; Sieczkowska, S.; Stelmaszczyk-Emmel, A.; Demkow, U.; Ciepiela, O. The influence of agents differentiating HL-60 cells toward granulocyte-like cells on their ability to release neutrophil extracellular traps. Immunol. Cell. Biol. 2018, 96, 413–425. [Google Scholar] [CrossRef]
- Hoppenbrouwers, T.; Autar, A.S.A.; Sultan, A.R.; Abraham, T.E.; van Cappellen, W.A.; Houtsmuller, A.B.; van Wamel, W.J.B.; van Beusekom, H.M.M.; van Neck, J.W.; de Maat, M.P.M. In vitro induction of NETosis: Comprehensive live imaging comparison and systematic review. PLoS ONE 2017, 12, e0176472. [Google Scholar] [CrossRef] [Green Version]
- Kahlenberg, J.M.; Carmona-Rivera, C.; Smith, C.K.; Kaplan, M.J. Neutrophil extracellular trap-associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J. Immunol. 2013, 190, 1217–1226. [Google Scholar] [CrossRef] [Green Version]
- Silvestre-Roig, C.; Braster, Q.; Wichapong, K.; Lee, E.Y.; Teulon, J.M.; Berrebeh, N.; Winter, J.; Adrover, J.M.; Santos, G.S.; Froese, A.; et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature 2019, 569, 236–240. [Google Scholar] [CrossRef]
- Yousefi, S.; Mihalache, C.; Kozlowski, E.; Schmid, I.; Simon, H.U. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009, 16, 1438–1444. [Google Scholar] [CrossRef] [PubMed]
- Bertin, F.R.; Rys, R.N.; Mathieu, C.; Laurance, S.; Lemarie, C.A.; Blostein, M.D. Natural killer cells induce neutrophil extracellular trap formation in venous thrombosis. J. Thromb. Haemost. 2019, 17, 403–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatfield, S.M.; Grebe, K.; Whitehead, L.W.; Rogers, K.L.; Nebl, T.; Murphy, J.M.; Wicks, I.P. Monosodium urate crystals generate nuclease-resistant neutrophil extracellular traps via a distinct molecular pathway. J. Immunol. 2018, 200, 1802–1816. [Google Scholar] [CrossRef] [Green Version]
- Schauer, C.; Janko, C.; Munoz, L.E.; Zhao, Y.; Kienhofer, D.; Frey, B.; Lell, M.; Manger, B.; Rech, J.; Naschberger, E.; et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat. Med. 2014, 20, 511–517. [Google Scholar] [CrossRef]
- Warnatsch, A.; Ioannou, M.; Wang, Q.; Papayannopoulos, V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 2015, 349, 316–320. [Google Scholar] [CrossRef] [Green Version]
- Menegazzo, L.; Ciciliot, S.; Poncina, N.; Mazzucato, M.; Persano, M.; Bonora, B.; Albiero, M.; Vigili de Kreutzenberg, S.; Avogaro, A.; Fadini, G.P. NETosis is induced by high glucose and associated with type 2 diabetes. Acta Diabetol. 2015, 52, 497–503. [Google Scholar] [CrossRef]
- Wong, S.L.; Demers, M.; Martinod, K.; Gallant, M.; Wang, Y.; Goldfine, A.B.; Kahn, C.R.; Wagner, D.D. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 2015, 21, 815–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Zhou, X.; Yin, Y.; Mai, Y.; Wang, D.; Zhang, X. Hyperglycemia induces neutrophil extracellular traps formation through an NADPH oxidase-dependent pathway in diabetic retinopathy. Front Immunol. 2018, 9, 3076. [Google Scholar] [CrossRef]
- Hosseinzadeh, A.; Thompson, P.R.; Segal, B.H.; Urban, C.F. Nicotine induces neutrophil extracellular traps. J. Leuko Biol. 2016, 100, 1105–1112. [Google Scholar] [CrossRef]
- Lee, J.; Luria, A.; Rhodes, C.; Raghu, H.; Lingampalli, N.; Sharpe, O.; Rada, B.; Sohn, D.H.; Robinson, W.H.; Sokolove, J. Nicotine drives neutrophil extracellular traps formation and accelerates collagen-induced arthritis. Rheumatology (Oxford) 2017, 56, 644–653. [Google Scholar] [CrossRef] [Green Version]
- Jorch, S.K.; Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 2017, 23, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Boeltz, S.; Amini, P.; Anders, H.J.; Andrade, F.; Bilyy, R.; Chatfield, S.; Cichon, I.; Clancy, D.M.; Desai, J.; Dumych, T.; et al. To NET or not to NET: current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death Differ. 2019, 26, 395–408. [Google Scholar] [CrossRef] [Green Version]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef]
- Hakkim, A.; Fuchs, T.A.; Martinez, N.E.; Hess, S.; Prinz, H.; Zychlinsky, A.; Waldmann, H. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat. Chem. Biol. 2011, 7, 75–77. [Google Scholar] [CrossRef] [PubMed]
- Douda, D.N.; Khan, M.A.; Grasemann, H.; Palaniyar, N. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc. Natl. Acad. Sci. USA 2015, 112, 2817–2822. [Google Scholar] [CrossRef] [Green Version]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef] [Green Version]
