Vitamin B3-Based Biologically Active Compounds as Inhibitors of Human Cholinesterases

 , ,
, , 



Abstract

1. Introduction
2. Results
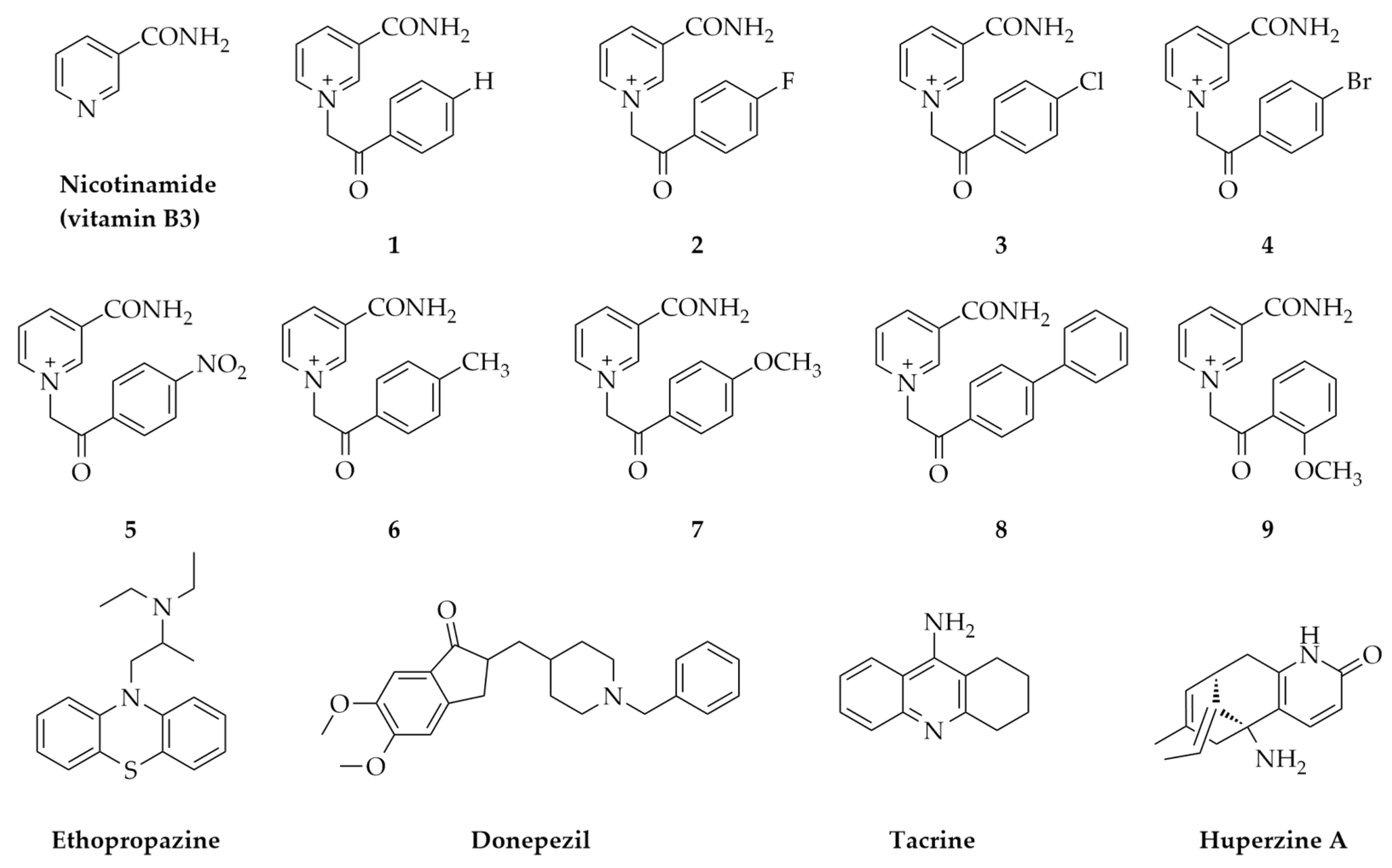
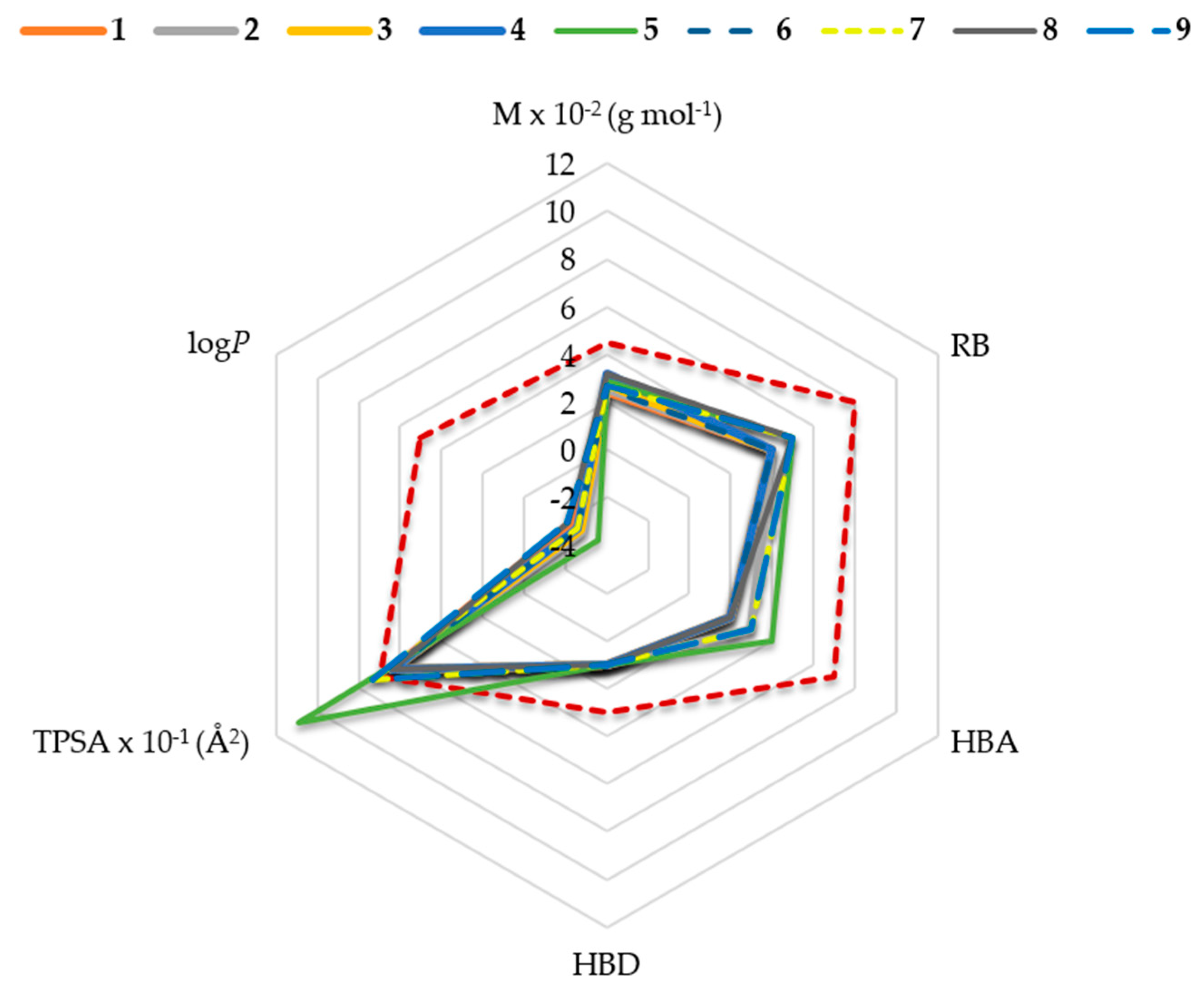
2.1. In Silico Physicochemical Characteristics of Nicotinamide Derivatives
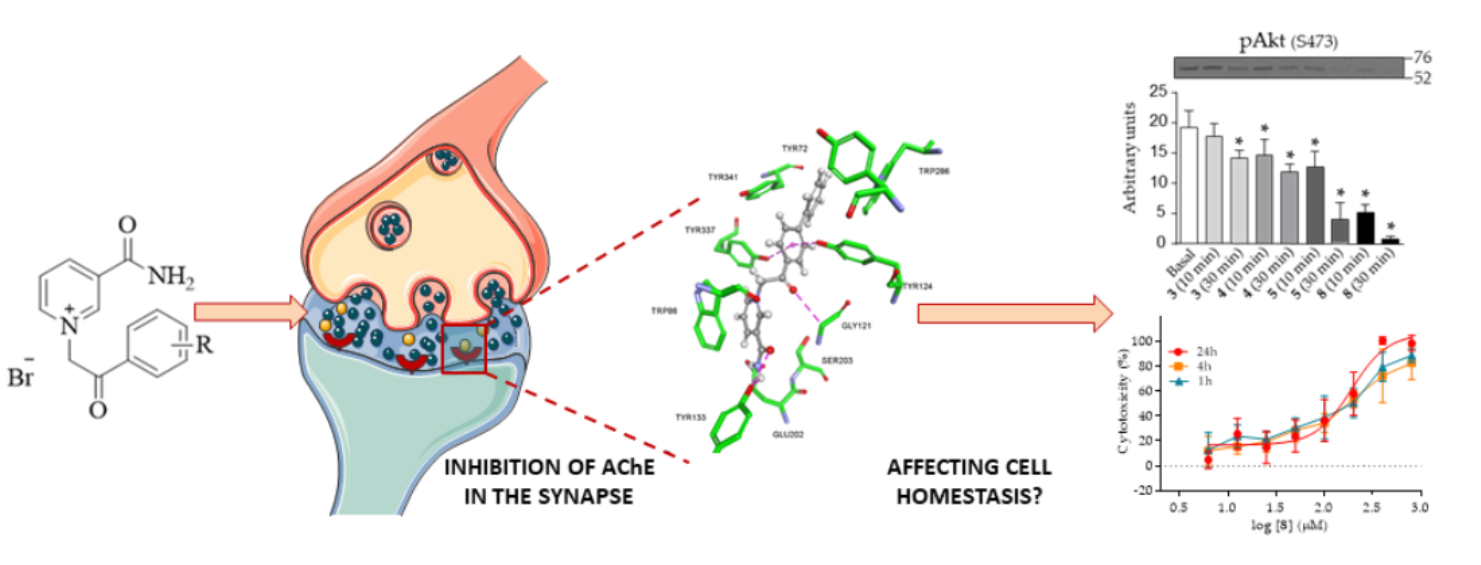
2.2. Reversible Inhibition of AChE and BChE by Nicotinamide Derivatives
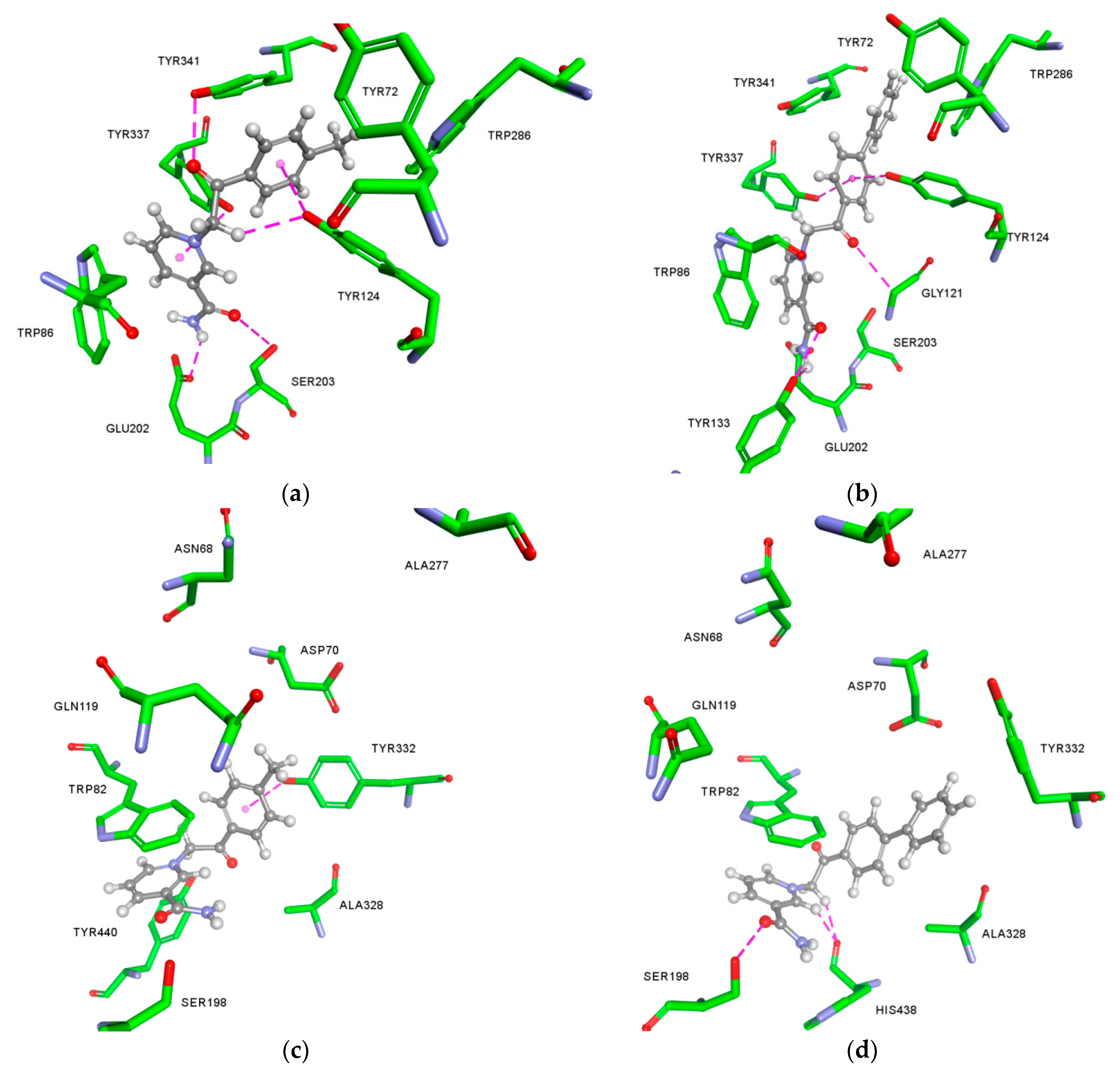
2.3. Molecular Docking Studies
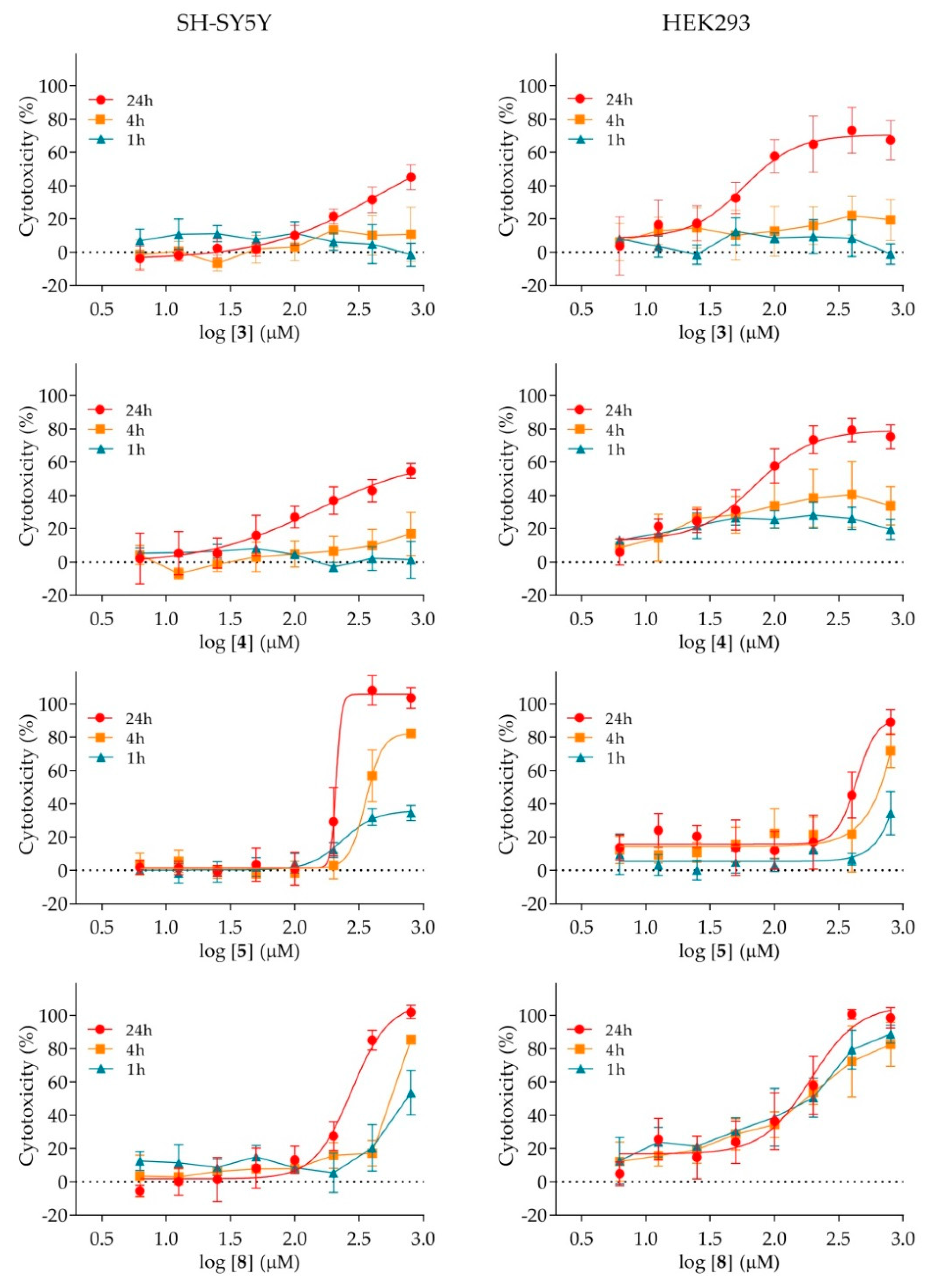
2.4. Cytotoxicity of Nicotinamide Derivatives
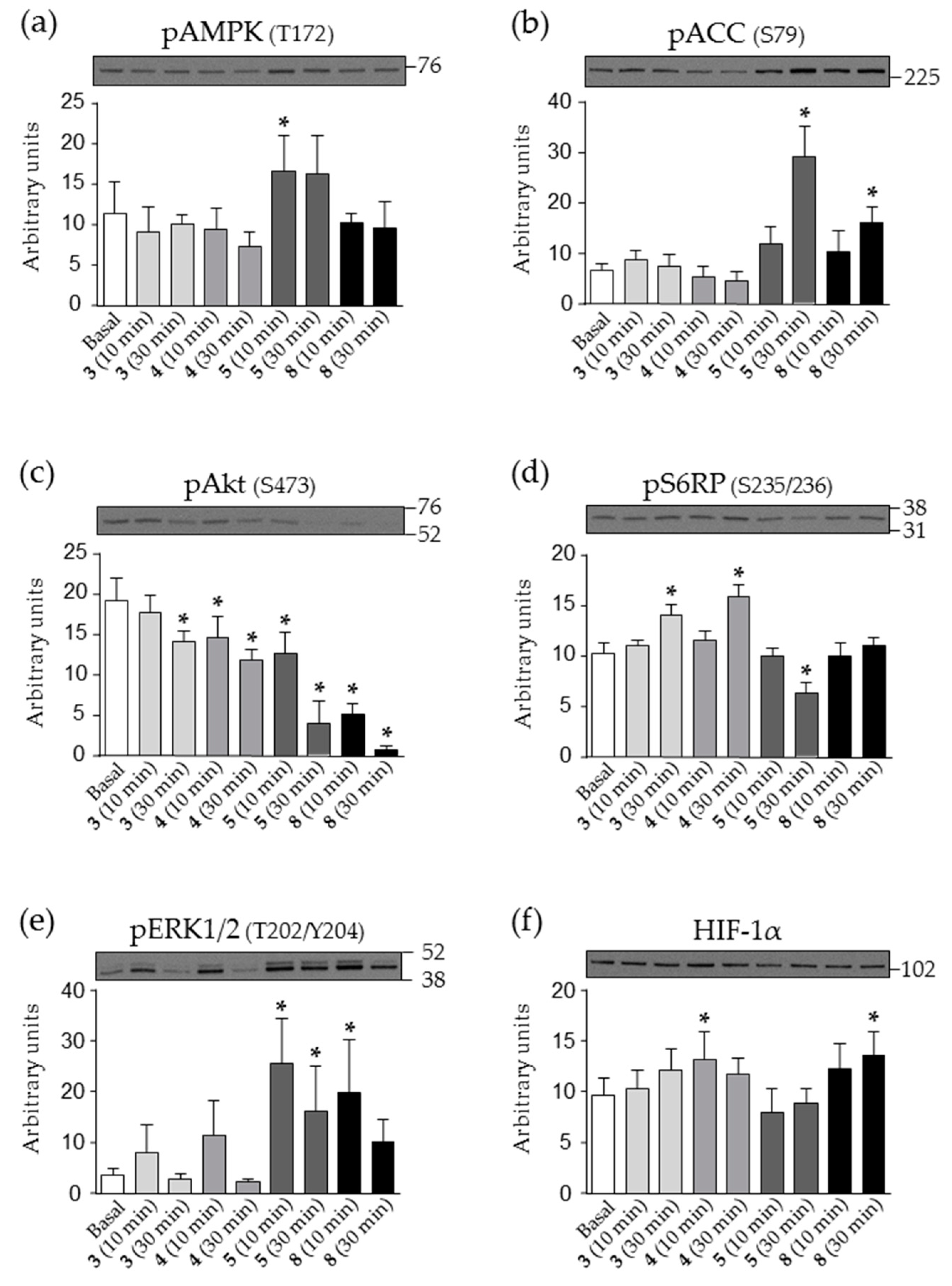
2.5. Effect of Nicotinamide Derivatives on Intracellular Signalling in HEK293 Cells
3. Discussion
4. Materials and Methods
4.1. Nicotinamide Derivatives and Their Physicochemical Characteristics In Silico
4.2. Reagents and Enzymes
4.3. AChE and BChE Reversible Inhibition
4.4. Molecular Docking
4.5. Cytotoxicity Assay
4.6. Analysis of Intracellular Signalling by Western Blot (WB)
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACC | acetyl-CoA carboxylase |
| AChE | acetylcholinesterase |
| AD | Alzheimer’s disease |
| Akt | protein kinase B |
| AMPK | AMP-activated protein kinase |
| ATCh | acetylthiocholine |
| BChE | butyrylcholinesterase |
| DMEM | Dulbecco′s Modified Eagle′s Medium |
| DTNB | 5,5′-dithiobis(2-nitrobenzoic acid |
| EDTA | ethylenediaminetetraacetic acid |
| EMEM | Eagle’s Minimum Essential Medium |
| ERK | extracellular signal-regulated kinase |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
