Transcriptional Regulation of Inflammasomes


Abstract
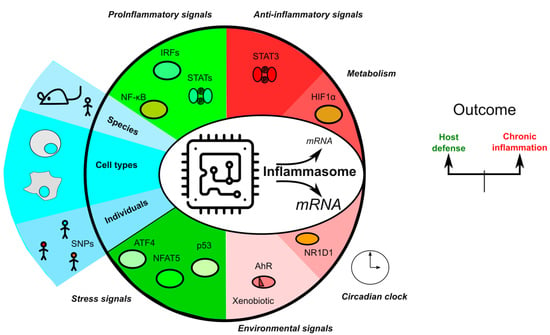
1. Introduction
2. Overview of Inflammasome Complexes
3. Overview of Transcription Regulation
4. Transcriptional Regulation of Inflammasome Sensors
4.1. NLRP1
4.2. NLRP3
4.3. NAIP and NLRC4
4.4. AIM2
4.5. Pyrin
5. Transcriptional Regulation of ASC
6. Transcriptional Regulation of Inflammatory Caspases
6.1. Caspase-1
6.2. Caspase-4
6.3. Caspase-11
6.4. Caspase-5
7. Transcriptional Regulation of Downstream Targets
7.1. GSDMD
7.2. IL-18 and IL-18BP
7.3. IL-1b and IL-1RA
7.3.1. Roles of the Lineage-Specific/Pioneer TFs PU.1 and C/EBPβ
7.3.2. Signal-Dependent TFs (NF-κB)
7.3.3. Metabolic Regulation of IL1b
7.3.4. Negative Regulation of IL1b
7.3.5. IL1b Regulation in T Cells
7.3.6. IL1b Regulation in DCs
7.3.7. IL1RN
8. Conclusions
Funding
Conflicts of Interest
Abbreviations
| AhR | Aryl hydrocarbon receptor |
| AIM2 | Absent in melanoma 2 |
| AP-1 | Activator protein-1 |
| ARE | Antioxidant responsive element |
| ASC | Apoptosis-associated speck-like protein containing a card |
| ATF | Activating transcription factor |
| BLIMP1 | B lymphocyte-induced maturation protein-1 |
| BMDM | Bone marrow-derived macrophage |
| BCL6 | B cell lymphoma 6 |
| Brd4 | Bromodomain-containing protein 4 |
| CHOP | C/ebp homologous protein |
| CRE | Camp-response element |
| CREB | Camp response element-binding protein |
| DC | Dendritic cell |
| DSS | Dextran sodium sulfate |
| EBV | Epstein–Barr virus |
| EICE | Ets-irf composite element |
| ENU | N-ethyl-n-nitrosourea |
| ERV | Endogenous retrovirus |
| ETS | Erythroblast transformation specific |
| GAS | Gamma-activated site |
| GFI1 | Growth factor independence 1 |
| GRE1 | Gli-responsive element |
| HAMPs | Homeostasis-altering molecular processes |
| HAT | Histone acetyl transferase |
| HIF1α | Hypoxia-inducible factor 1α |
| HRE | Hypoxia response element |
| IFNAR | Interferon-α/β receptor |
| IRE-1 | Inositol-requiring enzyme 1 |
| IRF | Interferon regulatory factor |
| ISRE | Ifn-stimulated response element |
| LTR | Long terminal repeat |
| NAIP | Neuronal apoptosis inhibitory protein |
| NFAT5 | Nuclear factor of activated t cells 5 |
| NF-κB | Nuclear factor κb |
| NLR | Nucleotide-binding domain and leucine-rich repeat containing |
| NLRP | Nlr family pyrin domain containing |
| Nod2 | Nucleotide-binding oligomerization domain 2 |
| NR1D1 | Nuclear receptor subfamily 1 group d member 1 |
| Nrf2 | Nf-e2-related factor 2 |
| ORE | Osmotic response element |
| PAMP | Pathogen-associated molecular pattern |
| PARP1 | Poly [ADP-ribose] polymerase 1 |
| PERK | Pkr-like er protein kinase |
| P-TEFb SBE | Positive transcription elongation factor b STAT-Binding Element |
| SREBP-1a | Sterol regulatory element binding protein-1a |
| STAT1 | Signal Transducer and Activator of Transcription 1 |
| SWI/SNF | Switch/sucrose nonfermenting |
| TCA | Tricarboxylic acid |
| TF | Transcription factor |
| TLR | Toll-like receptor |
| TNF | Tumor necrosis factor |
| TSS | Transcription start site |
| UPR | Unfolded protein response |
| XRE | Xenobiotic response elements |
References
- Paludan, S.R.; Pradeu, T.; Masters, S.L.; Mogensen, T.H. Constitutive immune mechanisms: Mediators of host defence and immune regulation. Nat. Rev. Immunol. 2020, 1–14. [Google Scholar] [CrossRef]
- Song, N.; Li, T. Regulation of NLRP3 Inflammasome by Phosphorylation. Front. Immunol. 2018, 9, 2305. [Google Scholar] [CrossRef] [PubMed]
- Samir, P.; Kanneganti, T.-D. Hidden Aspects of Valency in Immune System Regulation. Trends Immunol. 2019, 40, 1082–1094. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Castejon, G. Control of the inflammasome by the ubiquitin system. FEBS J. 2019, 287, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Moretti, J.; Blander, J.M. Increasing complexity of NLRP3 inflammasome regulation. J. Leukoc. Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Taabazuing, C.Y.; Griswold, A.R.; Bachovchin, D.A. The NLRP1 and CARD8 inflammasomes. Immunol. Rev. 2020, 297, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.-J.; Tu, L.; Huang, X.-M.; Huang, J.; Qiu, N.; Xie, G.-H.; Liao, J.-X.; Du, W.; Zhang, Y.-Y.; Tian, J.-Y. LncRNA MALAT1 facilitates inflammasome activation via epigenetic suppression of Nrf2 in Parkinson’s disease. Mol. Brain 2020, 13, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Cao, L.; Zhou, R.; Yang, X.; Wu, M. The lncRNA Neat1 promotes activation of inflammasomes in macrophages. Nat. Commun. 2019, 10, 1–17. [Google Scholar] [CrossRef]
- Zhong, F.L.; Robinson, K.; Teo, D.E.T.; Tan, K.-Y.; Lim, C.; Harapas, C.R.; Yu, C.-H.; Xie, W.H.; Sobota, R.M.; Au, V.B.; et al. Human DPP9 represses NLRP1 inflammasome and protects against autoinflammatory diseases via both peptidase activity and FIIND domain binding. J. Biol. Chem. 2018, 293, 18864–18878. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nat. Cell Biol. 2014, 514, 187–192. [Google Scholar] [CrossRef]
- Bitto, N.J.; Baker, P.J.; Dowling, J.K.; Wray-McCann, G.; De Paoli, A.; Tran, L.S.; Leung, P.L.; Stacey, K.J.; Mansell, A.; Masters, S.L.; et al. Membrane vesicles from Pseudomonas aeruginosa activate the noncanonical inflammasome through caspase-5 in human monocytes. Immunol. Cell Biol. 2018, 96, 1120–1130. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nat. Cell Biol. 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.-C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nat. Cell Biol. 2016, 535, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.; Xia, S.; Liu, X.; Lieberman, J.; Wu, H. Cryo-EM structure of the gasdermin A3 membrane pore. Nat. Cell Biol. 2018, 557, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.L.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Rühl, S.; Broz, P. Caspase-11 activates a canonical NLRP3 inflammasome by promoting K+efflux. Eur. J. Immunol. 2015, 45, 2927–2936. [Google Scholar] [CrossRef]
- Schmid-Burgk, J.L.; Gaidt, M.M.; Schmidt, T.; Ebert, T.S.; Bartok, E.; Hornung, V. Caspase-4 mediates non-canonical activation of the NLRP3 inflammasome in human myeloid cells. Eur. J. Immunol. 2015, 45, 2911–2917. [Google Scholar] [CrossRef]
- Broz, P.; Monack, D.M. Molecular mechanisms of inflammasome activation during microbial infections. Immunol. Rev. 2011, 243, 174–190. [Google Scholar] [CrossRef]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nat. Cell Biol. 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Pathak, S.; McDermott, M.F.; Savic, S. Autoinflammatory diseases: Update on classification diagnosis and management. J. Clin. Pathol. 2016, 70, 1–8. [Google Scholar] [CrossRef]
- McDaniel, M.M.; Kottyan, L.C.; Singh, H.; Pasare, C. Suppression of Inflammasome Activation by IRF8 and IRF4 in cDCs Is Critical for T Cell Priming. Cell Rep. 2020, 31, 107604. [Google Scholar] [CrossRef]
- Benaoudia, S.; Martin, A.; Gamez, M.P.; Gay, G.; Lagrange, B.; Cornut, M.; Krasnykov, K.; Claude, J.; Bourgeois, C.F.; Hughes, S.; et al. A genome-wide screen identifies IRF2 as a key regulator of caspase-4 in human cells. EMBO Rep. 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Lee, B.L.; Stowe, I.B.; Kornfeld, O.S.; O’Rourke, K.; Mirrashidi, K.M.; Haley, B.; Watanabe, C.; Roose-Girma, M.; Modrusan, Z.; et al. IRF2 transcriptionally induces GSDMD expression for pyroptosis. Sci. Signal. 2019, 12, eaax4917. [Google Scholar] [CrossRef] [PubMed]
- Aachoui, Y.; Kajiwara, Y.; Leaf, I.A.; Mao, D.; Ting, J.P.-Y.; Coers, J.; Aderem, A.; Buxbaum, J.D.; Miao, E.A. Canonical Inflammasomes Drive IFN-γ to Prime Caspase-11 in Defense against a Cytosol-Invasive Bacterium. Cell Host Microbe 2015, 18, 320–332. [Google Scholar] [CrossRef]
- Karki, R.; Lee, E.; Place, D.; Samir, P.; Mavuluri, J.; Sharma, B.R.; Balakrishnan, A.; Malireddi, R.S.; Geiger, R.; Zhu, Q.; et al. IRF8 Regulates Transcription of Naips for NLRC4 Inflammasome Activation. Cell 2018, 173, 920–933. [Google Scholar] [CrossRef] [PubMed]
- Guarda, G.; Braun, M.; Staehli, F.; Tardivel, A.; Mattmann, C.; Förster, I.; Farlik, M.; Decker, T.; Du Pasquier, R.A.; Romero, P.; et al. Type I Interferon Inhibits Interleukin-1 Production and Inflammasome Activation. Immunology 2011, 34, 213–223. [Google Scholar] [CrossRef]
- Heinz, S.; Romanoski, C.E.; Benner, C.; Glass, C.K. The selection and function of cell type-specific enhancers. Nat. Rev. Mol. Cell Biol. 2015, 16, 144–154. [Google Scholar] [CrossRef]
- Sandstrom, A.; Mitchell, P.S.; Goers, L.; Mu, E.W.; Lesser, C.F.; Vance, R.E. Functional degradation: A mechanism of NLRP1 inflammasome activation by diverse pathogen enzymes. Science 2019, 364, eaau1330. [Google Scholar] [CrossRef]
- Boyden, E.D.; Dietrich, W.F. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat. Genet. 2006, 38, 240–244. [Google Scholar] [CrossRef]
- Masters, S.L.; Gerlic, M.; Metcalf, D.; Preston, S.; Pellegrini, M.; O’Donnell, J.A.; McArthur, K.; Baldwin, T.M.; Chevrier, S.; Nowell, C.J.; et al. NLRP1 Inflammasome Activation Induces Pyroptosis of Hematopoietic Progenitor Cells. Immunology 2012, 37, 1009–1023. [Google Scholar] [CrossRef]
- Im, S.-S.; Yousef, L.; Blaschitz, C.; Liu, J.Z.; Edwards, R.A.; Young, S.G.; Raffatellu, M.; Osborne, T.F. Linking Lipid Metabolism to the Innate Immune Response in Macrophages through Sterol Regulatory Element Binding Protein-1a. Cell Metab. 2011, 13, 540–549. [Google Scholar] [CrossRef]
- Murphy, A.J.; Kraakman, M.J.; Kammoun, H.L.; Dragoljevic, D.; Lee, M.K.; Lawlor, K.E.; Wentworth, J.M.; VasanthaKumar, A.; Gerlic, M.; Whitehead, L.W.; et al. IL-18 Production from the NLRP1 Inflammasome Prevents Obesity and Metabolic Syndrome. Cell Metab. 2016, 23, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Fenini, G.; Grossi, S.; Contassot, E.; Biedermann, T.; Reichmann, E.; French, L.E.; Beer, H.-D. Genome Editing of Human Primary Keratinocytes by CRISPR/Cas9 Reveals an Essential Role of the NLRP1 Inflammasome in UVB Sensing. J. Investig. Dermatol. 2018, 138, 2644–2652. [Google Scholar] [CrossRef]
- Sand, J.; Haertel, E.; Biedermann, T.; Contassot, E.; Reichmann, E.; French, L.E.; Werner, S.; Beer, H.-D. Expression of inflammasome proteins and inflammasome activation occurs in human, but not in murine keratinocytes. Cell Death Dis. 2018, 9, 24. [Google Scholar] [CrossRef]
- D’Osualdo, A.; Anania, V.G.; Yu, K.; Lill, J.R.; Kaufman, R.J.; Matsuzawa, S.-I.; Reed, J.C. Transcription Factor ATF4 Induces NLRP1 Inflammasome Expression during Endoplasmic Reticulum Stress. PLoS ONE 2015, 10, e0130635. [Google Scholar] [CrossRef]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; Macdonald, K.L.; Speert, D.P.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting Edge: NF-κB Activating Pattern Recognition and Cytokine Receptors License NLRP3 Inflammasome Activation by Regulating NLRP3 Expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Eigenbrod, T.; Núñez, G. Cutting Edge: TNF-α Mediates Sensitization to ATP and Silica via the NLRP3 Inflammasome in the Absence of Microbial Stimulation. J. Immunol. 2009, 183, 792–796. [Google Scholar] [CrossRef]
- Juliana, C.; Fernandes-Alnemri, T.; Kang, S.; Farias, A.; Qin, F.; Alnemri, E.S. Non-transcriptional Priming and Deubiquitination Regulate NLRP3 Inflammasome Activation. J. Biol. Chem. 2012, 287, 36617–36622. [Google Scholar] [CrossRef] [PubMed]
- Fernandes-Alnemri, T.; Kang, S.; Anderson, C.; Sagara, J.; Fitzgerald, K.A.; Alnemri, E.S. Cutting Edge: TLR Signaling Licenses IRAK1 for Rapid Activation of the NLRP3 Inflammasome. J. Immunol. 2013, 191, 3995–3999. [Google Scholar] [CrossRef] [PubMed]
- Ghonime, M.G.; Shamaa, O.R.; Das, S.; Eldomany, R.A.; Fernandes-Alnemri, T.; Alnemri, E.S.; Gavrilin, M.A.; Wewers, M.D. Inflammasome Priming by Lipopolysaccharide Is Dependent upon ERK Signaling and Proteasome Function. J. Immunol. 2014, 192, 3881–3888. [Google Scholar] [CrossRef]
- Qiao, Y.; Wang, P.; Qi, J.; Zhang, L.; Gao, C. TLR-induced NF-κB activation regulates NLRP3 expression in murine macrophages. FEBS Lett. 2012, 586, 1022–1026. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Ahn, H.; Yu, S.; Ahn, J.-H.; Ko, H.-J.; Kweon, M.-N.; Hong, E.-J.; An, B.-S.; Lee, E.; Lee, G.-S. IκBζ controls NLRP3 inflammasome activation via upregulation of the Nlrp3 gene. Cytokine 2020, 127, 154983. [Google Scholar] [CrossRef]
- Hildebrand, D.G.; Alexander, E.; Hörber, S.; Lehle, S.; Obermayer, K.; Münck, N.-A.; Rothfuss, O.; Frick, J.-S.; Morimatsu, M.; Schmitz, I.; et al. IκBζ Is a Transcriptional Key Regulator of CCL2/MCP-1. J. Immunol. 2013, 190, 4812–4820. [Google Scholar] [CrossRef] [PubMed]
- Lecoeur, H.; Prina, E.; Rosazza, T.; Kokou, K.; N’Diaye, P.; Aulner, N.; Varet, H.; Bussotti, G.; Xing, Y.; Milon, G.; et al. Targeting Macrophage Histone H3 Modification as a Leishmania Strategy to Dampen the NF-κB/NLRP3-Mediated Inflammatory Response. Cell Rep. 2020, 30, 1870–1882. [Google Scholar] [CrossRef]
- Pourcet, B.; Zecchin, M.; Ferri, L.; Beauchamp, J.; Sitaula, S.; Billon, C.; Delhaye, S.; Vanhoutte, J.; Mayeuf-Louchart, A.; Thorel, Q.; et al. Nuclear Receptor Subfamily 1 Group D Member 1 Regulates Circadian Activity of NLRP3 Inflammasome to Reduce the Severity of Fulminant Hepatitis in Mice. Gastroenterology 2018, 154, 1449–1464. [Google Scholar] [CrossRef]
- Wang, S.; Lin, Y.; Yuan, X.; Li, F.; Guo, L.; Wu, B. REV-ERBα integrates colon clock with experimental colitis through regulation of NF-κB/NLRP3 axis. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Huai, W.; Zhao, R.; Song, H.; Zhao, J.; Zhang, L.; Zhang, L.; Gao, C.; Han, L.; Zhao, W. Aryl hydrocarbon receptor negatively regulates NLRP3 inflammasome activity by inhibiting NLRP3 transcription. Nat. Commun. 2014, 5, 4738. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Xiong, X.-Q.; Zang, Y.-H.; Tong, Y.; Zhou, B.; Chen, Q.; Li, Y.-H.; Gao, X.-Y.; Kang, Y.-M.; Zhu, G.-Q. BCL6 attenuates renal inflammation via negative regulation of NLRP3 transcription. Cell Death Dis. 2017, 8, e3156. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Meng, Q.; Liang, S.; Ma, Y.; Li, R.; Li, G.; Zeng, H. The transcription factor GFI1 negatively regulates NLRP3 inflammasome activation in macrophages. FEBS Lett. 2014, 588, 4513–4519. [Google Scholar] [CrossRef]
- Ma, P.; Zha, S.; Shen, X.; Zhao, Y.; Li, L.; Yang, L.; Lei, M.; Liu, W. NFAT5 mediates hypertonic stress-induced atherosclerosis via activating NLRP3 inflammasome in endothelium. Cell Commun. Signal. 2019, 17, 1–13. [Google Scholar] [CrossRef]
- Erlich, Z.; Shlomovitz, I.; Edry-Botzer, L.; Cohen, H.; Frank, D.; Wang, H.; Lew, A.M.; Lawlor, K.E.; Zhan, Y.; Vince, J.E.; et al. Macrophages, rather than DCs, are responsible for inflammasome activity in the GM-CSF BMDC model. Nat. Immunol. 2019, 20, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhao, Y.; Shi, J.; Shao, F. Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proc. Natl. Acad. Sci. USA 2013, 110, 14408–14413. [Google Scholar] [CrossRef] [PubMed]
- Kortmann, J.; Brubaker, S.W.; Monack, D.M. Cutting Edge: Inflammasome Activation in Primary Human Macrophages Is Dependent on Flagellin. J. Immunol. 2015, 195, 815–819. [Google Scholar] [CrossRef]
- Kofoed, E.M.; Vance, R.E. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nat. Cell Biol. 2011, 477, 592–595. [Google Scholar] [CrossRef]
- Zhao, Y.; Shi, J.; Shi, X.; Wang, Y.; Wang, F.; Shao, F. Genetic functions of the NAIP family of inflammasome receptors for bacterial ligands in mice. J. Exp. Med. 2016, 213, 647–656. [Google Scholar] [CrossRef]
- Langlais, D.; Barreiro, L.B.; Gros, P. The macrophage IRF8/IRF1 regulome is required for protection against infections and is associated with chronic inflammation. J. Exp. Med. 2016, 213, 585–603. [Google Scholar] [CrossRef] [PubMed]
- Fortier, A.; Doiron, K.; Saleh, M.; Grinstein, S.; Gros, P. Restriction of Legionella pneumophila Replication in Macrophages Requires Concerted Action of the Transcriptional Regulators Irf1 and Irf8 and Nod-Like Receptors Naip5 and Nlrc4. Infect. Immun. 2009, 77, 4794–4805. [Google Scholar] [CrossRef]
- Kim, S.; Bagadia, P.; Anderson, D.A.; Liu, T.-T.; Huang, X.; Theisen, D.J.; O’Connor, K.W.; Ohara, R.A.; Iwata, A.; Murphy, T.L.; et al. High Amount of Transcription Factor IRF8 Engages AP1-IRF Composite Elements in Enhancers to Direct Type 1 Conventional Dendritic Cell Identity. Immunity 2020, 53, 759–774. [Google Scholar] [CrossRef]
- Fernandes-Alnemri, T.; Yu, J.-W.; Datta, P.; Wu, J.; Alnemri, E.S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nat. Cell Biol. 2009, 458, 509–513. [Google Scholar] [CrossRef]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.G.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nat. Cell Biol. 2009, 458, 514–518. [Google Scholar] [CrossRef]
- Di Micco, A.; Frera, G.; Lugrin, J.; Jamilloux, Y.; Hsu, E.-T.; Tardivel, A.; De Gassart, A.; Zaffalon, L.; Bujisic, B.; Siegert, S.; et al. AIM2 inflammasome is activated by pharmacological disruption of nuclear envelope integrity. Proc. Natl. Acad. Sci. USA 2016, 113, E4671–E4680. [Google Scholar] [CrossRef] [PubMed]
- Man, S.M.; Karki, R.; Malireddi, R.S.; Neale, G.; Vogel, P.; Yamamoto, M.; Lamkanfi, M.; Kanneganti, T.-D. The transcription factor IRF1 and guanylate-binding proteins target activation of the AIM2 inflammasome by Francisella infection. Nat. Immunol. 2015, 16, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Meunier, E.; Wallet, P.; Dreier, R.F.; Costanzo, S.; Anton, L.; Rühl, S.; Dussurgey, S.; Dick, M.S.; Kistner, A.; Rigard, M.; et al. Guanylate-binding proteins promote activation of the AIM2 inflammasome during infection with Francisella novicida. Nat. Immunol. 2015, 16, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Gaidt, M.M.; Ebert, T.S.; Chauhan, D.; Ramshorn, K.; Pinci, F.; Zuber, S.; O’Duill, F.; Schmid-Burgk, J.L.; Hoss, F.; Buhmann, R.; et al. The DNA Inflammasome in Human Myeloid Cells Is Initiated by a STING-Cell Death Program Upstream of NLRP3. Cell 2017, 171, 1110–1124. [Google Scholar] [CrossRef]
- Fisch, D.; Bando, H.; Clough, B.; Hornung, V.; Yamamoto, M.; Shenoy, A.R.; Frickel, E. Human GBP 1 is a microbe-specific gatekeeper of macrophage apoptosis and pyroptosis. EMBO J. 2019, 38, e100926. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Sagulenko, V.; Zamoshnikova, A.; Richards, A.A.; Cridland, J.A.; Irvine, K.M.; Stacey, K.J.; Sweet, M.J. Acute lipopolysaccharide priming boosts inflammasome activation independently of inflammasome sensor induction. Immunobiology 2012, 217, 1325–1329. [Google Scholar] [CrossRef]
- Dombrowski, Y.; Peric, M.; Koglin, S.; Kammerbauer, C.; Göss, C.; Anz, D.; Simanski, M.; Gläser, R.; Harder, J.; Hornung, V.; et al. Cytosolic DNA Triggers Inflammasome Activation in Keratinocytes in Psoriatic Lesions. Sci. Transl. Med. 2011, 3, 82ra38. [Google Scholar] [CrossRef]
- Doody, G.M.; Care, M.A.; Burgoyne, N.J.; Bradford, J.R.; Bota, M.; Bonifer, C.; Westhead, D.R.; Tooze, R.M. An extended set of PRDM1/BLIMP1 target genes links binding motif type to dynamic repression. Nucleic Acids Res. 2010, 38, 5336–5350. [Google Scholar] [CrossRef]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science 2016, 351, 1083–1087. [Google Scholar] [CrossRef]
- Xu, H.; Yang, J.; Gao, W.; Li, L.; Li, P.; Zhang, L.; Gong, Y.-N.; Peng, X.; Xi, J.J.; Chen, S.; et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nat. Cell Biol. 2014, 513, 237–241. [Google Scholar] [CrossRef]
- Jamilloux, Y.; Magnotti, F.; Belot, A.; Henry, T. The pyrin inflammasome: From sensing RhoA GTPases-inhibiting toxins to triggering autoinflammatory syndromes. Pathog. Dis. 2018, 76. [Google Scholar] [CrossRef] [PubMed]
- Gavrilin, M.A.; Mitra, S.; Seshadri, S.; Nateri, J.; Berhe, F.; Hall, M.W.; Wewers, M.D. Pyrin Critical to Macrophage IL-1β Response toFrancisellaChallenge. J. Immunol. 2009, 182, 7982–7989. [Google Scholar] [CrossRef] [PubMed]
- Centola, M.; Wood, G.; Frucht, D.M.; Galon, J.; Aringer, M.; Farrell, C.; Kingma, D.W.; Horwitz, M.E.; Mansfield, E.; Holland, S.M.; et al. The gene for familial Mediterranean fever, MEFV, is expressed in early leukocyte development and is regulated in response to inflammatory mediators. Blood 2000, 95, 3223–3231. [Google Scholar] [CrossRef] [PubMed]
- Grandemange, S.; Aksentijevich, I.; Jeru, I.; Gül, A.; Touitou, I. The regulation of MEFV expression and its role in health and familial Mediterranean fever. Genes Immun. 2011, 12, 497–503. [Google Scholar] [CrossRef]
- Papin, S.; Cazeneuve, C.; Duquesnoy, P.; Jéru, I.; Sahali, D.; Amselem, S. The Tumor Necrosis Factor α-dependent Activation of the Human Mediterranean Fever (MEFV) Promoter Is Mediated by a Synergistic Interaction between C/EBPβ and NFκB p65. J. Biol. Chem. 2003, 278, 48839–48847. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Malik, A.; Guy, C.; Vogel, P.; Kanneganti, T.-D. TNF/TNFR axis promotes pyrin inflammasome activation and distinctly modulates pyrin inflammasomopathy. J. Clin. Investig. 2018, 129, 150–162. [Google Scholar] [CrossRef]
- Deyoung, K.L.; E Ray, M.; A Su, Y.; Anzick, S.L.; Johnstone, R.W.; A Trapani, J.; Meltzer, P.S.; Trent, J.M. Cloning a novel member of the human interferon-inducible gene family associated with control of tumorigenicity in a model of human melanoma. Oncogene 1997, 15, 453–457. [Google Scholar] [CrossRef]
- E Conway, K.; McConnell, B.B.; E Bowring, C.; Donald, C.D.; Warren, S.T.; Vertino, P.M. TMS1, a novel proapoptotic caspase recruitment domain protein, is a target of methylation-induced gene silencing in human breast cancers. Cancer Res. 2000, 60, 6236–6242. [Google Scholar]
- Ghosh, S.; Wallerath, C.; Covarrubias, S.; Hornung, V.; Carpenter, S.B.; Fitzgerald, K.A.; Wallareth, C. The PYHIN Protein p205 Regulates the Inflammasome by Controlling Asc Expression. J. Immunol. 2017, 199, 3249–3260. [Google Scholar] [CrossRef]
- Brunette, R.L.; Young, J.M.; Whitley, D.G.; Brodsky, I.E.; Malik, H.S.; Stetson, D.B. Extensive evolutionary and functional diversity among mammalian AIM2-like receptors. J. Exp. Med. 2012, 209, 1969–1983. [Google Scholar] [CrossRef]
- Ming, X.; Li, W.; Maeda, Y.; Blumberg, B.; Raval, S.; Cook, S.D.; Dowling, P.C. Caspase-1 expression in multiple sclerosis plaques and cultured glial cells. J. Neurol. Sci. 2002, 197, 9–18. [Google Scholar] [CrossRef]
- Giamarellos-Bourboulis, E.J.; Van De Veerdonk, F.L.; Mouktaroudi, M.; Raftogiannis, M.; Antonopoulou, A.; Joosten, L.A.; Pickkers, P.; Savva, A.; Georgitsi, M.; Van Der Meer, J.W.M.; et al. Inhibition of caspase-1 activation in gram-negative sepsis and experimental endotoxemia. Crit. Care 2011, 15, R27. [Google Scholar] [CrossRef] [PubMed]
- Fairfax, B.P.; Humburg, P.; Makino, S.; Naranbhai, V.; Wong, D.; Lau, E.; Jostins, L.; Plant, K.; Andrews, R.; McGee, C.; et al. Innate Immune Activity Conditions the Effect of Regulatory Variants upon Monocyte Gene Expression. Science 2014, 343, 1246949. [Google Scholar] [CrossRef]
- Dai, C.; Krantz, S.B. Interferon gamma induces upregulation and activation of caspases 1, 3, and 8 to produce apoptosis in human erythroid progenitor cells. Blood 1999, 93, 3309–3316. [Google Scholar] [CrossRef] [PubMed]
- Tamura, T.; Ishihara, M.; Lamphier, M.S.; Tanaka, N.; Oishi, I.; Aizawa, S.; Matsuyama, T.; Mak, T.W.; Taki, S.; Taniguchi, T. An IRF-1-dependent pathway of DNA damage-induced apoptosis in mitogen-activated T lymphocytes. Nat. Cell Biol. 1995, 376, 596–599. [Google Scholar] [CrossRef] [PubMed]
- Cuesta, N.; Nhu, Q.M.; Zudaire, E.; Polumuri, S.; Cuttitta, F.; Vogel, S.N. IFN regulatory factor-2 regulates macrophage apoptosis through a STAT1/3- and caspase-1-dependent mechanism. J. Immunol. (Baltim. MD 1950) 2007, 178, 3602–3611. [Google Scholar] [CrossRef]
- Lv, D.; Zhang, K.; Li, R. Interferon regulatory factor 8 regulates caspase-1 expression to facilitate Epstein-Barr virus reactivation in response to B cell receptor stimulation and chemical induction. PLoS Pathog. 2018, 14, e1006868. [Google Scholar] [CrossRef]
- Thygesen, S.J.; Stacey, K.J. IRF 1 and IRF 2 regulate the non-canonical inflammasome. EMBO Rep. 2019, 20, e48891. [Google Scholar] [CrossRef]
- Yang, H.-J.; Wang, M.; Wang, L.; Cheng, B.-F.; Lin, X.-Y.; Feng, Z.-W. NF-κB Regulates Caspase-4 Expression and Sensitizes Neuroblastoma Cells to Fas-Induced Apoptosis. PLoS ONE 2015, 10, e0117953. [Google Scholar] [CrossRef]
- Schauvliege, R.; Vanrobaeys, J.; Schotte, P.; Beyaert, R. Caspase-11 Gene Expression in Response to Lipopolysaccharide and Interferon-γ Requires Nuclear Factor-κB and Signal Transducer and Activator of Transcription (STAT) 1. J. Biol. Chem. 2002, 277, 41624–41630. [Google Scholar] [CrossRef]
- Crowley, S.M.; Han, X.; Allaire, J.M.; Stahl, M.; Rauch, I.; Knodler, L.A.; Vallance, B.A. Intestinal restriction of Salmonella Typhimurium requires caspase-1 and caspase-11 epithelial intrinsic inflammasomes. PLoS Pathog. 2020, 16, e1008498. [Google Scholar] [CrossRef] [PubMed]
- Endo, M.; Mori, M.; Akira, S.; Gotoh, T. C/EBP Homologous Protein (CHOP) Is Crucial for the Induction of Caspase-11 and the Pathogenesis of Lipopolysaccharide-Induced Inflammation. J. Immunol. 2006, 176, 6245–6253. [Google Scholar] [CrossRef] [PubMed]
- Eleazer, R.; Fondufe-Mittendorf, Y.N. The multifaceted role of PARP1 in RNA biogenesis. Wiley Interdiscip. Rev. RNA 2020, e12607. [Google Scholar] [CrossRef] [PubMed]
- Paul, O.H.; Covic, M.; Hasan, S.; Imhof, R.; Hottiger, M.O. The Enzymatic and DNA Binding Activity of PARP-1 Are Not Required for NF-κB Coactivator Function. J. Biol. Chem. 2001, 276, 45588–45597. [Google Scholar] [CrossRef]
- Yoo, L.; Hong, S.; Shin, K.S.; Kang, S.J. PARP-1 regulates the expression of caspase-11. Biochem. Biophys. Res. Commun. 2011, 408, 489–493. [Google Scholar] [CrossRef]
- Frank, A.K.; Leu, J.I.-J.; Zhou, Y.; Devarajan, K.; Nedelko, T.; Klein-Szanto, A.; Hollstein, M.C.; Murphy, M.E. The Codon 72 Polymorphism of p53 Regulates Interaction with NF-κB and Transactivation of Genes Involved in Immunity and Inflammation. Mol. Cell. Biol. 2011, 31, 1201–1213. [Google Scholar] [CrossRef][Green Version]
- Gupta, S.; Radha, V.; Furukawa, Y.; Swarup, G. Direct Transcriptional Activation of Human Caspase-1 by Tumor Suppressor p53. J. Biol. Chem. 2001, 276, 10585–10588. [Google Scholar] [CrossRef]
- Sadasivam, S.; Gupta, S.; Radha, V.; Batta, K.; Kundu, T.K.; Swarup, G. Caspase-1 activator Ipaf is a p53-inducible gene involved in apoptosis. Oncogene 2004, 24, 627–636. [Google Scholar] [CrossRef]
- Hagar, J.A.; Powell, D.A.; Aachoui, Y.; Ernst, R.K.; Miao, E.A. Cytoplasmic LPS Activates Caspase-11: Implications in TLR4-Independent Endotoxic Shock. Science 2013, 341, 1250–1253. [Google Scholar] [CrossRef]
- Kayagaki, N.; Wong, M.T.; Stowe, I.B.; Ramani, S.R.; Gonzalez, L.C.; Akashi-Takamura, S.; Miyake, K.; Zhang, J.; Lee, W.P.; Muszyński, A.; et al. Noncanonical Inflammasome Activation by Intracellular LPS Independent of TLR4. Science 2013, 341, 1246–1249. [Google Scholar] [CrossRef]
- Kajiwara, Y.; Schiff, T.; Voloudakis, G.; Sosa, M.A.G.; Elder, G.; Bozdagi, O.; Buxbaum, J.D. A Critical Role for Human Caspase-4 in Endotoxin Sensitivity. J. Immunol. 2014, 193, 335–343. [Google Scholar] [CrossRef]
- Lin, X.Y.; Choi, M.S.K.; Porter, A.G. Expression Analysis of the Human Caspase-1 Subfamily Reveals Specific Regulation of theCASP5Gene by Lipopolysaccharide and Interferon-γ. J. Biol. Chem. 2000, 275, 39920–39926. [Google Scholar] [CrossRef] [PubMed]
- Casson, C.N.; Yu, J.; Reyes, V.M.; Taschuk, F.O.; Yadav, A.; Copenhaver, A.M.; Nguyen, H.T.; Collman, R.G.; Shin, S. Human caspase-4 mediates noncanonical inflammasome activation against gram-negative bacterial pathogens. Proc. Natl. Acad. Sci. USA 2015, 112, 6688–6693. [Google Scholar] [CrossRef] [PubMed]
- Eckhart, L.; Kittel, C.; Gawlas, S.; Gruber, F.; Mildner, M.; Jilma, B.; Tschachler, E. Identification of a novel exon encoding the amino-terminus of the predominant caspase-5 variants. Biochem. Biophys. Res. Commun. 2006, 348, 682–688. [Google Scholar] [CrossRef]
- Salskov-Iversen, M.L.; Johansen, C.; Kragballe, K.; Iversen, L. Caspase-5 Expression Is Upregulated in Lesional Psoriatic Skin. J. Investig. Dermatol. 2011, 131, 670–676. [Google Scholar] [CrossRef]
- Li, Y.; Guo, X.; Hu, C.; Du, Y.; Guo, C.; Wang, D.; Zhao, W.; Huang, G.; Li, C.; Lu, Q.; et al. Type I IFN operates pyroptosis and necroptosis during multidrug-resistant A. baumannii infection. Cell Death Differ. 2018, 25, 1304–1318. [Google Scholar] [CrossRef]
- Liu, Z.; Gan, L.; Xu, Y.; Luo, D.; Ren, Q.; Wu, S.; Sun, C. Melatonin alleviates inflammasome-induced pyroptosis through inhibiting NF-κB/GSDMD signal in mice adipose tissue. J. Pineal Res. 2017, 63, e12414. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Immunological and Inflammatory Functions of the Interleukin-1 Family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Kang, H.S.; Paik, S.G.; Pyun, K.H.; Anderson, K.L.; E Torbett, B.; Choi, I. Roles of IFN consensus sequence binding protein and PU.1 in regulating IL-18 gene expression. J. Immunol. 1999, 163, 2000–2007. [Google Scholar]
- Verweyen, E.; Holzinger, D.; Weinhage, T.; Hinze, C.; Wittkowski, H.; Pickkers, P.; Albeituni, S.; Verbist, K.; Nichols, K.E.; Schulert, G.; et al. Synergistic Signaling of TLR and IFNα/β Facilitates Escape of IL-18 Expression from Endotoxin Tolerance. Am. J. Respir. Crit. Care Med. 2020, 201, 526–539. [Google Scholar] [CrossRef]
- Zhu, Q.; Kanneganti, T.-D. Cutting Edge: Distinct Regulatory Mechanisms Control Proinflammatory Cytokines IL-18 and IL-1β. J. Immunol. 2017, 198, 4210–4215. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.; Eidenschenk, C.; Ota, N.; Wong, K.; Lohmann, U.; Kühl, A.A.; Wang, X.; Manzanillo, P.; Li, Y.; Rutz, S.; et al. Interleukin-22 Induces Interleukin-18 Expression from Epithelial Cells during Intestinal Infection. Immunity 2015, 42, 321–331. [Google Scholar] [CrossRef]
- Levy, M.; Thaiss, C.A.; Zeevi, D.; Dohnalová, L.; Zilberman-Schapira, G.; Mahdi, J.A.; David, E.; Savidor, A.; Korem, T.; Herzig, Y.; et al. Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell 2015, 163, 1428–1443. [Google Scholar] [CrossRef] [PubMed]
- Hurgin, V.; Novick, D.; Rubinstein, M. The promoter of IL-18 binding protein: Activation by an IFN-/-induced complex of IFN regulatory factor 1 and CCAAT/enhancer binding protein. Proc. Natl. Acad. Sci. USA 2002, 99, 16957–16962. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.; Paulukat, J.; Pfeilschifter, J.; Mühl, H. Molecular mechanisms of IL-18BP regulation in DLD-1 cells: Pivotal direct action of the STAT1/GAS axis on the promoter level. J. Cell. Mol. Med. 2008, 13, 1987–1994. [Google Scholar] [CrossRef]
- Kominato, Y.; Galson, D.; Waterman, W.R.; Webb, A.C.; E Auron, P. Monocyte expression of the human prointerleukin 1 beta gene (IL1B) is dependent on promoter sequences which bind the hematopoietic transcription factor Spi-1/PU.1. Mol. Cell. Biol. 1995, 15, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Minderjahn, J.; Schmidt, A.; Fuchs, A.; Schill, R.; Raithel, J.; Babina, M.; Schmidl, C.; Gebhard, C.; Schmidhofer, S.; Mendes, K.; et al. Mechanisms governing the pioneering and redistribution capabilities of the non-classical pioneer PU.1. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Plachetka, A.; Chayka, O.; Wilczek, C.; Melnik, S.; Bonifer, C.; Klempnauer, K.-H. C/EBPβ Induces Chromatin Opening at a Cell-Type-Specific Enhancer. Mol. Cell. Biol. 2008, 28, 2102–2112. [Google Scholar] [CrossRef][Green Version]
- Liang, M.D.; Zhang, Y.; McDevit, D.; Marecki, S.; Nikolajczyk, B.S. The Interleukin-1β Gene Is Transcribed from a Poised Promoter Architecture in Monocytes. J. Biol. Chem. 2006, 281, 9227–9237. [Google Scholar] [CrossRef]
- Zhang, Y.; Saccani, S.; Shin, H.; Nikolajczyk, B.S. Dynamic Protein Associations Define Two Phases of IL-1β Transcriptional Activation. J. Immunol. 2008, 181, 503–512. [Google Scholar] [CrossRef]
- Serkkola, E.; Hurme, M. Synergism between protein-kinase C and cAMP-dependent pathways in the expression of the interleukin-1beta gene is mediated via the activator-protein-1 (AP-1) enhancer activity. JBIC J. Biol. Inorg. Chem. 1993, 213, 243–249. [Google Scholar] [CrossRef]
- Unlu, S.; Kumar, A.; Waterman, W.R.; Tsukada, J.; Wang, K.Z.; Galson, D.L.; Auron, P.E. Phosphorylation of IRF8 in a pre-associated complex with Spi-1/PU.1 and non-phosphorylated Stat1 is critical for LPS induction of the IL1B gene. Mol. Immunol. 2007, 44, 3364–3379. [Google Scholar] [CrossRef]
- Shirakawa, F.; Saito, K.; A Bonagura, C.; Galson, D.L.; Fenton, M.J.; Webb, A.C.; E Auron, P. The human prointerleukin 1 beta gene requires DNA sequences both proximal and distal to the transcription start site for tissue-specific induction. Mol. Cell. Biol. 1993, 13, 1332–1344. [Google Scholar] [CrossRef] [PubMed]
- Adamik, J.; Wang, K.Z.Q.; Unlu, S.; Su, A.-J.A.; Tannahill, G.M.; Galson, D.L.; O’Neill, L.A.; Auron, P.E. Distinct Mechanisms for Induction and Tolerance Regulate the Immediate Early Genes Encoding Interleukin 1β and Tumor Necrosis Factor α. PLoS ONE 2013, 8, e70622. [Google Scholar] [CrossRef]
- Hiscott, J.; Marois, J.; Garoufalis, J.; D’Addario, M.; Roulston, A.; Kwan, I.; Pepin, N.; Lacoste, J.; Nguyen, H.; Bensi, G. Characterization of a functional NF-kappa B site in the human interleukin 1 beta promoter: Evidence for a positive autoregulatory loop. Mol. Cell. Biol. 1993, 13, 6231–6240. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, J.; Saito, K.; Waterman, W.R.; Webb, A.C.; E Auron, P. Transcription factors NF-IL6 and CREB recognize a common essential site in the human prointerleukin 1 beta gene. Mol. Cell. Biol. 1994, 14, 7285–7297. [Google Scholar] [CrossRef][Green Version]
- Chandra, G.; Cogswell, J.P.; Miller, L.R.; Godlevski, M.M.; Stinnett, S.W.; Noel, S.L.; Kadwell, S.H.; A Kost, T.; Gray, J.G. Cyclic AMP signaling pathways are important in IL-1 beta transcriptional regulation. J. Immunol. 1995, 155, 4535–4543. [Google Scholar]
- Chen, Y.; Zhuang, S.; Cassenaer, S.; Casteel, D.E.; Gudi, T.; Boss, G.R.; Pilz, R.B. Synergism between Calcium and Cyclic GMP in Cyclic AMP Response Element-Dependent Transcriptional Regulation Requires Cooperation between CREB and C/EBP-β. Mol. Cell. Biol. 2003, 23, 4066–4082. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.-G.; Kang, S.-K.; Noh, S.-H.; Park, K.-K.; Chang, Y.-C.; Lee, Y.-C.; Kim, C.-H. PGE2 induces IL-1β gene expression in mouse osteoblasts through a cAMP–PKA signaling pathway. Int. Immunopharmacol. 2004, 4, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Zasłona, Z.; Pålsson-McDermott, E.M.; Menon, D.; Haneklaus, M.; Flis, E.; Prendeville, H.; Corcoran, S.E.; Peters-Golden, M.; O’Neill, L.A. The Induction of Pro–IL-1β by Lipopolysaccharide Requires Endogenous Prostaglandin E2Production. J. Immunol. 2017, 198, 3558–3564. [Google Scholar] [CrossRef]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.T.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nat. Cell Biol. 2013, 496, 238–242. [Google Scholar] [CrossRef]
- Ménégaut, L.; Thomas, C.; Jalil, A.; Julla, J.B.; Magnani, C.; Ceroi, A.; Basmaciyan, L.; Dumont, A.; Le Goff, W.; Mathew, M.J.; et al. Interplay between Liver X Receptor and Hypoxia Inducible Factor 1α Potentiates Interleukin-1β Production in Human Macrophages. Cell Rep. 2020, 31, 107665. [Google Scholar] [CrossRef]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef] [PubMed]
- Myles, I.A.; Fontecilla, N.M.; Valdez, P.A.; Vithayathil, P.J.; Naik, S.; Belkaid, Y.; Ouyang, W.; Datta, S.K. Signaling via the IL-20 receptor inhibits cutaneous production of IL-1β and IL-17A to promote infection with methicillin-resistant Staphylococcus aureus. Nat. Immunol. 2013, 14, 804–811. [Google Scholar] [CrossRef] [PubMed]
- Doitsh, G.; Galloway, N.L.K.; Geng, X.; Yang, Z.; Monroe, K.M.; Zepeda, O.; Hunt, P.W.; Hatano, H.; Sowinski, S.; Muñoz-Arias, I.; et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nat. Cell Biol. 2013, 505, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Martin, B.N.; Wang, C.; Zhang, C.-J.; Kang, Z.; Gulen, M.F.; Zepp, J.A.; Zhao, J.; Bian, G.; Do, J.-S.; Min, B.; et al. T cell–intrinsic ASC critically promotes TH17-mediated experimental autoimmune encephalomyelitis. Nat. Immunol. 2016, 17, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Arbore, G.; West, E.E.; Spolski, R.; Robertson, A.A.B.; Klos, A.; Rheinheimer, C.; Dutow, P.; Woodruff, T.M.; Yu, Z.X.; O’Neill, L.A.; et al. T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4+ T cells. Science 2016, 352, aad1210. [Google Scholar] [CrossRef] [PubMed]
- Pulugulla, S.H.; Packard, T.A.; Galloway, N.L.; Grimmett, Z.W.; Doitsh, G.; Adamik, J.; Galson, D.L.; Greene, W.C.; Auron, P.E. Distinct mechanisms regulate IL1B gene transcription in lymphoid CD4 T cells and monocytes. Cytokine 2018, 111, 373–381. [Google Scholar] [CrossRef]
- Wan, C.-K.; Li, P.; Spolski, R.; Oh, J.; Andraski, A.B.; Du, N.; Yu, Z.-X.; Dillon, C.P.; Green, D.R.; Leonard, W.J. IL-21-mediated non-canonical pathway for IL-1β production in conventional dendritic cells. Nat. Commun. 2015, 6, 7988. [Google Scholar] [CrossRef][Green Version]
- Huang, X.; Feng, Z.; Jiang, Y.; Li, J.; Xiang, Q.; Guo, S.; Yang, C.; Fei, L.; Guo, G.; Zheng, L.; et al. VSIG4 mediates transcriptional inhibition of Nlrp3 and Il-1β in macrophages. Sci. Adv. 2019, 5, eaau7426. [Google Scholar] [CrossRef]
- Hirsch, E.; Irikura, V.M.; Paul, S.M.; Hirsh, D. Functions of interleukin 1 receptor antagonist in gene knockout and overproducing mice. Proc. Natl. Acad. Sci. USA 1996, 93, 11008–11013. [Google Scholar] [CrossRef]
- Arend, W.P.; Smith, M.F.; Janson, R.W.; Joslin, F.G. IL-1 receptor antagonist and IL-1 beta production in human monocytes are regulated differently. J. Immunol. 1991, 147, 1530–1536. [Google Scholar] [PubMed]
- La, E.; Fischer, S.M. Transcriptional Regulation of Intracellular IL-1 Receptor Antagonist Gene by IL-1α in Primary Mouse Keratinocytes. J. Immunol. 2001, 166, 6149–6155. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Mier, J.W.; Vogel, W.; E Aulitzky, W.; Wiedermann, C.J.; Vannier, E.; Huber, C.; A Dinarello, C. Induction of circulating IL-1 receptor antagonist by IFN treatment. J. Immunol. 1993, 150, 4687–4692. [Google Scholar] [PubMed]
- Tamassia, N.; Castellucci, M.; Rossato, M.; Gasperini, S.; Bosisio, D.; Giacomelli, M.; Badolato, R.; Cassatella, M.A.; Bazzoni, F. Uncovering an IL-10-dependent NF-KB recruitment to the IL-1ra promoter that is impaired in STAT3 functionally defective patients. FASEB J. 2009, 24, 1365–1375. [Google Scholar] [CrossRef]
- Camilli, G.; Bohm, M.; Piffer, A.C.; Lavenir, R.; Williams, D.L.; Neven, B.; Grateau, G.; Georgin-Lavialle, S.; Quintin, J. β-Glucan–induced reprogramming of human macrophages inhibits NLRP3 inflammasome activation in cryopyrinopathies. J. Clin. Investig. 2020, 130, 4561–4573. [Google Scholar] [CrossRef]
- Quintin, J.; Saeed, S.; Martens, J.H.; Giamarellos-Bourboulis, E.J.; Ifrim, D.C.; Logie, C.; Jacobs, L.; Jansen, T.; Kullberg, B.-J.; Wijmenga, C.; et al. Candida albicans Infection Affords Protection against Reinfection via Functional Reprogramming of Monocytes. Cell Host Microbe 2012, 12, 223–232. [Google Scholar] [CrossRef]
- Christ, A.; Günther, P.; Lauterbach, M.A.; Duewell, P.; Biswas, D.; Pelka, K.; Scholz, C.J.; Oosting, M.; Haendler, K.; Baßler, K.; et al. Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming. Cell 2018, 172, 162–175. [Google Scholar] [CrossRef]
- Grandemange, S.; Sanchez, E.; Louis-Plence, P.; Mau-Them, F.T.; Bessis, D.; Coubes, C.; Frouin, E.; Seyger, M.; Girard, M.; Puechberty, J.; et al. A new autoinflammatory and autoimmune syndrome associated with NLRP1 mutations: NAIAD (NLRP1-associated autoinflammation with arthritis and dyskeratosis). Ann. Rheum. Dis. 2016, 76, 1191–1198. [Google Scholar] [CrossRef]
- Zhong, F.L.; Mamaï, O.; Sborgi, L.; Boussofara, L.; Hopkins, R.; Robinson, K.; Szeverényi, I.; Takeichi, T.; Balaji, R.; Lau, A.; et al. Germline NLRP1 Mutations Cause Skin Inflammatory and Cancer Susceptibility Syndromes via Inflammasome Activation. Cell 2016, 167, 187–202. [Google Scholar] [CrossRef]
- Weiss, E.S.; Girard-Guyonvarc’H, C.; Holzinger, D.; De Jesus, A.A.; Tariq, Z.; Picarsic, J.; Schiffrin, E.J.; Foell, D.; Grom, A.A.; Ammann, S.; et al. Interleukin-18 diagnostically distinguishes and pathogenically promotes human and murine macrophage activation syndrome. Blood 2018, 131, 1442–1455. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.N.; Lachmann, H.J.; McDermott, M.F. Interleukin-1–Receptor Antagonist in the Muckle–Wells Syndrome. N. Engl. J. Med. 2003, 348, 2583–2584. [Google Scholar] [CrossRef]
- Notarnicola, C.; Boizet-Bonhoure, B.; Barbara, P.D.S.; A Osta, M.; Cattan, D.; Touitou, I. Characterization of new mutations in the 5′-flanking region of the familial Mediterranean fever gene. Genes Immun. 2009, 10, 273–279. [Google Scholar] [CrossRef][Green Version]
- Kruidenier, L.; Chung, C.-W.; Cheng, Z.; Liddle, J.; Che, K.; Joberty, G.; Bantscheff, M.; Bountra, C.; Bridges, A.; Diallo, H.; et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nat. Cell Biol. 2012, 488, 404–408. [Google Scholar] [CrossRef]






{kind=link}
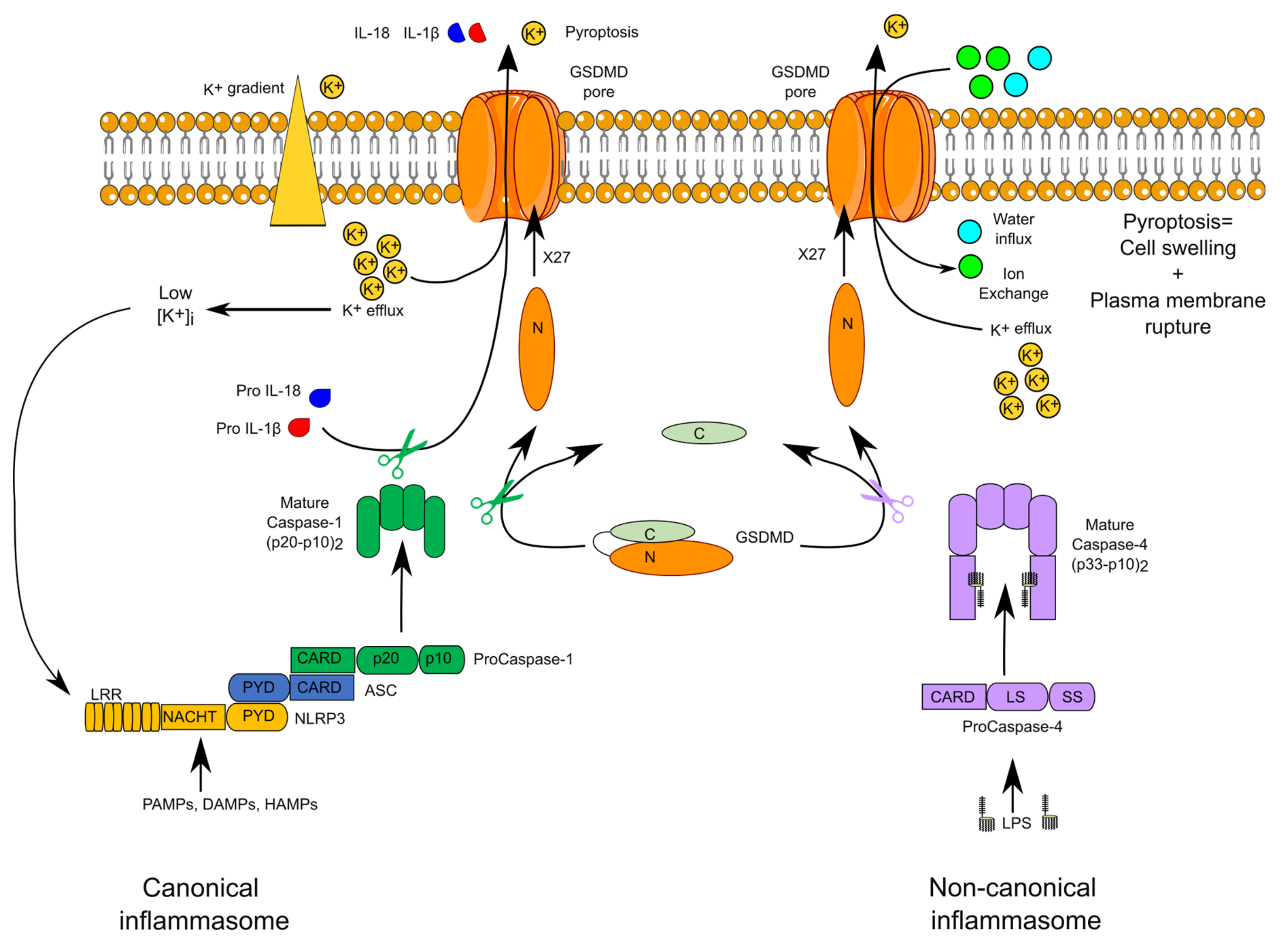{kind=link}
{kind=link}
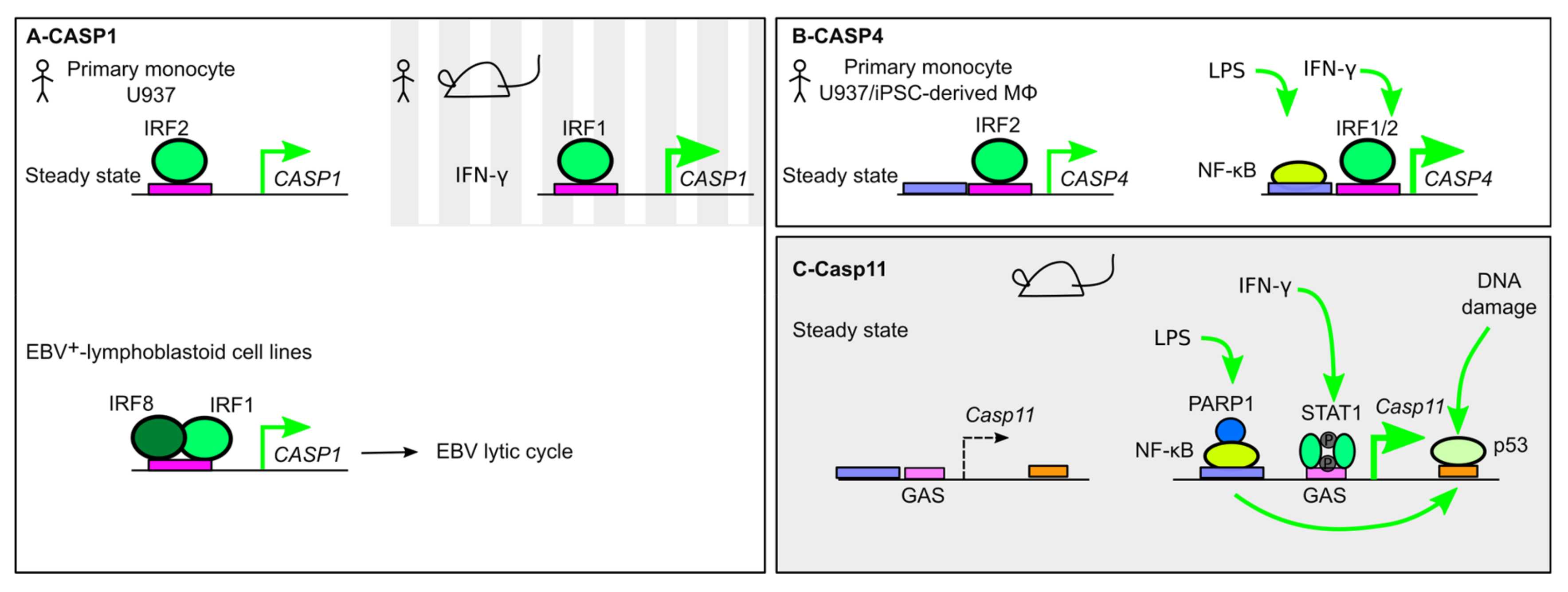{kind=link}
{kind=link}
{kind=link}
| TF | Context | Target | Species | Outcome |
|---|---|---|---|---|
| AhR | Xenobiotics, metabolites | Nlrp3 | m | Anti-inflammatory |
| ATF4 | ER stress | NLRP1 | h | Pro-inflammatory |
| BLIMP1 | Human steady state | AIM2 | h | Negative regulation |
| C/EBPβ | Cell differentiation | MEFV, IL1B, IL1RN | h | Cell-specific expression |
| CHOP | ER stress | Casp11 | m | LPS-induced lung inflammation |
| CREB | PGE2 signaling | IL1B | h | Pro-inflammatory |
| GFI1 | Negative feedback loop | Nlrp3 | m | Anti-inflammatory |
| HIF1α | Metabolism/Hypoxia | IL1B | h, m | Pro-inflammatory |
| IRF1 | IFN-Υ treatment | CASP1 | h, m | Pro-inflammatory |
| IRF1 | IFN-Υ treatment | IL18BP | h | Anti-inflammatory |
| IRF1/2 | IFN-Υ treatment | AIM2 | h | Pro-inflammatory |
| IRF2 | Steady state | CASP4 | h | Inflammasome competence |
| IRF2 | Steady state | GSDMD | h, m | Inflammasome competence |
| IRF4 | cDC1-steady state | Nlrc4, Il1b, Pycard | m | Antigen presentation |
| IRF8 | cDC2-steady state | Nlrp3, Nlrc4, Pycard, Il1b | m | Antigen presentation |
| IRF8 | BMDM-steady state | Naip2, 5, 6, Nlrc4 | m | Resistance to Salmonella |
| IRF8 | EBV + lymphoblastoid cells | CASP1 | h | EBV lytic cycle |
| ISGF3 | type I IFN response | Il18 | m | LPS-mediated induction |
| LXRα | Metabolism | IL1B | h | Pro-inflammatory |
| NF-κB | Pro-inflammatory signals | NLRP3, MEFV, CASP4, CASP5, Casp11, Gsdmd, IL18, IL1B, IL1RN | h, m | Kinetics of inflammasome response |
| NFAT5 | Osmotic stress | Nlrp3 | m | Pro-inflammatory |
| NR1D1 | Circadian clock | Nlrp3 | m | Circadian oscillation |
| NRF2 | Oxidative stress | Il1b | m | Pro-inflammatory |
| p53 | DNA damage | Casp11, CASP1, NLRC4 | m, h | Pro-inflammatory |
| PU.1 | Cell differentiation | IL-18, IL1B, IL1RN | h | Cell-specific expression |
| SREBP-1A | NF-κB activation | Nlrp1a | m | Metabolic inflammation |
| STAT1 | IFN-Υ treatment | AIM2 | h | Induction |
| STAT1 | IFN-Υ treatment | IL18BP | h | Anti-inflammatory |
| STAT3 | IL-10, IL-20R family | Il1b | m | Anti-inflammatory |
| STAT3 | IL-21 in DC | Il1b | m | Resistance to pneumonia Virus of mice |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cornut, M.; Bourdonnay, E.; Henry, T. Transcriptional Regulation of Inflammasomes. Int. J. Mol. Sci. 2020, 21, 8087. https://doi.org/10.3390/ijms21218087
Cornut M, Bourdonnay E, Henry T. Transcriptional Regulation of Inflammasomes. International Journal of Molecular Sciences. 2020; 21(21):8087. https://doi.org/10.3390/ijms21218087
Chicago/Turabian StyleCornut, Maxence, Emilie Bourdonnay, and Thomas Henry. 2020. "Transcriptional Regulation of Inflammasomes" International Journal of Molecular Sciences 21, no. 21: 8087. https://doi.org/10.3390/ijms21218087
APA StyleCornut, M., Bourdonnay, E., & Henry, T. (2020). Transcriptional Regulation of Inflammasomes. International Journal of Molecular Sciences, 21(21), 8087. https://doi.org/10.3390/ijms21218087