- Erpenbeck, L.; Schon, M.P. Neutrophil extracellular traps: protagonists of cancer progression? Oncogene 2017, 36, 2483–2490. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.W.; Monteleone, M.; Boucher, D.; Sollberger, G.; Ramnath, D.; Condon, N.D.; von Pein, J.B.; Broz, P.; Sweet, M.J.; Schroder, K. Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaar6676. [Google Scholar] [CrossRef] [Green Version]
- Sollberger, G.; Choidas, A.; Burn, G.L.; Habenberger, P.; Di Lucrezia, R.; Kordes, S.; Menninger, S.; Eickhoff, J.; Nussbaumer, P.; Klebl, B.; et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaar6689. [Google Scholar] [CrossRef] [Green Version]
- Thiam, H.R.; Wong, S.L.; Qiu, R.; Kittisopikul, M.; Vahabikashi, A.; Goldman, A.E.; Goldman, R.D.; Wagner, D.D.; Waterman, C.M. NETosis proceeds by cytoskeleton and endomembrane disassembly and PAD4-mediated chromatin decondensation and nuclear envelope rupture. Proc. Natl Acad. Sci. USA 2020, 117, 7326–7337. [Google Scholar] [CrossRef] [Green Version]
- Tatsiy, O.; McDonald, P.P. Physiological stimuli induce PAD4-dependent, ROS-independent NETosis, with early and late events controlled by discrete signaling pathways. Front Immunol. 2018, 9, 2036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neeli, I.; Radic, M. Current challenges and limitations in antibody-based detection of citrullinated histones. Front Immunol. 2016, 7, 528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, G.; Wang, D.; Gu, J.; Shen, Q.; Gross, S.S.; Wang, Y. Neutral loss of isocyanic acid in peptide CID spectra: a novel diagnostic marker for mass spectrometric identification of protein citrullination. J. Am. Soc. Mass Spec. 2009, 20, 723–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilvawala, R.; Thompson, P.R. Peptidyl arginine deiminases: detection and functional analysis of protein citrullination. Curr. Opin. Struct. Biol. 2019, 59, 205–215. [Google Scholar] [CrossRef]
- Bicker, K.L.; Subramanian, V.; Chumanevich, A.A.; Hofseth, L.J.; Thompson, P.R. Seeing citrulline: development of a phenylglyoxal-based probe to visualize protein citrullination. J. Am. Chem. Soc. 2012, 134, 17015–17018. [Google Scholar] [CrossRef] [Green Version]
- Tilvawala, R.; Nguyen, S.H.; Maurais, A.J.; Nemmara, V.V.; Nagar, M.; Salinger, A.J.; Nagpal, S.; Weerapana, E.; Thompson, P.R. The rheumatoid arthritis-associated citrullinome. Cell Chem. Biol. 2018, 25, 691–704. [Google Scholar] [CrossRef] [Green Version]
- Sorvillo, N.; Mizurini, D.M.; Coxon, C.; Martinod, K.; Tilvawala, R.; Cherpokova, D.; Salinger, A.J.; Seward, R.J.; Staudinger, C.; Weerapana, E.; et al. Plasma peptidylarginine deiminase IV promotes VWF-platelet string formation and accelerates thrombosis after vessel injury. Circ. Res. 2019, 125, 507–519. [Google Scholar] [CrossRef]
- Joly, B.S.; Coppo, P.; Veyradier, A. Thrombotic thrombocytopenic purpura. Blood 2017, 129, 2836–2846. [Google Scholar] [CrossRef] [Green Version]
- Smeets, M.W.J.; Mourik, M.J.; Niessen, H.W.M.; Hordijk, P.L. Stasis promotes erythrocyte adhesion to von Willebrand factor. Arter. Thromb. Vasc. Biol. 2017, 37, 1618–1627. [Google Scholar] [CrossRef] [Green Version]
- Maugeri, N.; Campana, L.; Gavina, M.; Covino, C.; De Metrio, M.; Panciroli, C.; Maiuri, L.; Maseri, A.; D’Angelo, A.; Bianchi, M.E.; et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J. Thromb. Haemost. 2014, 12, 2074–2088. [Google Scholar] [CrossRef]
- Vogel, S.; Bodenstein, R.; Chen, Q.; Feil, S.; Feil, R.; Rheinlaender, J.; Schaffer, T.E.; Bohn, E.; Frick, J.S.; Borst, O.; et al. Platelet-derived HMGB1 is a critical mediator of thrombosis. J. Clin. Investig. 2015, 125, 4638–4654. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, G.T.; Green, E.R.; Mecsas, J. Neutrophils to the ROScue: Mechanisms of NADPH oxidase activation and bacterial resistance. Front Cell Infect. Microbiol. 2017, 7, 373. [Google Scholar] [CrossRef]
- Chen, K.; Nishi, H.; Travers, R.; Tsuboi, N.; Martinod, K.; Wagner, D.D.; Stan, R.; Croce, K.; Mayadas, T.N. Endocytosis of soluble immune complexes leads to their clearance by FcgammaRIIIB but induces neutrophil extracellular traps via FcgammaRIIA in vivo. Blood 2012, 120, 4421–4431. [Google Scholar] [CrossRef] [Green Version]
- Aleman, O.R.; Mora, N.; Cortes-Vieyra, R.; Uribe-Querol, E.; Rosales, C. Differential use of human neutrophil Fcgamma receptors for inducing neutrophil extracellular trap formation. J. Immunol. Res. 2016, 2016, 2908034. [Google Scholar] [CrossRef] [Green Version]
- Lood, C.; Arve, S.; Ledbetter, J.; Elkon, K.B. TLR7/8 activation in neutrophils impairs immune complex phagocytosis through shedding of FcgRIIA. J. Exp. Med. 2017, 214, 2103–2119. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, S.; Opneja, A.; Nayak, L. The role of neutrophils in thrombosis. Thromb. Res. 2018, 170, 87–96. [Google Scholar] [CrossRef]
- Pertiwi, K.R.; van der Wal, A.C.; Pabittei, D.R.; Mackaaij, C.; van Leeuwen, M.B.; Li, X.; de Boer, O.J. Neutrophil Extracellular Traps Participate in All Different Types of Thrombotic and Haemorrhagic Complications of Coronary Atherosclerosis. Thromb. Haemost. 2018, 118, 1078–1087. [Google Scholar] [CrossRef]
- Mangold, A.; Alias, S.; Scherz, T.; Hofbauer, T.; Jakowitsch, J.; Panzenbock, A.; Simon, D.; Laimer, D.; Bangert, C.; Kammerlander, A.; et al. Coronary Neutrophil Extracellular Trap Burden and Deoxyribonuclease Activity in ST-Elevation Acute Coronary Syndrome Are Predictors of ST-Segment Resolution and Infarct Size. Circ. Res. 2015, 116, 1182–1192. [Google Scholar] [CrossRef] [Green Version]
- Savchenko, A.S.; Martinod, K.; Seidman, M.A.; Wong, S.L.; Borissoff, J.I.; Piazza, G.; Libby, P.; Goldhaber, S.Z.; Mitchell, R.N.; Wagner, D.D. Neutrophil extracellular traps form predominantly during the organizing stage of human venous thromboembolism development. J. Thromb. Haemost. 2014, 12, 860–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massberg, S.; Grahl, L.; von Bruehl, M.L.; Manukyan, D.; Pfeiler, S.; Goosmann, C.; Brinkmann, V.; Lorenz, M.; Bidzhekov, K.; Khandagale, A.B.; et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 2010, 16, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Brill, A.; Fuchs, T.A.; Savchenko, A.S.; Thomas, G.M.; Martinod, K.; De Meyer, S.F.; Bhandari, A.A.; Wagner, D.D. Neutrophil extracellular traps promote deep vein thrombosis in mice. J. Thromb. Haemost. 2012, 10, 136–144. [Google Scholar] [CrossRef] [Green Version]
- Quillard, T.; Franck, G.; Mawson, T.; Folco, E.; Libby, P. Mechanisms of erosion of atherosclerotic plaques. Curr. Opin. Lipidol. 2017, 28, 434–441. [Google Scholar] [CrossRef]
- Naruko, T.; Ueda, M.; Haze, K.; van der Wal, A.C.; van der Loos, C.M.; Itoh, A.; Komatsu, R.; Ikura, Y.; Ogami, M.; Shimada, Y.; et al. Neutrophil infiltration of culprit lesions in acute coronary syndromes. Circulation 2002, 106, 2894–2900. [Google Scholar] [CrossRef] [Green Version]
- Megens, R.T.; Vijayan, S.; Lievens, D.; Doring, Y.; van Zandvoort, M.A.; Grommes, J.; Weber, C.; Soehnlein, O. Presence of luminal neutrophil extracellular traps in atherosclerosis. Thromb. Haemost. 2012, 107, 597–598. [Google Scholar] [CrossRef]
- Yunoki, K.; Naruko, T.; Komatsu, R.; Shirai, N.; Nakagawa, M.; Sugioka, K.; Ikura, Y.; Kusano, K.F.; Itoh, A.; Haze, K.; et al. Relation of elevated levels of plasma myeloperoxidase to impaired myocardial microcirculation after reperfusion in patients with acute myocardial infarction. Am. J. Cardiol. 2010, 105, 922–929. [Google Scholar] [CrossRef]
- Libby, P. Mechanisms of acute coronary syndromes and their implications for therapy. N. Engl. J. Med. 2013, 368, 2004–2013. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.Q.; Dong, L.; Hatsukami, T.; Phan, B.A.; Chu, B.; Moore, A.; Lane, T.; Neradilek, M.B.; Polissar, N.; Monick, D.; et al. MR imaging of carotid plaque composition during lipid-lowering therapy a prospective assessment of effect and time course. Jacc Cardiovasc. Imag. 2011, 4, 977–986. [Google Scholar] [CrossRef] [Green Version]
- Takarada, S.; Imanishi, T.; Kubo, T.; Tanimoto, T.; Kitabata, H.; Nakamura, N.; Tanaka, A.; Mizukoshi, M.; Akasaka, T. Effect of statin therapy on coronary fibrous-cap thickness in patients with acute coronary syndrome: assessment by optical coherence tomography study. Atherosclerosis 2009, 202, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Jernberg, T.; Hasvold, P.; Henriksson, M.; Hjelm, H.; Thuresson, M.; Janzon, M. Cardiovascular risk in post-myocardial infarction patients: nationwide real world data demonstrate the importance of a long-term perspective. Euro. Heart J. 2015, 36, 1163–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quillard, T.; Araujo, H.A.; Franck, G.; Shvartz, E.; Sukhova, G.; Libby, P. TLR2 and neutrophils potentiate endothelial stress, apoptosis, and detachment: implications for superficial erosion. Euro. Heart J. 2015, 36, 1394–1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franck, G.; Mawson, T.; Sausen, G.; Salinas, M.; Masson, G.S.; Cole, A.; Beltrami-Moreira, M.; Chatzizisis, Y.; Quillard, T.; Tesmenitsky, Y.; et al. Flow Perturbation Mediates Neutrophil Recruitment and Potentiates Endothelial Injury via TLR2 in Mice: Implications for Superficial Erosion. Circ. Res. 2017, 121, 31–42. [Google Scholar] [CrossRef]
- Goldmann, B.U.; Rudolph, V.; Rudolph, T.K.; Holle, A.K.; Hillebrandt, M.; Meinertz, T.; Baldus, S. Neutrophil activation precedes myocardial injury in patients with acute myocardial infarction. Free Radic. Biol. Med. 2009, 47, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Borissoff, J.I.; Joosen, I.A.; Versteylen, M.O.; Brill, A.; Fuchs, T.A.; Savchenko, A.S.; Gallant, M.; Martinod, K.; Ten Cate, H.; Hofstra, L.; et al. Elevated levels of circulating DNA and chromatin are independently associated with severe coronary atherosclerosis and a prothrombotic state. Arter. Thromb. Vasc. Biol. 2013, 33, 2032–2040. [Google Scholar] [CrossRef] [Green Version]
- Ferrante, G.; Nakano, M.; Prati, F.; Niccoli, G.; Mallus, M.T.; Ramazzotti, V.; Montone, R.A.; Kolodgie, F.D.; Virmani, R.; Crea, F. High levels of systemic myeloperoxidase are associated with coronary plaque erosion in patients with acute coronary syndromes: a clinicopathological study. Circulation 2010, 122, 2505–2513. [Google Scholar] [CrossRef] [Green Version]
- Barrett, T.J.; Distel, E.; Murphy, A.J.; Hu, J.; Garshick, M.S.; Ogando, Y.; Liu, J.; Vaisar, T.; Heinecke, J.W.; Berger, J.S.; et al. Apolipoprotein AI promotes atherosclerosis regression in diabetic mice by suppressing myelopoiesis and plaque inflammation. Circulation 2019, 140, 1170–1184. [Google Scholar] [CrossRef]
- Qi, H.; Yang, S.; Zhang, L. Neutrophil Extracellular Traps and Endothelial Dysfunction in Atherosclerosis and Thrombosis. Front Immunol. 2017, 8, 928. [Google Scholar] [CrossRef]
- Meegan, J.E.; Yang, X.; Beard, R.S., Jr.; Jannaway, M.; Chatterjee, V.; Taylor-Clark, T.E.; Yuan, S.Y. Citrullinated histone 3 causes endothelial barrier dysfunction. Biochem. Biophys. Res. Commun. 2018, 503, 1498–1502. [Google Scholar] [CrossRef]
- Saffarzadeh, M.; Juenemann, C.; Queisser, M.A.; Lochnit, G.; Barreto, G.; Galuska, S.P.; Lohmeyer, J.; Preissner, K.T. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS ONE 2012, 7, e32366. [Google Scholar] [CrossRef]
- Evrard, S.M.; Lecce, L.; Michelis, K.C.; Nomura-Kitabayashi, A.; Pandey, G.; Purushothaman, K.R.; d’Escamard, V.; Li, J.R.; Hadri, L.; Fujitani, K.; et al. Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat. Commun. 2016, 7, 11853. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.Y.; Qin, L.; Baeyens, N.; Li, G.; Afolabi, T.; Budatha, M.; Tellides, G.; Schwartz, M.A.; Simons, M. Endothelial-to-mesenchymal transition drives atherosclerosis progression. J. Clin. Inves. 2015, 125, 4514–4528. [Google Scholar] [CrossRef] [Green Version]
- Wesseling, M.; Sakkers, T.R.; de Jager, S.C.A.; Pasterkamp, G.; Goumans, M.J. The morphological and molecular mechanisms of epithelial/endothelial-to-mesenchymal transition and its involvement in atherosclerosis. Vasc. Pharm. 2018, 106, 1–8. [Google Scholar] [CrossRef]
- Pieterse, E.; Rother, N.; Garsen, M.; Hofstra, J.M.; Satchell, S.C.; Hoffmann, M.; Loeven, M.A.; Knaapen, H.K.; van der Heijden, O.W.H.; Berden, J.H.M.; et al. Neutrophil Extracellular Traps Drive Endothelial-to-Mesenchymal Transition. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1371–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itabe, H.; Kato, R.; Sawada, N.; Obama, T.; Yamamoto, M. The significance of oxidized low-density lipoprotein in body fluids as a marker related to diseased condimns. Curr. Med. Chem. 2019, 26, 1576–1593. [Google Scholar] [CrossRef]
- Lee, J.Y.; Lim, S.; Park, S.; Moon, M.H. Characterization of oxidized phospholipids in oxidatively modified low density lipoproteins by nanoflow liquid chromatography-tandem mass spectrometry. J. Chromato. A 2013, 1288, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Sasabe, N.; Keyamura, Y.; Obama, T.; Inoue, N.; Masuko, Y.; Igarashi, Y.; Aiuchi, T.; Kato, R.; Yamaguchi, T.; Kuwata, H.; et al. Time course changes in phosphatidylcholine profile during oxidative modification of low-density lipoprotein. Lipids Health Dis. 2014, 13, 48. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Zhang, W.; Gu, X.; Chen, X.; Hong, L.; Laird, J.M.; Salomon, R.G. Lysophosphatidylcholine is generated by spontaneous deacylation of oxidized phospholipids. Chem Res. Toxicol. 2011, 24, 111–118. [Google Scholar] [CrossRef] [Green Version]