| HBA | hydrogen bond acceptors |
| HBD | hydrogen bond donors |
| HIF-1α | hypoxia-inducible factor-1α |
| IC50 | half maximal inhibitory concentration |
| logP | lipophilicity |
| M | molecular weight |
| mTOR | mammalian target of rapamycin |
| NAD | nicotinamide adenine dinucleotide |
| PBS | phosphate buffer saline |
| RB | number of rotating bonds |
| S6RP | S6 ribosomal protein |
| TPSA | topological polar surface area |
Appendix A
Appendix A.1. Molecular Docking
Appendix A.2. The Predicted Non-Bonding Interactions for Modelled Complexes by Molecular Docking

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model Complex | Amino Acid | Type of Non-Bonding Interaction |
|---|---|---|
| 6-AChE | Ser203 | Hydrogen bond |
| Tyr341 | Hydrogen bond, π-π stacked, π-alkyl | |
| Glu202 | Hydrogen bond | |
| Tyr124 | Hydrogen bond (x2), π-π T-shaped | |
| Trp86 | π-cation (x2), π-π stacked (x2) | |
| Tyr337 | Hydrogen bond, π-π stacked | |
| Trp286 | π-alkyl | |
| 8-AChE | Tyr133 | Hydrogen bond (x2) |
| Glu202 | Hydrogen bond | |
| Gly121 | Hydrogen bond | |
| Trp86 | π-cation (x2), π-π stacked (x2) | |
| Tyr124 | Hydrogen bond, π-π T-shaped | |
| Tyr337 | Hydrogen bond, π-π T-shaped | |
| Trp286 | π-π stacked | |
| Tyr341 | π-π T-shaped | |
| 6-BChE | Trp82 | π-cation (x2), π-π stacked (x2), π-π T-shaped |
| Tyr332 | Hydrogen bond, π-π stacked, π-alkyl | |
| Tyr440 | π-π stacked | |
| Ala328 | π-alkyl | |
| 8-BChE | Ser198 | Hydrogen bond |
| His438 | Hydrogen bond (x2) | |
| Trp82 | π-cation (x2), π-π stacked (x2), π-π T-shaped (x2) | |
