1. Introduction
Nonalcoholic fatty liver disease (NAFLD), an etiology of chronic liver disease, involves hepatic steatosis, steatohepatitis, and cirrhosis and is associated with metabolic disorders including insulin resistance, type 2 diabetes, and obesity [
1]. Hepatic steatosis, a hallmark of NAFLD, is caused by an imbalance in triglyceride (TG) synthesis, including de novo lipogenesis (DNL), the uptake of free fatty acids from adipose tissue or diet, and TG removal via fatty acid oxidation, very low-density lipoprotein (VLDL) secretion, and lipophagy [
2]. Thus, excess DNL can contribute to the development of NAFLD through the overproduction of fatty acids [
2]. DNL is stimulated by lipogenic transcription factors including sterol regulatory element-binding protein-1c (SREBP-1c; gene
SREBF1), carbohydrate responsive element binding protein (ChREBP), and liver X receptor (LXR; gene
NR1H3), which positively regulate the expression of lipogenic genes including fatty acid synthase (
FAS), acetyl CoA carboxylase (
ACC), and stearoyl-CoA desaturase 1 (
SCD1). Therefore, the regulation of DNL is a potential therapeutic method for NAFLD. Recently, studies on the role of epigenetics in the regulation of hepatic lipogenesis have been performed [
3]. However, epigenetic regulation of lipogenesis through histone methylation is poorly understood.
LXRs are ligand-activated transcription factors that play an important role in promoting lipogenesis [
4]. There are two subtypes of LXRs: LXRα and LXRβ. LXRα is abundantly expressed in the liver, adipose tissue, and kidney, whereas LXRβ is expressed ubiquitously. Ligand-activated LXRs form obligate heterodimers with retinoid X receptors (RXRs) and regulate expression of target genes containing LXR response elements (LXRE). LXRα directly stimulates the expression of LXRα-dependent lipogenic genes, including
FAS,
ACC, and
SCD1, through binding to LXRE in their promoter regions. In addition, LXRα upregulates the expression of SREBP-1c, a critical regulator of lipogenesis, through binding to the LXRE in the promoter region, thereby promoting the expression of its downstream lipogenic genes, including
FAS, ACC, and
SCD1 [
4]. Thus, the LXRα-induced activation of SREBP1c plays an important role in promoting hepatic lipogenesis. Accordingly, activation of LXRα increases hepatic TG accumulation and results in hepatic steatosis [
5]. Although recent studies have revealed several coactivators of LXRα [
4], histone modifiers involved in the epigenetic regulation of LXRα-dependent lipogenesis are not well understood.
Histone methylation marks are responsible for the epigenetic regulation of chromatin structure through addition or removal of methyl groups from lysine residues of histone tails [
6]. Mono-, di-, and tri-methylation marks and their respective demethylation of lysine residues within histones H3 and H4 act as epigenetic switches that can either activate or repress transcription. Histone H3 tri-methylation at lysine 4 (H3K4me3) and lysine 36 (H3K36me3) activate transcription, whereas tri-methylation or di-methylation at lysine 9 (H3K9me3 or H3K9me2) and tri-methylation at lysine 27 (H3K27me3) generally repress transcription [
6]. Histone demethylases remove methyl groups from modified histones, thereby activating or repressing gene transcription. The histone demethylase Jumonji domain-containing protein 2B (JMJD2B, also known as KDM4B) is a Jumonji (Jmj)—containing histone demethylase that removes repressive di- and tri-methylation marks at lysine 9 (H3K9me2/3) on histone H3, converting the marks to the monomethylated state; thus, JMJD2B functions as a transcription activator [
7,
8]. It has been reported that JMJD2B regulates cellular responses via cell differentiation [
9], cell proliferation [
10], DNA damage [
11], steroid hormone [
12], and energy status [
13]. Notably, JMJD2B expression is enhanced in various cancers and plays an important role in the promotion of tumorigenesis [
14].
Previously, we demonstrated the functional role of JMJD2B in hepatic steatosis [
15]. JMJD2B upregulated the expression of peroxisome proliferator-activated receptor γ2 (
PPARγ2) and its steatosis target genes including
CD36 and fatty acid-binding protein (
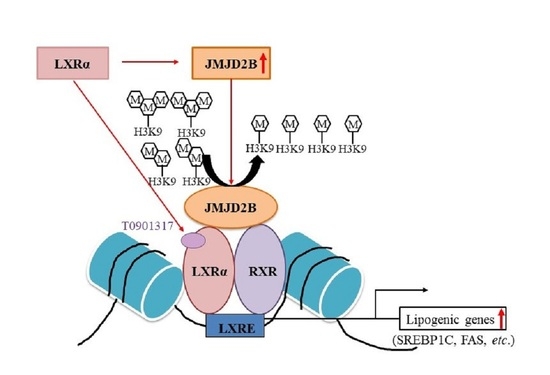
FABP), which are necessary for fatty acid uptake, thus resulting in the development of hepatic steatosis. Meanwhile, we also observed that JMJD2B expression increased in HepG2 cells and mice treated with the LXRα agonist T0901317, coinciding with the upregulated expression of LXRα-dependent lipogenic genes, suggesting that JMJD2B is involved in LXRα-dependent lipogenesis. Therefore, in the current study, we investigated whether JMJD2B plays a role in the LXRα-mediated stimulation of lipogenesis, besides the promotion of fatty acid uptake via upregulation of PPARγ2 expression. Here, we provide evidence that JMJD2B is an epigenetic co-activator of LXRα, and functions to stimulate LXRα-dependent lipogenesis.
3. Discussion
Previously, we reported that JMJD2B enhances PPARγ2 expression by removing the repressive histone marks H3K9me2 and H3K9me3 in the promoter of PPARγ2, stimulating the expression of
PPARγ2 and its steatosis target genes such as
CD36 and
FABP, resulting in the development of hepatic steatosis [
15]. In the current study, we speculated that JMJD2B might also play an epigenetic role in LXRα-activated lipogenesis, leading to the development of NAFLD. Our current data demonstrate that JMJD2B induces LXRα-dependent lipogenesis by removing repressive histone marks H3K9me2 and H3K9me3 near LXREs of lipogenic gene promoters. These results suggest that JMJD2B is an epigenetic coactivator of LXRα to stimulate LXRα-dependent lipogenic genes.
The earliest stage of NAFLD is hepatic steatosis, which reflects TG accumulation in hepatocytes as a result of increased de novo lipogenesis and increased uptake of free fatty acids that exceeds the rate of fatty acid oxidation, VLDL secretion, and lipophagy [
2]. Recently, several studies have reported a link between the regulation of hepatic steatosis and histone modifications [
15,
16,
17,
18]. Histone deacetylase 3 (HDAC3) is an epigenetic regulator associated with hepatic steatosis [
16,
17]. HDAC3 is recruited to the promoter of PPARγ2 by retinoic acid receptor-related orphan receptor-α and represses PPARγ2 expression by deacetylation, thereby downregulating the expression of its target fatty acid uptake genes and preventing hepatic steatosis [
16]. In addition, HDAC3 is also co-recruited with prospero-related homeobox 1 protein to lipid metabolism genes by hepatocyte nuclear factor 4α and downregulates the expression of genes involved in TG synthesis and lipolysis [
17]. A recent study also demonstrated that histone H3K4 methyltransferase MLL3/4 is recruited to the PPARγ responsible element of PPARγ2 and its target steatosis genes, stimulating their expression and resulting in hepatic steatosis [
18]. Furthermore, our previous study demonstrated that the histone H3K9 demethylase JMJD2B stimulates the expression of PPARγ2 and its target genes, resulting in hepatic steatosis [
15]. These reports have focused on the development of hepatic steatosis by the epigenetic regulation of fatty acid uptake-related genes such as
PPARγ2 and
CD36 by histone modification enzymes.
In addition to increased fatty acid uptake into hepatocytes, DNL also contributes significantly to TG accumulation in the pathogenesis of NAFLD [
2]. Recently, it has been reported that the H3K9me2 demethylase, JMJD1C, is recruited to lipogenic promoter regions by USF-1 and demethylates H3K9me2, thus stimulating lipogenic gene expression [
19]. This suggests that JMJD1C plays an important epigenetic regulatory role in the activation of lipogenesis. DNL occurs through the activation of SREBP1, ChREBP, and LXRα, which stimulate the expression of lipogenic genes, including
FAS,
ACC, and
SCD1 [
2]. Among the lipogenic transcription factors, LXRα enhances SREBP1c expression by binding to the LXRE on its gene promoter, which subsequently induces the expression of lipogenic genes [
3]. In addition, LXRα also stimulates the expression of lipogenic genes directly through binding to LXREs on their promoters. Thus, LXRα plays a critical role in lipogenesis [
4]. The expression of LXRα and its downstream lipogenic genes are enhanced in liver biopsies from NAFLD patients. Thus, understanding the regulation of LXRα-dependent lipogenesis could provide an important therapeutic strategy against NAFLD.
Recently, the epigenetic regulation of LXRα-dependent lipogenesis by histone acetylation has been reported [
20,
21]. Histone deacetylase 5 (HDAC5) interacts with LXRα and inhibits its transcriptional activity by deacetylation at LXREs, leading to the reduction of LXRα-dependent lipogenesis [
20]. HDAC5 expression level was reduced in HFD-fed obese mice, indicating that HDAC5 downregulation might contribute to the development of NAFLD. Furthermore, HDAC3 also downregulates the expression of LXRα in hepatic cells [
21]. Accordingly, HDAC5 and HDAC3 can act as co-repressors of LXRα and are candidate therapeutic targets for the treatment of NAFLD. However, the epigenetic regulation of LXRα-dependent lipogenesis by histone methylation is poorly understood. In the current study, we investigated the epigenetic role of the histone demethylase JMJD2B in LXRα-dependent lipogenesis.
We observed that JMJD2B expression was enhanced in LXRα agonist-treated mice and HepG2 cells, as well as in HFD-fed mice and palmitate-treated HepG2 cells, concomitant with upregulated expression of LXRα-activated lipogenic genes, suggesting that JMJD2B is involved in LXRα-dependent lipogenesis. To explore whether JMJD2B participates in LXRα-dependent lipogenesis, we performed a loss-of-function study in HepG2 cells. Knockdown of JMJD2B using siRNA significantly reduced LXRα agonist-induced expression of LXRα and its target lipogenic genes, including SREBF1, FAS, ACC, and SCD1, suggesting that JMJD2B plays a role in LXRα-dependent lipogenesis. We then assessed the direct effects of JMJD2B on LXRα-mediated lipogenesis in HepG2 cells and mice. Adenovirus-mediated overexpression of JMJD2B in HepG2 cells enhanced the expression of LXRα and its target lipogenic genes, concomitant with increased intracellular TG accumulation. Furthermore, in vivo overexpression of JMJD2B using adenovirus also stimulated the expression of LXRα-dependent lipogenic genes and resulted in hepatic steatosis in HFD-fed mice. Taken together, these results indicate that JMJD2B stimulates LXRα-dependent lipogenesis, contributing to the development of hepatic steatosis.
JMJD2B is a histone demethylase responsible for converting the repressive histone marks H3K9me2 and H3K9me3 into H3K9me [
8]. To characterize the mechanism by which JMJD2B stimulates LXRα-dependent lipogenesis, we investigated whether JMJD2B affects the enrichment of histone H3K9me2/3 near the LXRE on
SREBF1 promoter. To this end, we performed chromatin immunoprecipitation experiments in JMJD2B-overexpressing HepG2 cells. ChIP-PCR revealed that overexpression of JMJD2B increased both JMJD2B and LXRα enrichment at the LXRE of
SREBF1, indicating that JMJD2B is recruited to the LXRE region together with LXRα. Consistently, overexpression of JMJD2B reduced H3K9me2 and H3K9me3 enrichment in the same location, indicating that JMJD2B removes the repressive histone marks H3K9me2 and H3K9me3 at LXREs of the
SREBF1 promoter. These results were consistent with those in LXRα agonist-treated HepG2 cells, which showed an increased enrichment of JMJD2B and LXRα in the vicinity of LXREs on the
SREBF1 promoter coupled with reduced levels of histone H3K9me2 and H3K9me3. These results suggest that the activation of LXRα induces the recruitment of JMJD2B to the LXRE of LXRα-dependent lipogenic genes, which subsequently leads to the removal of repressive histone marks H3K9me2 and H3K9me3 and results in the stimulation of LXRα-dependent lipogenesis.
Histone-modification enzymes including histone methyltransferases (or histone demethylases) and histone acetylases (histone deacetylases) are recruited to gene promoters by DNA-binding transcription factors or coactivators, and subsequently regulate gene expression through chromatin modification [
22]. A previous study demonstrated that the H3K4 methyltransferase MLL1 was recruited to the LXRE on the
FAS promoter by activating the signal cointegrator, and thereby upregulated
FAS expression [
22]. Thus, in the current study, we attempted to characterize how JMJD2B was recruited to LXRE. We speculated that JMJD2B was recruited to the LXRE region through a direct interaction with LXRE-bound LXRα. Therefore, we performed a co-immunoprecipitation assay in HepG2 cells infected with Ad-JMJD2B and incubated in the presence or absence of T0901378. Coimmunoprecipitation assay revealed an interaction between JMJD2B and LXRα in only both Ad-JMJD2B-infected and T09013178-treated HepG2 cells, suggesting that JMJD2B could be recruited to LXRα-activated lipogenic promoters by interacting with ligand-activated LXRα on LXRE, thus stimulating LXRα-dependent lipogenic genes by removing repressive H3K9me2/me3 marks around the LXRE.
The activation of LXRα is dependent on chromatin modifications, providing more insight into the regulation of LXRα-dependent gene expression. Activation of LXRα facilitates histone modifications at the LXRE of the LXRα target promoter genes by recruiting histone modification enzymes via several co-regulators, inducing chromatin remodeling [
23]. LXRα significantly increases H3 and H4 acetylation, H3-S10 phosphorylation, and H3K4 methylation at the LXRE of
FAS [
23]. As mentioned previously, LXRα activation recruits H3K4 methyltransferases MLL1 to the LXRE of
FAS and enhances H3K4 trimethylation (H3K4me3) [
22], enhancing the expression of LXRα-dependent lipogenic genes. Our current data also suggest that LXRα activation recruits JMJD2B to the LXRE of
SREBF1 and reduces H3K9me2 and H3K9me3 marks at the LXRE of LXRα-dependent lipogenic genes. This suggests that LXRα activation induces JMJD2B-mediated removal of H3K9me2/me3 at LXREs and stimulates LXRα-dependent lipogenic genes.
Finally, we confirmed the inducible in vivo effects of JMJD2B on LXRα-dependent lipogenesis in mice infected with an Ad-JMJD2B. Overexpression of JMJD2B using recombinant Ad-JMJD2B induced hepatic TG accumulation, indicating that JMJD2B can promote the development of hepatic steatosis. The hepatic mRNA levels of NR1H3 and its target lipogenic genes were significantly increased in Ad-JMJD2B-injected mice, consistent with the results in JMJD2B-overexpressed HepG2 cells. Taken together, these results suggest that JMJD2B stimulates LXRα-dependent lipogenesis, leading to the development of hepatic steatosis. Thus, JMJD2B could play an epigenetic regulatory role in LXRα-activated lipogenesis, and its overexpression could contribute to the development of hepatic steatosis.
4. Materials and Methods
4.1. Reagents
Dulbecco’s modified Eagle’s medium (DMEM), penicillin–streptomycin, and fetal bovine serum (FBS) were obtained from HyClone Laboratories Inc. (Logan, UT, USA). Antibodies against LXRα, H3K9me2, and H3K9me3 were purchased from Millipore (Billerica, MA, USA). Antibodies against JMJD2B, FAS, SREBP1c, and β-actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Palmitate and T0901317 were purchased from Sigma–Aldrich (St. Louis, MO, USA).
4.2. Cell Culture and Treatment with T0901317 or Palmitate
The human hepatocellular carcinoma cell line, HepG2, was obtained from the American Type Culture Collection (Manassas, VA, USA). The HepG2 cells were cultured in DMEM supplemented with 10.0% heat-inactivated FBS and 100 U/mL penicillin–streptomycin at 37 °C in a humidified atmosphere with 5% CO2. Subsequently, the cells were seeded on plates and treated with 10 μM T0901317 and 100 μM palmitate for 24 h.
4.3. Triglyceride (TG) Measurement
TG levels from HepG2 cell lysates and liver tissues were determined as described previously [
15].
4.4. Transfection of HepG2 Cells with siRNAs
To deplete JMJD2B, a duplex of siRNA targeting JMJD2B (sense: 5′-CCAGUUCAGU AUCAAUUAAAGCCCG-3′, antisense: 5′-CGGGCUUUAAUUGAUACUGAACUGGAG-3′) was designed and synthesized by Integrated DNA Technologies (Coralville, IA, USA). HepG2 cells were transfected with the siRNAs using InterferinTM transfection reagent (Polyplus-Transfection Inc., New York, NY, USA).
4.5. Infection of HepG2 Cells with Recombinant Adenovirus
Adenovirus vectors encoding green fluorescent protein (GFP) or JMJD2B (Ad-GFP or Ad-JMJD2B) were purchased from Vector Biolabs (Malvern, PA, USA). HepG2 cells were infected with Ad-GFP or Ad-JMJD2B at a multiplicity of infection (MOI) ranging from 1 to 100 PFU/cell. Cells were washed and fresh medium was added. At 48 h post infection, the cells were harvested and analyzed.
4.6. Total RNA Preparation and Quantitative Real-Time Polymerase Chain Reaction (qPCR)
Total RNA was extracted from HepG2 cell lysates or liver tissues using TRIZOL
® reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. cDNA was generated from 1 μg of total RNA using the GoScript™ Reverse Transcription System (Promega, Madison, WI, USA) in accordance with the manufacturer’s protocol. Quantitative real-time PCR was performed using a SYBR green premixed Taq reaction mixture with gene-specific primers. Gene-specific primers used in this study are listed in
Table S1.
4.7. Western Blot Analysis
Equal amounts of protein (20 μg/lane) from HepG2 cell lysates or liver tissues were resolved by 10% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA, USA). The membranes were blocked in 5% non-fat skim milk and probed with primary antibodies. After washing with Tween 20/Tris-buffered saline (T-TBS), membranes were incubated with a horseradish peroxidase-conjugated secondary antibody (1:1000) at room temperature for 1 h. Membranes were then washed three times with T-TBS, and proteins were detected using an enhanced chemiluminescence (ECL) Western Blot Detection Kit (Amersham, Uppsala, Sweden).
4.8. Chromatin Immunoprecipitation (ChIP)-qPCR
HepG2 cells were fixed with 1% formaldehyde at room temperature for 10 min. The crosslinked chromatin was sheared by sonication into 400 bp fragments using a Bioruptor Sonicator (Diagenode, Denville, NJ, USA). Samples were immunoprecipitated using 1–2 μg antibodies against JMJD2B, and LXRα, H3K9me2, and H3K9me3, or a nonspecific IgG control in the presence of secondary antibody conjugated to Dynabeads (Invitrogen, Carlsbad, CA, USA). Purified DNA was subjected to qPCR using the following primers: SREBP1c LXRE region sense: 5′-GTAAACGGAGGGTTGGAGC-3′, SREBP1c LXRE region antisense: 5′-CTGAATGGGGTTGGGGTTA-3′. ChIP data were normalized to those of the control IgG and expressed as a percentage of the input.
4.9. Luciferase Reporter Assay
The luciferase reporters, 3XLXRE-Luc, SREBF1 promoter-Luc, and FAS promoter-Luc, were gifted by Prof. Lee (Seoul National University, Seoul, Korea). The human SREBF1 promoter from −1564 to +1 relative to the transcription initiation site, and mouse FAS promoter from −1594 to +65 were inserted into pGL3 basic luciferase vector (Promega), respectively. The expression vectors for human JMJD2B and LXRα were obtained from Addgene (La Jolla, CA, USA). The transient transfection of reporters and expression vectors was performed using transfection reagents jetPRIME (Polyplus-Transfection Inc., New York, NY, USA). Luciferase activity was measured with a luminometer and normalized to that of galactosidase.
4.10. Co-Immunoprecipitation (CoIP) Assay
HepG2 cells were infected with adenovirus expressing JMJD2B (Ad-JMJD2B) and then incubated in the presence or absence of T0901317 for 24 h. The cells were harvested and lysed in lysis buffer (25 mM Tris-HCL pH7.4, 150 mM NaCl, 1% NP-40, 1 mM EDTA, 5% glycerol) containing 0.5 mM PMSF and 1 × protease inhibitor (TransLab Biosciences, Daejon, Korea) for 30 min on ice. The lysates were then centrifuged at 13,000 rpm for 30 min at 4 °C. Amounts of 500 μg protein determined using the Bradford assay. Samples were incubated with antibody against JMJD2B (Santa Cruz, CA, USA) or nonspecific IgG control for overnight at 4 °C via gentle rocking. Protein A/G Plus-Agarose beads (Santa Cruz, CA, USA) were added to capture the immune complexes (protein antibody beads) via mixing for 1 h at 4 °C on rotator. Beads were washed three times in wash buffer (25 mM Tris-HCL pH 7.4, 150 mM NaCl, 1% NP-40, 1 mM EDTA, 5% glycerol) with 5 min rotation in between. The proteins were eluted from the beads with 1× SDS loading buffer at 95 °C for 5 min. Co-immunoprecipitated proteins were analyzed by immunoblotting with LXRα antibody.
4.11. Animal Experiments
To make a mouse model of fatty liver disease, C57BL/6J mice (8 weeks of age) were treated with T0901317 daily by oral gavage for 5 days or were fed an HFD for 12 weeks. Furthermore, to overexpress JMJD2B in mice, C57BL/6 mice (8 weeks of age) were injected with a total of 1 × 109 PFU recombinant adenovirus (Ad-GFP or Ad-JMJD2B) via tail vein injection. After injection, adenovirus-injected mice were fed an HFD for 2 weeks. At the end of the treatment period, the mice were sacrificed and the liver was immediately removed and frozen at −80 °C. All animal experiments were approved by Pusan National University Institutional Animal Care and Use Committee in accordance with the established ethical and scientific care procedures (PNU-2017-1483).
4.12. Statistical Analysis
The data shown in this study are expressed as mean ± SD. The data were analyzed using one-way ANOVA, and the differences between means were determined using the Tukey–Kramer post-hoc test. Values were considered statistically significant at p < 0.05.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