- Obama, T.; Kato, R.; Masuda, Y.; Takahashi, K.; Aiuchi, T.; Itabe, H. Analysis of modified apolipoprotein B-100 structures formed in oxidized low-density lipoprotein using LC-MS/MS. Proteomics 2007, 7, 2132–2141. [Google Scholar] [CrossRef]
- Levitan, I.; Volkov, S.; Subbaiah, P.V. Oxidized LDL: diversity, patterns of recognition, and pathophysiology. Antioxid. Redox. Signal. 2010, 13, 39–75. [Google Scholar] [CrossRef] [Green Version]
- Chellan, B.; Rojas, E.; Zhang, C.; Hofmann Bowman, M.A. Enzyme-modified non-oxidized LDL (ELDL) induces human coronary artery smooth muscle cell transformation to a migratory and osteoblast-like phenotype. Sci. Rep. 2018, 8, 11954. [Google Scholar] [CrossRef]
- Torzewski, M.; Suriyaphol, P.; Paprotka, K.; Spath, L.; Ochsenhirt, V.; Schmitt, A.; Han, S.R.; Husmann, M.; Gerl, V.B.; Bhakdi, S.; et al. Enzymatic modification of low-density lipoprotein in the arterial wall: a new role for plasmin and matrix metalloproteinases in atherogenesis. Arter. Thromb. Vasc. Biol. 2004, 24, 2130–2136. [Google Scholar] [CrossRef] [Green Version]
- Torzewski, M.; Klouche, M.; Hock, J.; Messner, M.; Dorweiler, B.; Torzewski, J.; Gabbert, H.E.; Bhakdi, S. Immunohistochemical demonstration of enzymatically modified human LDL and its colocalization with the terminal complement complex in the early atherosclerotic lesion. Arter. Thromb. Vasc. Biol. 1998, 18, 369–378. [Google Scholar] [CrossRef] [Green Version]
- Suriyaphol, P.; Fenske, D.; Zahringer, U.; Han, S.R.; Bhakdi, S.; Husmann, M. Enzymatically modified nonoxidized low-density lipoprotein induces interleukin-8 in human endothelial cells: role of free fatty acids. Circulation 2002, 106, 2581–2587. [Google Scholar] [CrossRef] [Green Version]
- Klouche, M.; May, A.E.; Hemmes, M.; Messner, M.; Kanse, S.M.; Preissner, K.T.; Bhakdi, S. Enzymatically modified, nonoxidized LDL induces selective adhesion and transmigration of monocytes and T-lymphocytes through human endothelial cell monolayers. Arter. Thromb. Vasc. Biol. 1999, 19, 784–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chellan, B.; Reardon, C.A.; Getz, G.S.; Hofmann Bowman, M.A. Enzymatically modified low-density lipoprotein promotes foam cell formation in smooth muscle cells via macropinocytosis and enhances receptor-mediated uptake of oxidized low-density lipoprotein. Arter. Thromb. Vasc. Biol. 2016, 36, 1101–1113. [Google Scholar] [CrossRef] [Green Version]
- Lux, C.A.; Koschinski, A.; Dersch, K.; Husmann, M.; Bhakdi, S. Hypersusceptibility of neutrophil granulocytes towards lethal action of free fatty acids contained in enzyme-modified atherogenic low density lipoprotein. Atherosclerosis 2009, 207, 116–122. [Google Scholar] [CrossRef]
- Yang, C.Y.; Raya, J.L.; Chen, H.H.; Chen, C.H.; Abe, Y.; Pownall, H.J.; Taylor, A.A.; Smith, C.V. Isolation, characterization, and functional assessment of oxidatively modified subfractions of circulating low-density lipoproteins. Arter. Thromb. Vasc. Biol. 2003, 23, 1083–1090. [Google Scholar] [CrossRef] [Green Version]
- Bancells, C.; Canals, F.; Benitez, S.; Colome, N.; Julve, J.; Ordonez-Llanos, J.; Sanchez-Quesada, J.L. Proteomic analysis of electronegative low-density lipoprotein. J. Lipid Res. 2010, 51, 3508–3515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bancells, C.; Villegas, S.; Blanco, F.J.; Benitez, S.; Gallego, I.; Beloki, L.; Perez-Cuellar, M.; Ordonez-Llanos, J.; Sanchez-Quesada, J.L. Aggregated electronegative low density lipoprotein in human plasma shows a high tendency toward phospholipolysis and particle fusion. J. Biol. Chem. 2010, 285, 32425–32435. [Google Scholar] [CrossRef] [Green Version]
- Ke, L.Y.; Chan, H.C.; Chen, C.C.; Lu, J.; Marathe, G.K.; Chu, C.S.; Chan, H.C.; Wang, C.Y.; Tung, Y.C.; McIntyre, T.M.; et al. Enhanced sphingomyelinase activity contributes to the apoptotic capacity of electronegative low-density lipoprotein. J. Med. Chem. 2016, 59, 1032–1040. [Google Scholar] [CrossRef]
- Benitez, S.; Sanchez-Quesada, J.L.; Ribas, V.; Jorba, O.; Blanco-Vaca, F.; Gonzalez-Sastre, F.; Ordonez-Llanos, J. Platelet-activating factor acetylhydrolase is mainly associated with electronegative low-density lipoprotein subfraction. Circulation 2003, 108, 92–96. [Google Scholar] [CrossRef] [Green Version]
- Gaubatz, J.W.; Gillard, B.K.; Massey, J.B.; Hoogeveen, R.C.; Huang, M.; Lloyd, E.E.; Raya, J.L.; Yang, C.Y.; Pownall, H.J. Dynamics of dense electronegative low density lipoproteins and their preferential association with lipoprotein phospholipase A(2). J. Lipid Res. 2007, 48, 348–357. [Google Scholar] [CrossRef] [Green Version]
- Benitez, S.; Camacho, M.; Arcelus, R.; Vila, L.; Bancells, C.; Ordonez-Llanos, J.; Sanchez-Quesada, J.L. Increased lysophosphatidylcholine and non-esterified fatty acid content in LDL induces chemokine release in endothelial cells. Relationship with electronegative LDL. Atherosclerosis 2004, 177, 299–305. [Google Scholar]
- Ke, L.Y.; Stancel, N.; Bair, H.; Chen, C.H. The underlying chemistry of electronegative LDL’s atherogenicity. Curr. Atheroscler. Rep. 2014, 16, 428. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Yang, J.H.; Burns, A.R.; Chen, H.H.; Tang, D.; Walterscheid, J.P.; Suzuki, S.; Yang, C.Y.; Sawamura, T.; Chen, C.H. Mediation of electronegative low-density lipoprotein signaling by LOX-1: a possible mechanism of endothelial apoptosis. Circ. Res. 2009, 104, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Jiang, T.; Yang, J.H.; Jiang, W.; Lu, J.; Marathe, G.K.; Pownall, H.J.; Ballantyne, C.M.; McIntyre, T.M.; Henry, P.D.; et al. Low-density lipoprotein in hypercholesterolemic human plasma induces vascular endothelial cell apoptosis by inhibiting fibroblast growth factor 2 transcription. Circulation 2003, 107, 2102–2108. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.C.; Lee, A.S.; Lu, L.S.; Ke, L.Y.; Chen, W.Y.; Dong, J.W.; Lu, J.; Chen, Z.; Chu, C.S.; Chan, H.C.; et al. Human electronegative LDL induces mitochondrial dysfunction and premature senescence of vascular cells in vivo. Aging Cell 2018, e12792. [Google Scholar] [CrossRef] [Green Version]
- Chan, H.C.; Ke, L.Y.; Chu, C.S.; Lee, A.S.; Shen, M.Y.; Cruz, M.A.; Hsu, J.F.; Cheng, K.H.; Chan, H.C.; Lu, J.; et al. Highly electronegative LDL from patients with ST-elevation myocardial infarction triggers platelet activation and aggregation. Blood 2013, 122, 3632–3641. [Google Scholar] [CrossRef] [Green Version]
- Puig, N.; Montolio, L.; Camps-Renom, P.; Navarra, L.; Jimenez-Altayo, F.; Jimenez-Xarrie, E.; Sanchez-Quesada, J.L.; Benitez, S. Electronegative LDL Promotes Inflammation and Triglyceride Accumulation in Macrophages. Cells 2020, 9, 583. [Google Scholar] [CrossRef] [Green Version]
- Sawada, N.; Obama, T.; Koba, S.; Takaki, T.; Iwamoto, S.; Aiuchi, T.; Kato, R.; Kikuchi, M.; Hamazaki, Y.; Itabe, H. Circulating oxidized LDL, increased in patients with acute myocardial infarction, is accompanied by heavily modified HDL. J. Lipid Res. 2020, 61, 816–829. [Google Scholar] [CrossRef] [Green Version]
- Itabe, H.; Yamamoto, H.; Imanaka, T.; Shimamura, K.; Uchiyama, H.; Kimura, J.; Sanaka, T.; Hata, Y.; Takano, T. Sensitive detection of oxidatively modified low density lipoprotein using a monoclonal antibody. J. Lipid Res. 1996, 37, 45–53. [Google Scholar]
- Itabe, H.; Suzuki, K.; Tsukamoto, Y.; Komatsu, R.; Ueda, M.; Mori, M.; Higashi, Y.; Takano, T. Lysosomal accumulation of oxidized phosphatidylcholine-apolipoprotein B complex in macrophages: intracellular fate of oxidized low density lipoprotein. Biochim. Biophys. Acta 2000, 1487, 233–245. [Google Scholar] [CrossRef]
- Tani, M.; Kawakami, A.; Mizuno, Y.; Imase, R.; Ito, Y.; Kondo, K.; Ishii, H.; Yoshida, M. Small dense LDL enhances THP-1 macrophage foam cell formation. J. Atheroscler. Thromb. 2011, 18, 698–704. [Google Scholar] [CrossRef] [Green Version]
- Hoogeveen, R.C.; Gaubatz, J.W.; Sun, W.; Dodge, R.C.; Crosby, J.R.; Jiang, J.; Couper, D.; Virani, S.S.; Kathiresan, S.; Boerwinkle, E.; et al. Small dense low-density lipoprotein-cholesterol concentrations predict risk for coronary heart disease: the Atherosclerosis Risk In Communities (ARIC) study. Arter. Thromb. Vasc. Biol. 2014, 34, 1069–1077. [Google Scholar] [CrossRef] [Green Version]
- Watts, G.F.; Chan, D.C. Atherogenic Dyslipoproteinemia and Management of ASCVD: Will New Indices Untie the Gordian Knot? J. Am. Coll. Cardiol. 2020, 75, 2136–2139. [Google Scholar] [CrossRef] [PubMed]
- Syvänne, M.; Taskinen, M.-R. Lipids and lipoproteins as coronary risk factors in non-insulin-dependent diabetes mellitus. Lancet 1997, 350, S20–S23. [Google Scholar] [CrossRef]
- Packard, C.J.; Demant, T.; Stewart, J.P.; Bedford, D.; Caslake, M.J.; Schwertfeger, G.; Bedynek, A.; Shepherd, J.; Seidel, D. Apolipoprotein B metabolism and the distribution of VLDL and LDL subfractions. J. Lipid Res. 2000, 41, 305–318. [Google Scholar] [PubMed]
- Koba, S.; Hirano, T.; Murayama, S.; Kotani, T.; Tsunoda, F.; Iso, Y.; Ban, Y.; Kondo, T.; Suzuki, H.; Katagiri, T. Small dense LDL phenotype is associated with postprandial increases of large VLDL and remnant-like particles in patients with acute myocardial infarction. Atherosclerosis 2003, 170, 131–140. [Google Scholar] [CrossRef]
- Galeano, N.F.; Milne, R.; Marcel, Y.L.; Walsh, M.T.; Levy, E.; Ngu’yen, T.D.; Gleeson, A.; Arad, Y.; Witte, L.; Al-Haideri, M.; et al. Apoprotein B structure and receptor recognition of triglyceride-rich low density lipoprotein (LDL) is modified in small LDL but not in triglyceride-rich LDL of normal size. J. Biol. Chem. 1994, 269, 511–519. [Google Scholar]
- Galeano, N.F.; Al-Haideri, M.; Keyserman, F.; Rumsey, S.C.; Deckelbaum, R.J. Small dense low density lipoprotein has increased affinity for LDL receptor-independent cell surface binding sites: A potential mechanism for increased atherogenicity. J. Lipid Res. 1998, 39, 1263–1273. [Google Scholar]
- Drechsler, M.; Megens, R.T.; van Zandvoort, M.; Weber, C.; Soehnlein, O. Hyperlipidemia-triggered neutrophilia promotes early atherosclerosis. Circulation 2010, 122, 1837–1845. [Google Scholar] [CrossRef] [PubMed]
- Doring, Y.; Drechsler, M.; Soehnlein, O.; Weber, C. Neutrophils in atherosclerosis: from mice to man. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 288–295. [Google Scholar] [CrossRef] [Green Version]
- Doring, Y.; Soehnlein, O.; Weber, C. Neutrophil extracellular traps in atherosclerosis and atherothrombosis. Circ. Res. 2017, 120, 736–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, R.; Mori, C.; Kitazato, K.; Arata, S.; Obama, T.; Mori, M.; Takahashi, K.; Aiuchi, T.; Takano, T.; Itabe, H. Transient increase in plasma oxidized LDL during the progression of atherosclerosis in apolipoprotein E knockout mice. Arter. Thromb. Vasc. Biol. 2009, 29, 33–39. [Google Scholar] [CrossRef]
- Obama, T.; Ohinata, H.; Takaki, T.; Iwamoto, S.; Sawada, N.; Aiuchi, T.; Kato, R.; Itabe, H. Cooperative action of oxidized low-density lipoproteins and neutrophils on endothelial inflammatory responses through neutrophil extracellular trap formation. Front Immunol. 2019, 10, 1899. [Google Scholar] [CrossRef] [Green Version]
- Jaipersad, A.S.; Lip, G.Y.; Silverman, S.; Shantsila, E. The role of monocytes in angiogenesis and atherosclerosis. J. Am. Coll. Cardiol. 2014, 63, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Cao, W.; Abdelgawwad, M.S.; Li, J.; Zheng, X.L. Apolipoprotein B100/low-density lipoprotein regulates proteolysis and functions of von Willebrand factor under arterial shear. Thromb. Haemost. 2019, 119, 1933–1946. [Google Scholar] [CrossRef]
- Awasthi, D.; Nagarkoti, S.; Kumar, A.; Dubey, M.; Singh, A.K.; Pathak, P.; Chandra, T.; Barthwal, M.K.; Dikshit, M. Oxidized LDL induced extracellular trap formation in human neutrophils via TLR-PKC-IRAK-MAPK and NADPH-oxidase activation. Free Rad. Biol. Med. 2016, 93, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Yotsumoto, S.; Muroi, Y.; Chiba, T.; Ohmura, R.; Yoneyama, M.; Magarisawa, M.; Dodo, K.; Terayama, N.; Sodeoka, M.; Aoyagi, R.; et al. Hyperoxidation of ether-linked phospholipids accelerates neutrophil extracellular trap formation. Sci. Rep. 2017, 7, 16026. [Google Scholar] [CrossRef]
- Sun, L.; Ishida, T.; Yasuda, T.; Kojima, Y.; Honjo, T.; Yamamoto, Y.; Yamamoto, H.; Ishibashi, S.; Hirata, K.; Hayashi, Y. RAGE mediates oxidized LDL-induced pro-inflammatory effects and atherosclerosis in non-diabetic LDL receptor-deficient mice. Cardiovasc. Res. 2009, 82, 371–381. [Google Scholar] [CrossRef] [Green Version]
- Collison, K.S.; Parhar, R.S.; Saleh, S.S.; Meyer, B.F.; Kwaasi, A.A.; Hammami, M.M.; Schmidt, A.M.; Stern, D.M.; Al-Mohanna, F.A. RAGE-mediated neutrophil dysfunction is evoked by advanced glycation end products (AGEs). J. Leuko Biol. 2002, 71, 433–444. [Google Scholar]
- Corriden, R.; Hollands, A.; Olson, J.; Derieux, J.; Lopez, J.; Chang, J.T.; Gonzalez, D.J.; Nizet, V. Tamoxifen augments the innate immune function of neutrophils through modulation of intracellular ceramide. Nat. Commun. 2015, 6, 8369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meher, A.K.; Spinosa, M.; Davis, J.P.; Pope, N.; Laubach, V.E.; Su, G.; Serbulea, V.; Leitinger, N.; Ailawadi, G.; Upchurch, G.R., Jr. Novel role of IL (interleukin)-1beta in neutrophil extracellular trap formation and abdominal aortic aneurysms. Arter. Thromb. Vasc. Biol. 2018, 38, 843–853. [Google Scholar] [CrossRef] [Green Version]
- Hollands, A.; Corriden, R.; Gysler, G.; Dahesh, S.; Olson, J.; Raza Ali, S.; Kunkel, M.T.; Lin, A.E.; Forli, S.; Newton, A.C.; et al. Natural product anacardic acid from cashew nut shells stimulates neutrophil extracellular trap production and bactericidal activity. BioChem 2016, 291, 13964–13973. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Yang, L.; Chang, N.; Hou, L.; Zhou, X.; Yang, L.; Li, L. Neutrophils undergo switch of apoptosis to NETosis during murine fatty liver injury via S1P receptor 2 signaling. Cell Death Dis. 2020, 11, 379. [Google Scholar] [CrossRef]
- Kurano, M.; Yatomi, Y. Sphingosine 1-Phosphate and Atherosclerosis. J. Atheroscler. Thromb. 2018, 25, 16–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neil, L.J.; Kaplan, M.J.; Carmona-Rivera, C. The role of neutrophils and neutrophil extracellular traps in vascular damage in systemic lupus erythematosus. J. Clin. Med. 2019, 8, 1325. [Google Scholar] [CrossRef] [Green Version]
- Deniset, J.F.; Kubes, P. Neutrophil heterogeneity: Bona fide subsets or polarization states? J. Leuko Biol. 2018, 103, 829–838. [Google Scholar] [CrossRef]
- Carmona-Rivera, C.; Kaplan, M.J. Low-density granulocytes: a distinct class of neutrophils in systemic autoimmunity. Semin. Immunopathol. 2013, 35, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, E.; Yalavarthi, S.; Berthier, C.C.; Hodgin, J.B.; Khandpur, R.; Lin, A.M.; Rubin, C.J.; Zhao, W.; Olsen, S.H.; Klinker, M.; et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J. Immunol. 2011, 187, 538–552. [Google Scholar] [CrossRef] [Green Version]
- Lopez, D.; Garcia-Valladares, I.; Palafox-Sanchez, C.A.; De La Torre, I.G.; Kobayashi, K.; Matsuura, E.; Lopez, L.R. Oxidized low-density lipoprotein/β2-glycoprotein I complexes and autoantibodies to oxLig-1/β2-glycoprotein I in patients with systemic lupus erythematosus and antiphospholipid syndrome. Am. J. Clin. Pathol. 2004, 121, 426–436. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, E.; Kobayashi, K.; Tabuchi, M.; Lopez, L.R. Oxidative modification of low-density lipoprotein and immune regulation of atherosclerosis. Prog. Lipid Res. 2006, 45, 466–486. [Google Scholar] [CrossRef]
- Matsuura, E.; Hughes, G.R.; Khamashta, M.A. Oxidation of LDL and its clinical implication. Autoimmun. Rev. 2008, 7, 558–566. [Google Scholar] [CrossRef]
- Zha, C.; Zhang, W.; Gao, F.; Xu, J.; Jia, R.; Cai, J.; Liu, Y. Anti-beta2GPI/beta2GPI induces neutrophil extracellular traps formation to promote thrombogenesis via the TLR4/MyD88/MAPKs axis activation. Neuropharmacology 2018, 138, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Sagar, D.; Gaddipati, R.; Ongstad, E.L.; Bhagroo, N.; An, L.L.; Wang, J.; Belkhodja, M.; Rahman, S.; Manna, Z.; Davis, M.A.; et al. LOX-1: A potential driver of cardiovascular risk in SLE patients. PLoS ONE 2020, 15, e0229184. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Obama, T.; Itabe, H. Neutrophils as a Novel Target of Modified Low-Density Lipoproteins and an Accelerator of Cardiovascular Diseases. Int. J. Mol. Sci. 2020, 21, 8312. https://doi.org/10.3390/ijms21218312
Obama T, Itabe H. Neutrophils as a Novel Target of Modified Low-Density Lipoproteins and an Accelerator of Cardiovascular Diseases. International Journal of Molecular Sciences. 2020; 21(21):8312. https://doi.org/10.3390/ijms21218312
Chicago/Turabian StyleObama, Takashi, and Hiroyuki Itabe. 2020. "Neutrophils as a Novel Target of Modified Low-Density Lipoproteins and an Accelerator of Cardiovascular Diseases" International Journal of Molecular Sciences 21, no. 21: 8312. https://doi.org/10.3390/ijms21218312
APA StyleObama, T., & Itabe, H. (2020). Neutrophils as a Novel Target of Modified Low-Density Lipoproteins and an Accelerator of Cardiovascular Diseases. International Journal of Molecular Sciences, 21(21), 8312. https://doi.org/10.3390/ijms21218312