| Asp70 | π-anion | |
| Ala328 | π-alkyl (x2) |
References
- Knowles, R.B.; Wyart, C.; Buldyrev, S.V.; Cruz, L.; Urbanc, B.; Hasselmo, M.E.; Stanley, H.E.; Hyman, B.T. Plaque-induced neurite abnormalities: Implications for disruption of neural networks in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 1999, 96, 5274–5279. [Google Scholar] [CrossRef] [PubMed]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Primers 2015, 1, 15056. [Google Scholar] [CrossRef] [PubMed]
- Moss, D.E. Improving anti-neurodegenerative benefits of acetylcholinesterase inhibitors in Alzheimer’s disease: Are Irreversible Inhibitors the Future? Int. J. Mol. Sci. 2020, 21, 3438. [Google Scholar] [CrossRef] [PubMed]
- McGleenon, B.M.; Dynan, K.B.; Passmore, A.P. Acetylcholinesterase inhibitors in Alzheimer’s disease. Br. J. Clin. Pharmacol. 1999, 48, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Hasin, Y.; Avidan, N.; Bercovich, D.; Korczyn, A.D.; Silman, I.; Beckmann, J.S.; Sussman, J.L. Analysis of genetic polymorphisms in acetylcholinesterase as reflected in different populations. Curr. Alzheimer Res. 2005, 2, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Kapková, P.; Alptüzün, V.; Frey, P.; Erciyas, E.; Holzgrabe, U. Search for dual function inhibitors for Alzheimer’s disease: Synthesis and biological activity of acetylcholinesterase inhibitors of pyridinium-type and their Aβ fibril formation inhibition capacity. Bioorg. Med. Chem. 2006, 14, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Więckowska, A.; Kołaczkowski, M.; Bucki, A.; Justyna, G.; Monika, M.; Krzysztof, W.; Paula, Z.; Agata, S.; Grzegorz, K.; Monika, G.-L.; et al. Novel multi-target-directed ligands for Alzheimer’s disease: Combining cholinesterase inhibitors and 5-HT6 receptor antagonists. Design, synthesis and biological evaluation. Eur. J. Med. Chem. 2016, 124, 63–81. [Google Scholar] [CrossRef]
- Ramos, E.; Palomino-Antolín, A.; Bartolini, M.; Iriepa, I.; Moraleda, I.; Diez-Iriepa, D.; Samadi, A.; Cortina, C.V.; Chioua, M.; Egea, J.; et al. QuinoxalineTacrine QT78, a Cholinesterase Inhibitor as a Potential Ligand for Alzheimer’s Disease Therapy. Molecules 2019, 24, 1503. [Google Scholar] [CrossRef]
- Mehta, M.; Adem, A.; Sabbagh, M. New acetylcholinesterase inhibitors for Alzheimer’s disease. Int. J. Alzheimers Dis. 2012, 2012, 728983. [Google Scholar] [CrossRef]
- Galimberti, D.; Scarpini, E. Old and new acetylcholinesterase inhibitors for Alzheimer’s disease. Expert Opin. Investig. Drugs. 2016, 25, 1181–1187. [Google Scholar] [CrossRef]
- Darvesh, S. Butyrylcholinesterase as a diagnostic and therapeutic target for Alzheimer’s disease. Curr. Alzheimer Res. 2016, 13, 1173–1177. [Google Scholar] [CrossRef] [PubMed]
- Jasiecki, J.; Wasąg, B. Butyrylcholinesterase protein ends in the pathogenesis of Alzheimer’s disease-Could BCHE genotyping be helpful in Alzheimer’s therapy? Biomolecules 2019, 9, 592. [Google Scholar] [CrossRef] [PubMed]
- Masson, P.; Carletti, E.; Nachon, F. Structure, activities and biomedical applications of human butyrylcholinesterase. Protein Pept Lett. 2009, 16, 1215–1224. [Google Scholar] [CrossRef]
- Nordberg, A.; Ballard, C.; Bullock, R.; Darreh-Shori, T.; Somogyi, M. A review of butyrylcholinesterase as a therapeutic target in the treatment of Alzheimer’s disease. Prim. Care Companion CNS Disord 2013, 15. PCC.12r01412. [Google Scholar] [CrossRef] [PubMed]
- Liston, D.R.; Nielsen, J.A.; Villalobos, A.; Chapin, D.; Jones, S.B.; Hubbard, S.T.; Shalaby, I.A.; Ramirez, A.; Nason, D.; White, W.F. Pharmacology of selective acetylcholinesterase inhibitors: Implications for use in Alzheimer’s disease. Eur. J. Pharmacol. 2004, 486, 9–17. [Google Scholar] [CrossRef]
- Venneri, A.; Lane, R. Effects of cholinesterase inhibition on brain white matter volume in Alzheimer’s disease. Neuroreport 2009, 20, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Gilhus, N.E.; Verschuuren, J.J. Myasthenia gravis: Subgroup classification and therapeutic strategies. Lancet Neurol. 2015, 14, 1023–1036. [Google Scholar] [CrossRef]
- Gilhus, N.E. Myasthenia Gravis. N. Engl. J. Med. 2016, 375, 2570–2581. [Google Scholar] [CrossRef]
- Gilhus, N.E. Myasthenia and the neuromuscular junction. Curr. Opin. Neurol. 2015, 25, 523–529. [Google Scholar] [CrossRef]
- Orhan, G.; Orhan, I.; Öztekin-Subutay, N.; Ak, F.; Şener, B. Contemporary anticholinesterase pharmaceuticals of natural origin and their synthetic analogues for the treatment of Alzheimer’s disease. Recent Pat. CNS Drug Discov. 2009, 4, 43–51. [Google Scholar] [CrossRef]
- Katalinić, M.; Rusak, G.; Domaćinović Barović, J.; Šinko, G.; Jelić, D.; Antolović, R.; Kovarik, Z. Structural aspects of flavonoids as inhibitors of human butyrylcholinesterase. Eur. J. Med. Chem. 2010, 45, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Tumiatti, V.; Minarini, A.; Bolognesi, M.L.; Milelli, A.; Rosini, M.; Melchiorre, C. Tacrine derivatives and Alzheimer’s disease. Curr. Med. Chem. 2010, 17, 1825–1838. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Kaur, M.; Kukreja, H.; Chugh, R.; Silakari, O.; Singh, D. Acetylcholinesterase inhibitors as Alzheimer therapy: From nerve toxins to neuroprotection. Eur. J. Med. Chem. 2013, 70, 165–188. [Google Scholar] [CrossRef] [PubMed]
- Bosak, A.; Opsenica, D.M.; Šinko, G.; Zlatar, M.; Kovarik, Z. Structural aspects of 4-aminoquinolines as reversible inhibitors of human acetylcholinesterase and butyrylcholinesterase. Chem. Biol. Inter. 2019, 308, 101–109. [Google Scholar] [CrossRef]
- Musilek, K.; Komloova, M.; Holas, O.; Hrabinova, M.; Pohanka, M.; Dohnal, V.; Nachon, F.; Dolezal, M.; Kuca, K. Preparation and in vitro screening of symmetrical bis-isoquinolinium cholinesterase inhibitors bearing various connecting linkage-implications for early Myasthenia gravis treatment. Eur. J. Med. Chem. 2011, 46, 811–818. [Google Scholar] [CrossRef]
- Kharlamova, A.D.; Lushchekina, S.V.; Petrov, K.A.; Kharlamova, A.D.; Lushchekina, S.V.; Petrov, K.A.; Kots, E.D.; Nachon, F.; Villard-Wandhammer, M.; Zueva, I.V.; et al. Slow-binding inhibition of acetylcholinesterase by an alkylammonium derivative of 6-methyluracil: Mechanism and possible advantages for myasthenia gravis treatment. Biochem. J. 2016, 473, 1225–1236. [Google Scholar] [CrossRef]
- Salloway, S.; Ferris, S.; Kluger, A.; Goldman, R.; Griesing, T.; Kumar, D.; Richardson, S.; Donepezil 401 Study Group. Efficacy of donepezil in mild cognitive impairment: A randomized placebo-controlled trial. Neurology 2004, 63, 651–657. [Google Scholar] [CrossRef]
- Petersen, R.C.; Thomas, R.G.; Grundman, M.; Petersen, R.C.; Thomas, R.G.; Grundman, M.; Bennett, D.; Doody, R.; Ferris, S.; Galasko, D.; et al. Vitamin E and donepezil for the treatment of mild cognitive impairment. N. Engl. J. Med. 2005, 352, 2379–2388. [Google Scholar] [CrossRef]
- Tariot, P.N.; Solomon, P.R.; Morris, J.C.; Kershaw, P.; Lilienfeld, S.; Ding, C. A 5-month, randomized, placebo-controlled trial of galantamine in AD. Neurology 2000, 54, 2269–2276. [Google Scholar] [CrossRef]
- Corey-Bloom, J.; Anand, R.; Veach, J. A randomized trial evaluating the efficacy and safety of ENA 713 (rivastigmine tartrate), a new acetylcholinesterase inhibitor, in patients with mild to moderately severe Alzheimer’s disease. Int. J. Geriatr. Psychopharmacol. 1998, 1, 55–65. [Google Scholar]
- Crismon, M.L. Tacrine: First drug approved for Alzheimer’s disease. Ann. Pharmacother. 1994, 28, 744–751. [Google Scholar] [CrossRef]
- Watkins, P.B.; Zimmerman, H.J.; Knapp, M.J.; Gracon, S.I.; Lewis, K.W. Hepatotoxic effects of tacrine administration in patients with Alzheimer’s disease. JAMA 1994, 271, 992–998. [Google Scholar] [CrossRef] [PubMed]
- Farmakidis, C.; Pasnoor, M.; Dimachkie, M.M.; Barohn, R.J. Treatment of Myasthenia Gravis. Neurol. Clin. 2018, 36, 311–337. [Google Scholar] [CrossRef]
- Zhan, Z.J.; Bian, H.L.; Wang, J.W.; Shan, W.G. Synthesis of physostigmine analogues and evaluation of their anticholinesterase activities. Bioorg. Med. Chem. Lett. 2010, 20, 1532–1534. [Google Scholar] [CrossRef]
- Anand, P.; Singh, B. A review on cholinesterase inhibitors for Alzheimer’s disease. Arch. Pharm. Res. 2013, 36, 375–399. [Google Scholar] [CrossRef]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimers Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef]
- Pankiewicz, K.W. Novel nicotinamide adenine dinucleotide analogues as potential anticancer agents: Quest for specific inhibition of inosine monophosphate dehydrogenase. Pharmacol. Ther. 1997, 76, 89–100. [Google Scholar] [CrossRef]
- Hwang, E.S.; Song, S.B. Nicotinamide is an inhibitor of SIRT1 in vitro, but can be a stimulator in cells. Cell Mol. Life Sci. 2017, 74, 3347–3362. [Google Scholar] [CrossRef]
- Song, S.B.; Park, J.S.; Chung, G.J.; Lee, I.H.; Hwang, E.S. Diverse therapeutic efficacies and more diverse mechanisms of nicotinamide. Metabolomics 2019, 15, 137. [Google Scholar] [CrossRef]
- Kamat, J.P.; Devasagayam, T.P. Nicotinamide (vitamin B3) as an effective antioxidant against oxidative damage in rat brain mitochondria. Redox Rep. 1999, 4, 179–184. [Google Scholar] [CrossRef]
- Esposito, E.; Aldrees, S.; Mastromonaco, C.; Zoroquiain, P.; Vila, N.; Logan, P.T.; Hari, S.; Burnier, M.N. