Amyotrophic Lateral Sclerosis Is Accompanied by Protein Derangements in the Olfactory Bulb-Tract Axis

 , ,
, ,  , and
, and 
Abstract
:1. Introduction
2. Results and Discussion
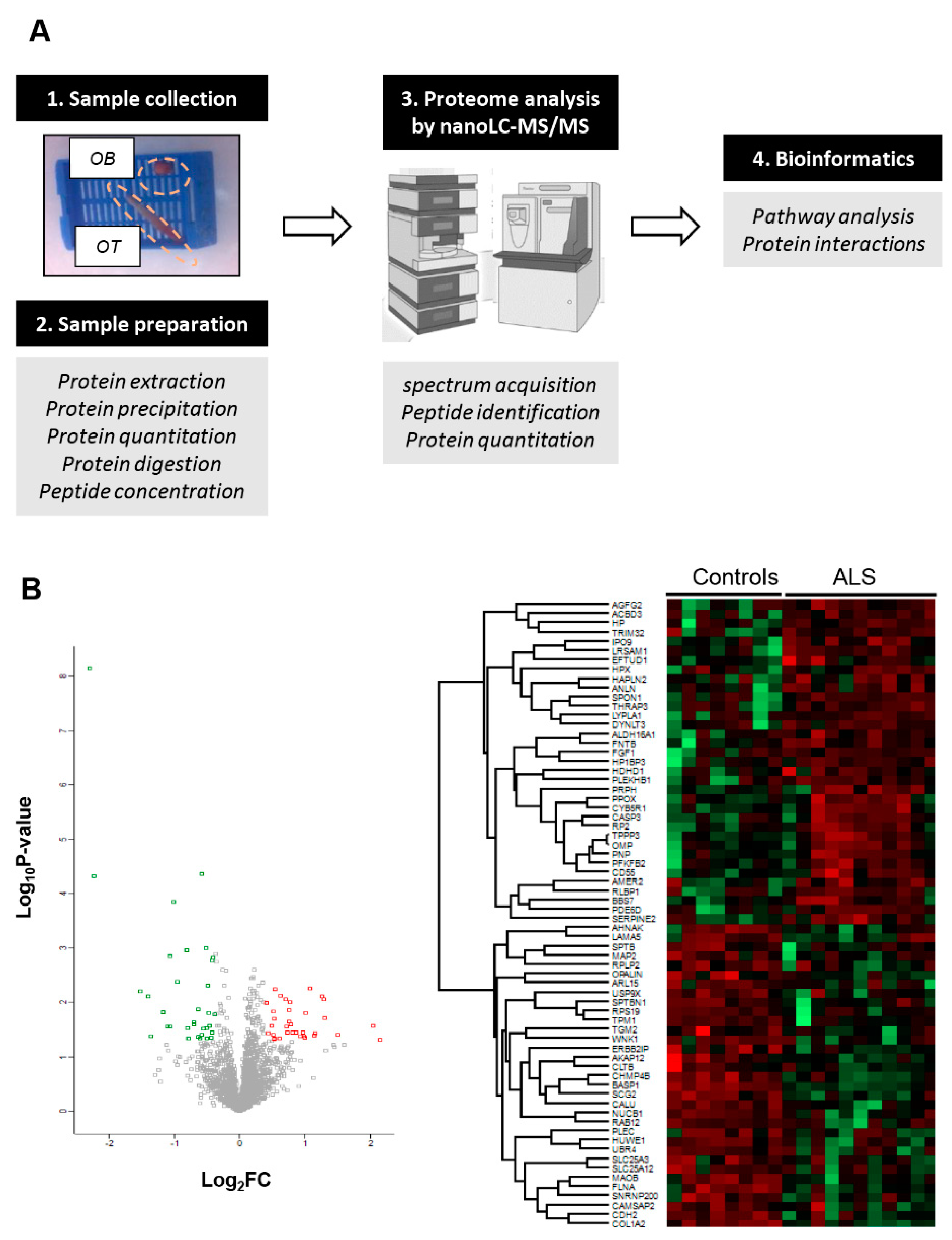
2.1. OB Proteome-Wide Analysis in Human ALS
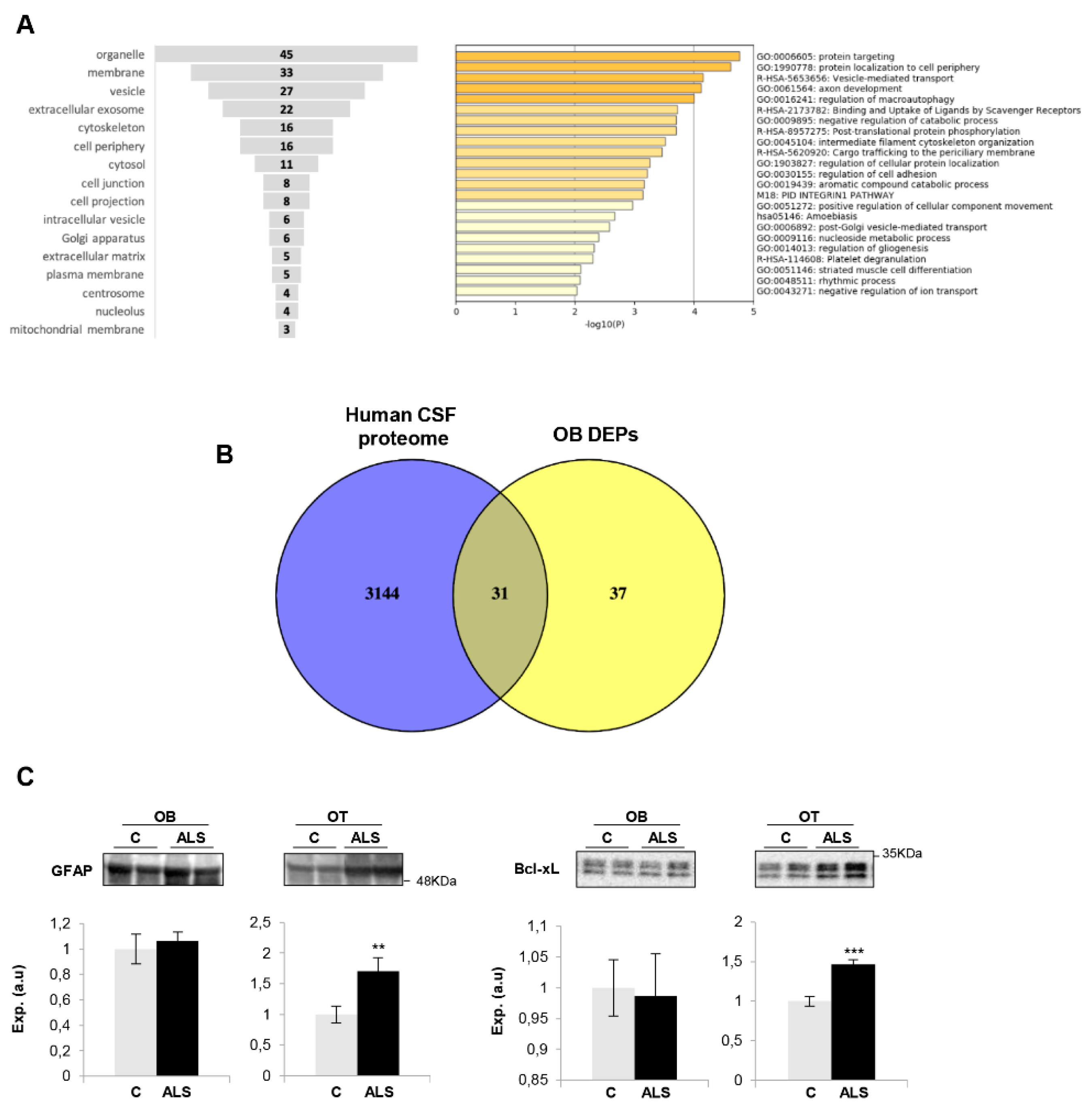
2.2. Functional Analysis for the Differential Ob Proteome Detected in ALS
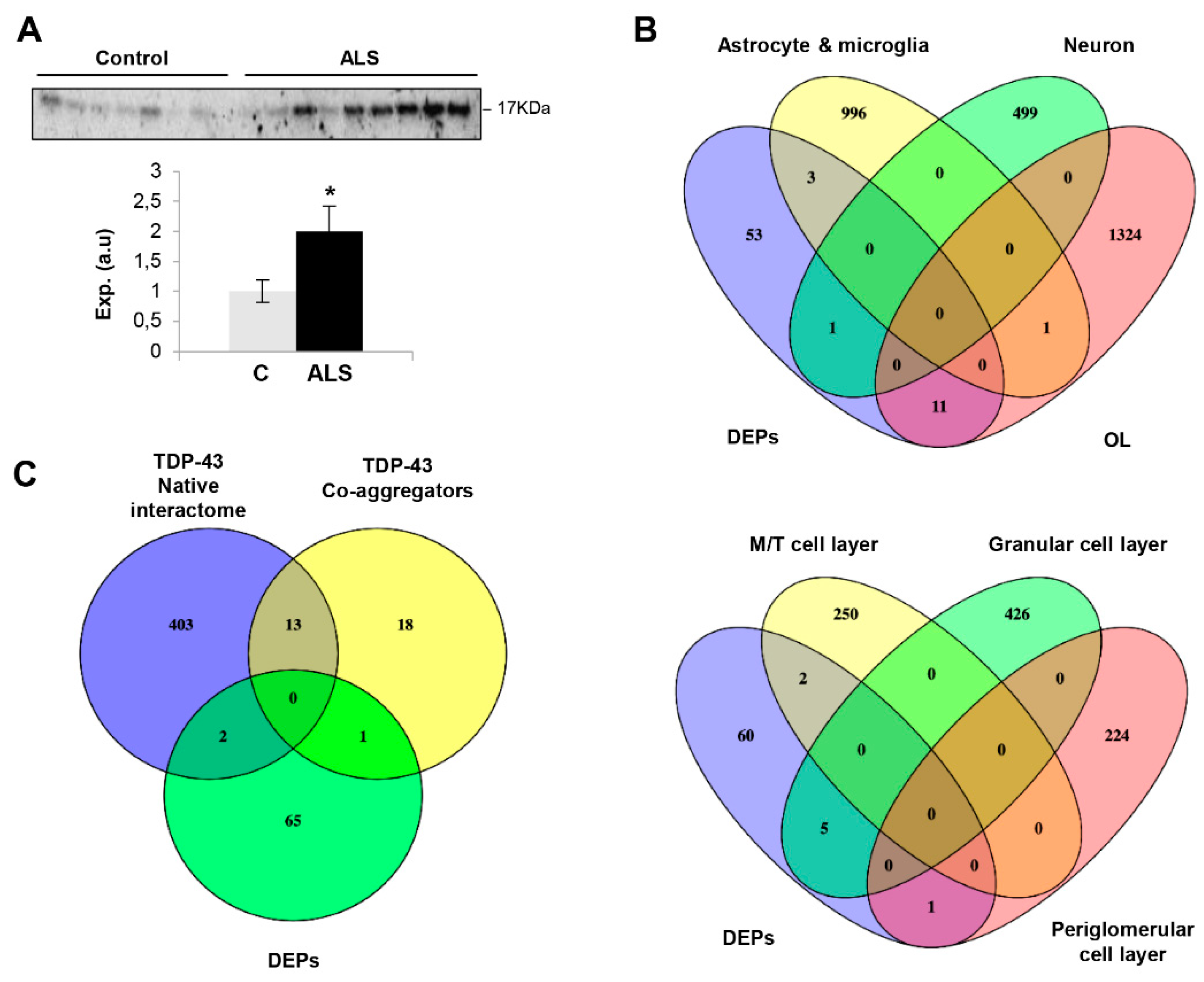
2.3. Imbalance in Survival Pathways Across the Olfactory Bulb-Tract Axis in ALS
3. Materials and Methods
3.1. Materials
3.2. Human Samples
3.3. Sample Preparation for Proteomic Analysis
3.4. Label Free LC-MS/MS
3.5. Protein Identification and Quantification
3.6. Bioinformatics
3.7. Western-Blotting
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALS | Amyotrophic lateral sclerosis |
| CAMK II | Calmodulin-dependent protein kinase II |
| ERK | Extracellular signal-regulated kinase |
| FDR | False discovery rate |
| MEK | Mitogen-activated protein kinase kinase |
| OB/OT | Olfactory bulb/olfactory tract |
| OMP | Olfactory marker protein |
| p38 MAPK | p38 mitogen-activated protein kinase |
| PKA | Protein kinase A |
| PKC | Protein kinase C |
| PMI | Post-mortem interval |
| SAPK/JNK | (stress-activated protein kinase/Jun-amino terminal kinase) |
| SEK1 | Mitogen-activated protein kinase Kinase 4 |
| TDP43 | TAR DNA-binding protein 43 |
References
- Brown, R.H., Jr.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 1602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiò, A.; Logroscino, G.; Traynor, B.; Collins, J.; Simeone, J.; Goldstein, L.; White, L. Global Epidemiology of Amyotrophic Lateral Sclerosis: A Systematic Review of the Published Literature. Neuroepidemiology 2013, 41, 118–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed]
- Swinnen, B.; Robberecht, W. The phenotypic variability of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2014, 10, 661–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renton, A.E.; Chio, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef]
- Andersen, P.M.; Al-Chalabi, A. Clinical genetics of amyotrophic lateral sclerosis: What do we really know? Nat. Rev. Neurol. 2011, 7, 603–615. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [Green Version]
- Yerbury, J.J.; Farrawell, N.E.; McAlary, L. Proteome Homeostasis Dysfunction: A Unifying Principle in ALS Pathogenesis. Trends Neurosci. 2020, 43, 274–284. [Google Scholar] [CrossRef]
- Robberecht, W.; Philips, T. The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci 2013, 14, 248–264. [Google Scholar] [CrossRef]
- Boeynaems, S.; Bogaert, E.; Van Damme, P.; Van Den Bosch, L. Inside out: The role of nucleocytoplasmic transport in ALS and FTLD. Acta Neuropathol. 2016, 132, 159–173. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.R.; King, O.D.; Shorter, J.; Gitler, A.D. Stress granules as crucibles of ALS pathogenesis. J. Cell Biol. 2013, 201, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, O.M.; Ghasemi, M.; Brown, R.H., Jr. Emerging mechanisms of molecular pathology in ALS. J. Clin. Invest. 2015, 125, 1767–1779. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; Calvo, A.; Chio, A.; Colville, S.; Ellis, C.M.; Hardiman, O.; Heverin, M.; Howard, R.S.; Huisman, M.H.B.; Keren, N.; et al. Analysis of amyotrophic lateral sclerosis as a multistep process: A population-based modelling study. Lancet. Neurol. 2014, 13, 1108–1113. [Google Scholar] [CrossRef]
- Burrell, J.R.; Kiernan, M.C.; Vucic, S.; Hodges, J.R. Motor neuron dysfunction in frontotemporal dementia. Brain 2011, 134, 2582–2594. [Google Scholar] [CrossRef] [Green Version]
- Abramzon, Y.A.; Fratta, P.; Traynor, B.J.; Chia, R. The Overlapping Genetics of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Front. Neurosci. 2020, 14, 42. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Almeida, S.; Lopez-Gonzalez, R. Dysregulated molecular pathways in amyotrophic lateral sclerosis–frontotemporal dementia spectrum disorder. EMBO J. 2017, 36, 2931–2950. [Google Scholar] [CrossRef] [PubMed]
- Strong, M.J.; Abrahams, S.; Goldstein, L.H.; Woolley, S.; McLaughlin, P.; Snowden, J.; Mioshi, E.; Roberts-South, A.; Benatar, M.; Hortobágyi, T.; et al. Amyotrophic lateral sclerosis—Frontotemporal spectrum disorder (ALS-FTSD): Revised diagnostic criteria. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 153–174. [Google Scholar] [CrossRef]
- Elian, M. Olfactory impairment in motor neuron disease: A pilot study. J. Neurol. Neurosurg. Psychiatry 1991, 54, 927–928. [Google Scholar] [CrossRef] [Green Version]
- Hawkes, C.H.; Shephard, B.C.; Geddes, J.F.; Body, G.D.; Martin, J.E. Olfactory disorder in motor neuron disease. Exp. Neurol. 1998, 150, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Doty, R.L. Olfactory dysfunction in neurodegenerative diseases: Is there a common pathological substrate? Lancet Neurol. 2017, 16, 478–488. [Google Scholar] [CrossRef]
- Viguera, C.; Wang, J.; Mosmiller, E.; Cerezo, A.; Maragakis, N.J. Olfactory dysfunction in amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 2018, 5, 976–981. [Google Scholar] [CrossRef]
- Pilotto, A.; Rossi, F.; Rinaldi, F.; Compostella, S.; Cosseddu, M.; Borroni, B.; Filosto, M.; Padovani, A. Exploring Olfactory Function and Its Relation with Behavioral and Cognitive Impairment in Amyotrophic Lateral Sclerosis Patients: A Cross-Sectional Study. Neurodegener. Dis. 2016, 16, 411–416. [Google Scholar] [CrossRef]
- Gunther, R.; Schrempf, W.; Hahner, A.; Hummel, T.; Wolz, M.; Storch, A.; Hermann, A. Impairment in Respiratory Function Contributes to Olfactory Impairment in Amyotrophic Lateral Sclerosis. Front. Neurol. 2018, 9, 79. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Escudero, V.; Rosales, M.; Munoz, J.L.; Scola, E.; Medina, J.; Khalique, H.; Garaulet, G.; Rodriguez, A.; Lim, F. Patient-derived olfactory mucosa for study of the non-neuronal contribution to amyotrophic lateral sclerosis pathology. J. Cell Mol. Med. 2015, 19, 1284–1295. [Google Scholar] [CrossRef] [Green Version]
- Takeda, T.; Iijima, M.; Uchihara, T.; Ohashi, T.; Seilhean, D.; Duyckaerts, C.; Uchiyama, S. TDP-43 Pathology Progression Along the Olfactory Pathway as a Possible Substrate for Olfactory Impairment in Amyotrophic Lateral Sclerosis. J. Neuropathol. Exp. Neurol. 2015, 74, 547–556. [Google Scholar] [CrossRef] [Green Version]
- Robertson, J.; Sanelli, T.; Xiao, S.; Yang, W.; Horne, P.; Hammond, R.; Pioro, E.P.; Strong, M.J. Lack of TDP-43 abnormalities in mutant SOD1 transgenic mice shows disparity with ALS. Neurosci. Lett. 2007, 420, 128–132. [Google Scholar] [CrossRef]
- Ringer, C.; Tune, S.; Bertoune, M.A.; Schwarzbach, H.; Tsujikawa, K.; Weihe, E.; Schutz, B. Disruption of calcitonin gene-related peptide signaling accelerates muscle denervation and dampens cytotoxic neuroinflammation in SOD1 mutant mice. Cell Mol. Life Sci. 2017, 74, 339–358. [Google Scholar] [CrossRef]
- Ringer, C.; Weihe, E.; Schutz, B. SOD1G93A Mutant Mice Develop a Neuroinflammation-Independent Dendropathy in Excitatory Neuronal Subsets of the Olfactory Bulb and Retina. J. Neuropathol. Exp. Neurol. 2017, 76, 769–778. [Google Scholar] [CrossRef] [Green Version]
- Munger, S.D.; Leinders-Zufall, T.; Zufall, F. Subsystem organization of the mammalian sense of smell. Annu Rev. Physiol. 2009, 71, 115–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brettschneider, J.; Del Tredici, K.; Toledo, J.B.; Robinson, J.L.; Irwin, D.J.; Grossman, M.; Suh, E.; Van Deerlin, V.M.; Wood, E.M.; Baek, Y.; et al. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann. Neurol. 2013, 74, 20–38. [Google Scholar] [CrossRef]
- Fatima, M.; Tan, R.; Halliday, G.M.; Kril, J.J. Spread of pathology in amyotrophic lateral sclerosis: Assessment of phosphorylated TDP-43 along axonal pathways. Acta Neuropathol. Commun. 2015, 3, 47. [Google Scholar] [CrossRef]
- Hedl, T.J.; San Gil, R.; Cheng, F.; Rayner, S.L.; Davidson, J.M.; De Luca, A.; Villalva, M.D.; Ecroyd, H.; Walker, A.K.; Lee, A. Proteomics Approaches for Biomarker and Drug Target Discovery in ALS and FTD. Front. Neurosci. 2019, 13, 548. [Google Scholar] [CrossRef] [Green Version]
- Iridoy, M.O.; Zubiri, I.; Zelaya, M.V.; Martinez, L.; Ausin, K.; Lachen-Montes, M.; Santamaria, E.; Fernandez-Irigoyen, J.; Jerico, I. Neuroanatomical Quantitative Proteomics Reveals Common Pathogenic Biological Routes between Amyotrophic Lateral Sclerosis (ALS) and Frontotemporal Dementia (FTD). Int. J. Mol. Sci. 2018, 20, 4. [Google Scholar] [CrossRef] [Green Version]
- Smethurst, P.; Sidle, K.C.; Hardy, J. Review: Prion-like mechanisms of transactive response DNA binding protein of 43 kDa (TDP-43) in amyotrophic lateral sclerosis (ALS). Neuropathol. Appl. Neurobiol. 2015, 41, 578–597. [Google Scholar] [CrossRef] [Green Version]
- Polymenidou, M.; Cleveland, D.W. The seeds of neurodegeneration: Prion-like spreading in ALS. Cell 2011, 147, 498–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rey, N.L.; Wesson, D.W.; Brundin, P. The olfactory bulb as the entry site for prion-like propagation in neurodegenerative diseases. Neurobiol. Dis. 2018, 109, 226–248. [Google Scholar] [CrossRef]
- Fernandez-Irigoyen, J.; Santamaria, E. Olfactory proteotyping: Towards the enlightenment of the neurodegeneration. Neural Regen Res. 2019, 14, 979–981. [Google Scholar] [CrossRef]
- Lachen-Montes, M.; Gonzalez-Morales, A.; Iloro, I.; Elortza, F.; Ferrer, I.; Gveric, D.; Fernandez-Irigoyen, J.; Santamaria, E. Unveiling the olfactory proteostatic disarrangement in Parkinson’s disease by proteome-wide profiling. Neurobiol. Aging 2019, 73, 123–134. [Google Scholar] [CrossRef]
- Lachen-Montes, M.; Gonzalez-Morales, A.; Zelaya, M.V.; Perez-Valderrama, E.; Ausin, K.; Ferrer, I.; Fernandez-Irigoyen, J.; Santamaria, E. Olfactory bulb neuroproteomics reveals a chronological perturbation of survival routes and a disruption of prohibitin complex during Alzheimer’s disease progression. Sci. Rep. 2017, 7, 9115. [Google Scholar] [CrossRef]
- Acquadro, E.; Caron, I.; Tortarolo, M.; Bucci, E.M.; Bendotti, C.; Corpillo, D. Human SOD1-G93A specific distribution evidenced in murine brain of a transgenic model for amyotrophic lateral sclerosis by MALDI imaging mass spectrometry. J. Proteome Res. 2014, 13, 1800–1809. [Google Scholar] [CrossRef] [PubMed]
- Togawa, J.; Ohi, T.; Yuan, J.H.; Takashima, H.; Furuya, H.; Takechi, S.; Fujitake, J.; Hayashi, S.; Ishiura, H.; Naruse, H.; et al. Atypical Familial Amyotrophic Lateral Sclerosis with Slowly Progressing Lower Extremities-predominant Late-onset Muscular Weakness and Atrophy. Intern. Med. 2019, 58, 1851–1858. [Google Scholar] [CrossRef] [Green Version]
- Oberstadt, M.; Classen, J.; Arendt, T.; Holzer, M. TDP-43 and Cytoskeletal Proteins in ALS. Mol. Neurobiol. 2018, 55, 3143–3151. [Google Scholar] [CrossRef]
- Oeckl, P.; Weydt, P.; Thal, D.R.; Weishaupt, J.H.; Ludolph, A.C.; Otto, M. Proteomics in cerebrospinal fluid and spinal cord suggests UCHL1, MAP2 and GPNMB as biomarkers and underpins importance of transcriptional pathways in amyotrophic lateral sclerosis. Acta Neuropathol. 2020, 139, 119–134. [Google Scholar] [CrossRef] [PubMed]
- Ekblom, J.; Aquilonius, S.M.; Jossan, S.S. Differential increases in catecholamine metabolizing enzymes in amyotrophic lateral sclerosis. Exp. Neurol. 1993, 123, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Pasinelli, P.; Houseweart, M.K.; Brown, R.H., Jr.; Cleveland, D.W. Caspase-1 and -3 are sequentially activated in motor neuron death in Cu,Zn superoxide dismutase-mediated familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2000, 97, 13901–13906. [Google Scholar] [CrossRef] [Green Version]
- Chou, S.M.; Taniguchi, A.; Wang, H.S.; Festoff, B.W. Serpin=serine protease-like complexes within neurofilament conglomerates of motoneurons in amyotrophic lateral sclerosis. J. Neurol. Sci. 1998, 160 (Suppl. 1), S73–S79. [Google Scholar] [CrossRef]
- Marques, R.F.; Engler, J.B.; Kuchler, K.; Jones, R.A.; Lingner, T.; Salinas, G.; Gillingwater, T.H.; Friese, M.A.; Duncan, K.E. Motor neuron translatome reveals deregulation of SYNGR4 and PLEKHB1 in mutant TDP-43 amyotrophic lateral sclerosis models. Hum. Mol. Genet. 2020. [Google Scholar] [CrossRef]
- Bahia El Idrissi, N.; Bosch, S.; Ramaglia, V.; Aronica, E.; Baas, F.; Troost, D. Complement activation at the motor end-plates in amyotrophic lateral sclerosis. J. Neuroinflamm. 2016, 13, 72. [Google Scholar] [CrossRef] [Green Version]
- Reisert, J.; Yau, K.W.; Margolis, F.L. Olfactory marker protein modulates the cAMP kinetics of the odour-induced response in cilia of mouse olfactory receptor neurons. J. Physiol. 2007, 585, 731–740. [Google Scholar] [CrossRef]
- Buiakova, O.I.; Baker, H.; Scott, J.W.; Farbman, A.; Kream, R.; Grillo, M.; Franzen, L.; Richman, M.; Davis, L.M.; Abbondanzo, S.; et al. Olfactory marker protein (OMP) gene deletion causes altered physiological activity of olfactory sensory neurons. Proc. Natl. Acad. Sci. USA 1996, 93, 9858–9863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakashima, N.; Nakashima, K.; Taura, A.; Takaku-Nakashima, A.; Ohmori, H.; Takano, M. Olfactory marker protein directly buffers cAMP to avoid depolarization-induced silencing of olfactory receptor neurons. Nat. Commun. 2020, 11, 2188. [Google Scholar] [CrossRef]
- Lee, A.C.; He, J.; Ma, M. Olfactory marker protein is critical for functional maturation of olfactory sensory neurons and development of mother preference. J. Neurosci. 2011, 31, 2974–2982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kass, M.D.; Moberly, A.H.; Rosenthal, M.C.; Guang, S.A.; McGann, J.P. Odor-specific, olfactory marker protein-mediated sparsening of primary olfactory input to the brain after odor exposure. J. Neurosci. 2013, 33, 6594–6602. [Google Scholar] [CrossRef] [Green Version]
- Albeanu, D.F.; Provost, A.C.; Agarwal, P.; Soucy, E.R.; Zak, J.D.; Murthy, V.N. Olfactory marker protein (OMP) regulates formation and refinement of the olfactory glomerular map. Nat. Commun. 2018, 9, 5073. [Google Scholar] [CrossRef] [Green Version]
- Tepe, B.; Hill, M.C.; Pekarek, B.T.; Hunt, P.J.; Martin, T.J.; Martin, J.F.; Arenkiel, B.R. Single-Cell RNA-Seq of Mouse Olfactory Bulb Reveals Cellular Heterogeneity and Activity-Dependent Molecular Census of Adult-Born Neurons. Cell Rep. 2018, 25, 2689–2703.e2683. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef]
- Ciryam, P.; Lambert-Smith, I.A.; Bean, D.M.; Freer, R.; Cid, F.; Tartaglia, G.G.; Saunders, D.N.; Wilson, M.R.; Oliver, S.G.; Morimoto, R.I.; et al. Spinal motor neuron protein supersaturation patterns are associated with inclusion body formation in ALS. Proc. Natl. Acad. Sci. USA 2017, 114, E3935–E3943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ethell, D.W. Disruption of cerebrospinal fluid flow through the olfactory system may contribute to Alzheimer’s disease pathogenesis. J. Alzheimers Dis. 2014, 41, 1021–1030. [Google Scholar] [CrossRef] [Green Version]
- Lachen-Montes, M.; Fernandez-Irigoyen, J.; Santamaria, E. Deconstructing the molecular architecture of olfactory areas using proteomics. Proteom. Clin. Appl. 2016. [Google Scholar] [CrossRef]
- Lachen-Montes, M.; Gonzalez-Morales, A.; Fernandez-Irigoyen, J.; Santamaria, E. Deployment of Label-Free Quantitative Olfactory Proteomics to Detect Cerebrospinal Fluid Biomarker Candidates in Synucleinopathies. Methods Mol. Biol. 2019, 2044, 273–289. [Google Scholar] [CrossRef]
- Macron, C.; Lavigne, R.; Nunez Galindo, A.; Affolter, M.; Pineau, C.; Dayon, L. Exploration of human cerebrospinal fluid: A large proteome dataset revealed by trapped ion mobility time-of-flight mass spectrometry. Data Brief. 2020, 31, 105704. [Google Scholar] [CrossRef]
- Lee, J.; Kannagi, M.; Ferrante, R.J.; Kowall, N.W.; Ryu, H. Activation of Ets-2 by oxidative stress induces Bcl-xL expression and accounts for glial survival in amyotrophic lateral sclerosis. FASEB J. 2009, 23, 1739–1749. [Google Scholar] [CrossRef]
- Palomo, V.; Nozal, V.; Rojas-Prats, E.; Gil, C.; Martinez, A. Protein kinase inhibitors for amyotrophic lateral sclerosis therapy. Br. J. Pharmacol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lachen-Montes, M.; Gonzalez-Morales, A.; Schvartz, D.; Zelaya, M.V.; Ausin, K.; Fernandez-Irigoyen, J.; Sanchez, J.C.; Santamaria, E. The olfactory bulb proteotype differs across frontotemporal dementia spectrum. J. Proteom. 2019, 201, 37–47. [Google Scholar] [CrossRef]
- Baczyk, M.; Alami, N.O.; Delestree, N.; Martinot, C.; Tang, L.; Commisso, B.; Bayer, D.; Doisne, N.; Frankel, W.; Manuel, M.; et al. Synaptic restoration by cAMP/PKA drives activity-dependent neuroprotection to motoneurons in ALS. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Krieger, C.; Lanius, R.A.; Pelech, S.L.; Shaw, C.A. Amyotrophic lateral sclerosis: The involvement of intracellular Ca2+ and protein kinase C. Trends Pharmacol. Sci. 1996, 17, 114–120. [Google Scholar] [CrossRef]
- Lanuza, M.A.; Santafe, M.M.; Garcia, N.; Besalduch, N.; Tomas, M.; Obis, T.; Priego, M.; Nelson, P.G.; Tomas, J. Protein kinase C isoforms at the neuromuscular junction: Localization and specific roles in neurotransmission and development. J. Anat. 2014, 224, 61–73. [Google Scholar] [CrossRef]
- Camerino, G.M.; Fonzino, A.; Conte, E.; De Bellis, M.; Mele, A.; Liantonio, A.; Tricarico, D.; Tarantino, N.; Dobrowolny, G.; Musaro, A.; et al. Elucidating the Contribution of Skeletal Muscle Ion Channels to Amyotrophic Lateral Sclerosis in search of new therapeutic options. Sci. Rep. 2019, 9, 3185. [Google Scholar] [CrossRef] [Green Version]
- Migheli, A.; Piva, R.; Atzori, C.; Troost, D.; Schiffer, D. c-Jun, JNK/SAPK kinases and transcription factor NF-kappa B are selectively activated in astrocytes, but not motor neurons, in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 1997, 56, 1314–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayala, V.; Granado-Serrano, A.B.; Cacabelos, D.; Naudi, A.; Ilieva, E.V.; Boada, J.; Caraballo-Miralles, V.; Llado, J.; Ferrer, I.; Pamplona, R.; et al. Cell stress induces TDP-43 pathological changes associated with ERK1/2 dysfunction: Implications in ALS. Acta Neuropathol. 2011, 122, 259–270. [Google Scholar] [CrossRef]
- Strong, M.J.; Kesavapany, S.; Pant, H.C. The pathobiology of amyotrophic lateral sclerosis: A proteinopathy? J. Neuropathol. Exp. Neurol. 2005, 64, 649–664. [Google Scholar] [CrossRef] [PubMed]
- Ackerley, S.; Grierson, A.J.; Banner, S.; Perkinton, M.S.; Brownlees, J.; Byers, H.L.; Ward, M.; Thornhill, P.; Hussain, K.; Waby, J.S.; et al. p38alpha stress-activated protein kinase phosphorylates neurofilaments and is associated with neurofilament pathology in amyotrophic lateral sclerosis. Mol. Cell Neurosci. 2004, 26, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.H.; Zhang, H.; Wagey, R.; Krieger, C.; Pelech, S.L. Protein kinase and protein phosphatase expression in amyotrophic lateral sclerosis spinal cord. J. Neurochem. 2003, 85, 432–442. [Google Scholar] [CrossRef]
- Strong, M.J.; Horobágyi, T.; Okamoto, K.; Kato, S. Amyotrophic lateral sclerosis, primary lateral sclerosis and spinal muscular atrophy. In Molecular Pathology of Dementia and Movement Disorders, 2nd ed.; 2011 International Society of Neuropathology; Dickson, D.W., Weller, R.O., Eds.; Blackwell Publish Ltd.: Oxford, UK, 2011; pp. 418–433. [Google Scholar]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Vizcaino, J.A.; Deutsch, E.W.; Wang, R.; Csordas, A.; Reisinger, F.; Rios, D.; Dianes, J.A.; Sun, Z.; Farrah, T.; Bandeira, N.; et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 2014, 32, 223–226. [Google Scholar] [CrossRef]
- Oughtred, R.; Stark, C.; Breitkreutz, B.J.; Rust, J.; Boucher, L.; Chang, C.; Kolas, N.; O’Donnell, L.; Leung, G.; McAdam, R.; et al. The BioGRID interaction database: 2019 update. Nucleic Acids Res. 2019, 47, D529–D541. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein ID | Protein Name | Gene | p Value | FC | Activity/Pathway |
|---|---|---|---|---|---|
| P80723 | Brain acid soluble protein 1 | BASP1 | 0.00 | 0.20 | transcription corepressor activity |
| P08123 | Collagen alpha-2(I) chain | COL1A2 | 0.00 | 0.21 | Integrin Pathway & Collagen trimerization |
| Q96PE5 | Opalin | OPALIN | 0.01 | 0.35 | oligodendrocyte terminal differentiation |
| Q7Z6Z7 | E3 ubiquitin-protein ligase HUWE1 | HUWE1 | 0.01 | 0.38 | proteasomal degradation |
| O15230 | Laminin subunit alpha-5 | LAMA5 | 0.04 | 0.39 | Integrin Pathway & signaling by GPCR |
| Q09666 | Neuroblast differentiation-associated protein AHNAK | AHNAK | 0.02 | 0.44 | Phospholipase-C Pathway |
| F5H7S3 | Tropomyosin alpha-1 chain | TPM1 | 0.03 | 0.47 | cytoskeletal protein binding |
| O43852 | Calumenin | CALU | 0.00 | 0.48 | calcium ion binding |
| Q5T4S7 | E3 ubiquitin-protein ligase UBR4 | UBR4 | 0.03 | 0.48 | ubiquitin-protein transferase activity |
| P13521 | Secretogranin-2 | SCG2 | 0.00 | 0.50 | chemoattractant activity |
| Q02818 | Nucleobindin-1 | NUCB1 | 0.00 | 0.52 | Golgi calcium homeostasis |
| P21333 | Filamin-A | FLNA | 0.00 | 0.57 | crosslink actin filaments |
| P39019 | 40S ribosomal protein S19 | RPS19 | 0.03 | 0.58 | pre-rRNA processing |
| Q6IQ22 | Ras-related protein Rab-12 | RAB12 | 0.05 | 0.58 | Vesicle trafficking |
| O75643 | U5 small nuclear ribonucleoprotein 200 kDa helicase | SNRNP200 | 0.02 | 0.61 | mRNA splicing |
| P11277 | Spectrin beta chain, erythrocytic | SPTB | 0.03 | 0.62 | actin filament binding |
| Q93008 | Probable ubiquitin carboxyl-terminal hydrolase FAF-X | USP9X | 0.01 | 0.64 | deubiquitinase, protein turnover |
| Q9NXU5 | ADP-ribosylation factor-like protein 15 | ARL15 | 0.04 | 0.64 | GTP binding |
| Q15149 | Plectin | PLEC | 0.05 | 0.66 | Cytoskeleton remodeling Neurofilaments |
| F5GWT4 | Serine/threonine-protein kinase WNK1 | WNK1 | 0.04 | 0.67 | regulation of electrolyte homeostasis |
| Q9H444 | Charged multivesicular body protein 4b | CHMP4B | 0.00 | 0.67 | sorting of endocytosed cell-surface receptors |
| Q08AD1 | Calmodulin-regulated spectrin-associated protein 2 | CAMSAP2 | 0.03 | 0.68 | regulator of neuronal polarity |
| Q02952 | A-kinase anchor protein 12 | AKAP12 | 0.00 | 0.70 | subcellular compartmentation of PKA/PKC |
| P21980 | Protein-glutamine gamma-glutamyltransferase 2 | TGM2 | 0.03 | 0.71 | cross-linking and conjugation of polyamines |
| P11137 | Microtubule-associated protein 2 | MAP2 | 0.04 | 0.71 | stabilization of microtubules |
| Q96RT1 | Protein LAP2 | ERBB2IP | 0.00 | 0.72 | Inhibits proinflammatory cytokine secretion |
| Q01082 | Spectrin beta chain, non-erythrocytic 1 | SPTBN1 | 0.02 | 0.72 | movement of the cytoskeleton |
| P05387 | 60S acidic ribosomal protein P2 | RPLP2 | 0.03 | 0.73 | protein synthesis |
| O75746 | Calcium-binding mitochondrial carrier protein Aralar1 | SLC25A12 | 0.04 | 0.74 | exchange of Asp for Glu in the mitochondria |
| P27338 | Amine oxidase [flavin-containing] B | MAOB | 0.00 | 0.75 | metabolism of neuroactive and vasoactive amines |
| Q00325 | Phosphate carrier protein, mitochondrial | SLC25A3 | 0.04 | 0.75 | regulation of mitochondrial permeability |
| P19022 | Cadherin-2 | CDH2 | 0.00 | 0.75 | cell-cell adhesion |
| P09497 | Clathrin light chain B | CLTB | 0.02 | 0.77 | vesicle biogenesis |
| P07093 | Glia-derived nexin | SERPINE2 | 0.01 | 1.33 | endopeptidase inhibitor activity |
| Q08623 | Pseudouridine-5-phosphatase | HDHD1 | 0.04 | 1.35 | pyrimidine nucleoside salvage |
| Q7Z2Z2 | Elongation factor Tu GTP-binding domain-containing protein 1 | EFTUD1 | 0.03 | 1.41 | translational activation of ribosomes |
| P00491 | Purine nucleoside phosphorylase | PNP | 0.01 | 1.43 | nucleoside binding |
| P49356 | Protein farnesyltransferase subunit beta | FNTB | 0.01 | 1.43 | farnesyltransferase activity |
| O60825 | 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 2 | PFKFB2 | 0.04 | 1.44 | Synthesis/degradation of fructose 2,6-bisP |
| Q9H3P7 | Golgi resident protein GCP60 | ACBD3 | 0.02 | 1.44 | maintenance of Golgi structure |
| P50336 | Protoporphyrinogen oxidase | PPOX | 0.05 | 1.45 | heme biosynthesis |
| Q9GZV7 | Hyaluronan and proteoglycan link protein 2 | HAPLN2 | 0.05 | 1.45 | establishment of blood-nerve barrier |
| Q96P70 | Importin-9 | IPO9 | 0.01 | 1.46 | nuclear protein import |
| P02790 | Hemopexin | HPX | 0.04 | 1.53 | heme transport |
| Q9BW30 | Tubulin polymerization-promoting protein family member 3 | TPPP3 | 0.01 | 1.54 | Regulator of microtubule dynamics |
| Q9UHQ9 | NADH-cytochrome b5 reductase 1 | CYB5R1 | 0.01 | 1.62 | desaturation/elongation of fatty acids |
| O75608 | Acyl-protein thioesterase 1 | LYPLA1 | 0.03 | 1.64 | phospholipase activity |
| Q5SSJ5 | Heterochromatin protein 1-binding protein 3 | HP1BP3 | 0.04 | 1.66 | heterochromatin organization |
| Q13049 | E3 ubiquitin-protein ligase TRIM32 | TRIM32 | 0.02 | 1.69 | ubiquitin-protein transferase activity |
| O95081 | Arf-GAP domain and FG repeat-containing protein 2 | AGFG2 | 0.01 | 1.70 | GTPase activator activity |
| Q8IWZ6 | Bardet-Biedl syndrome 7 protein | BBS7 | 0.01 | 1.70 | cilium assembly |
| F5GZH3 | Pleckstrin homology domain-containing family B member 1 | PLEKHB1 | 0.03 | 1.73 | cell differentiation |
| A6NGJ0 | Dynein light chain Tctex-type 3 | DYNLT3 | 0.04 | 1.76 | intracellular retrograde motility of vesicles |
| Q8IZ83 | Aldehyde dehydrogenase family 16 member A1 | ALDH16A1 | 0.04 | 1.82 | oxidoreductase activity |
| Q9NQW6 | Actin-binding protein anillin | ANLN | 0.04 | 1.91 | actomyosin contractile ring assembly |
| P05230 | Fibroblast growth factor 1 | FGF1 | 0.04 | 1.96 | Integrin binding |
| O43924 | GMP-Phosphodiesterase delta | PDE6D | 0.04 | 1.97 | ciliary targeting of farnesylated proteins |
| P42574 | Caspase-3;Caspase-3 subunit p17;Caspase-3 subunit p12 | CASP3 | 0.04 | 1.99 | apoptosis execution |
| Q9HCB6 | Spondin-1 | SPON1 | 0.02 | 2.01 | attachment of spinal cord & sensory neuron cells |
| Q6UWE0 | E3 ubiquitin-protein ligase LRSAM1 | LRSAM1 | 0.01 | 2.12 | ubiquitin-protein transferase activity |
| P12271 | Retinaldehyde-binding protein 1 | RLBP1 | 0.04 | 2.22 | retinoid metabolism |
| Q8N7J2 | APC membrane recruitment protein 2 | AMER2 | 0.04 | 2.23 | Wnt signaling pathway |
| O75695 | Protein XRP2 | RP2 | 0.01 | 2.40 | post-Golgi vesicle-mediated transport |
| H3BLV0 | Complement decay-accelerating factor | CD55 | 0.01 | 2.45 | regulation of the complement cascade |
| Q9Y2W1 | Thyroid hormone receptor-associated protein 3 | THRAP3 | 0.02 | 2.49 | regulation of mRNA splicing & transcription |
| P47874 | Olfactory marker protein | OMP | 0.04 | 2.84 | modulator of the olfactory signal-transduction |
| P00738 | Haptoglobin | HPR | 0.03 | 4.12 | antioxidant activity |
| P41219 | Peripherin | PRPH | 0.05 | 4.46 | Class-III neuronal intermediate filament |
| Groups | Age (years) | Onset | Sex | PMI | Neuropathological Diagnosis | AD Stages | OB Analysis | OT Analysis |
|---|---|---|---|---|---|---|---|---|
| Control | 65 | - | F | 3 h 45 m | Status cribosus | I/0 | Yes | No |
| 74 | - | M | 9 h 25 m | Lacunar infarction | III/A | Yes | No | |
| 45 | - | M | 18 h 30 m | Status cribosus | 0/0 | Yes | No | |
| 51 | - | F | 4 h | No lesions | 0/0 | Yes | Yes | |
| 67 | - | M | 5 h 50 m | Amyloid angiopathy | I/0 | Yes | Yes | |
| 59 | - | F | 5 h 30 m | Metastatic carcinoma | I/0 | Yes | Yes | |
| 60 | - | F | 12 h | Status cribosus | I/0 | Yes | Yes | |
| 75 | - | M | 5 h 30 m | Status cribosus | I/0 | Yes | Yes | |
| ALS | 57 | Bulbar | M | 4 h | ALS | IIA | Yes | Yes |
| 75 | Bulbar | F | 4 h 5 m | ALS | IIA | Yes | Yes | |
| 79 | Spinal | F | 2 h 10 m | ALS | IIA | Yes | Yes | |
| 57 | Bulbar | F | 10 h | ALS | I/0 | Yes | Yes | |
| 50 | Spinal | M | 10 h 10 m | ALS | I/0 | Yes | Yes | |
| 75 | Bulbar | M | 3 h | ALS | II/B | Yes | Yes | |
| 71 | Spinal | M | 3 h 25 m | ALS | I/0 | Yes | Yes | |
| 68 | Bulbar | F | 16 h 30 m | ALS | I/0 | Yes | Yes | |
| 63 | Spinal | F | 18 h | ALS | I/0 | Yes | Yes | |
| 53 | Bulbar | F | 10 h | ALS | 0/0 | Yes | Yes | |
| 71 | Bulbar | F | 18 h | ALS | I/A | Yes | Yes | |
| 45 | Spinal | F | 4 h | ALS | 0/0 | Yes | Yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lachén-Montes, M.; Mendizuri, N.; Ausin, K.; Andrés-Benito, P.; Ferrer, I.; Fernández-Irigoyen, J.; Santamaría, E. Amyotrophic Lateral Sclerosis Is Accompanied by Protein Derangements in the Olfactory Bulb-Tract Axis. Int. J. Mol. Sci. 2020, 21, 8311. https://doi.org/10.3390/ijms21218311
Lachén-Montes M, Mendizuri N, Ausin K, Andrés-Benito P, Ferrer I, Fernández-Irigoyen J, Santamaría E. Amyotrophic Lateral Sclerosis Is Accompanied by Protein Derangements in the Olfactory Bulb-Tract Axis. International Journal of Molecular Sciences. 2020; 21(21):8311. https://doi.org/10.3390/ijms21218311
Chicago/Turabian StyleLachén-Montes, Mercedes, Naroa Mendizuri, Karina Ausin, Pol Andrés-Benito, Isidro Ferrer, Joaquín Fernández-Irigoyen, and Enrique Santamaría. 2020. "Amyotrophic Lateral Sclerosis Is Accompanied by Protein Derangements in the Olfactory Bulb-Tract Axis" International Journal of Molecular Sciences 21, no. 21: 8311. https://doi.org/10.3390/ijms21218311
APA StyleLachén-Montes, M., Mendizuri, N., Ausin, K., Andrés-Benito, P., Ferrer, I., Fernández-Irigoyen, J., & Santamaría, E. (2020). Amyotrophic Lateral Sclerosis Is Accompanied by Protein Derangements in the Olfactory Bulb-Tract Axis. International Journal of Molecular Sciences, 21(21), 8311. https://doi.org/10.3390/ijms21218311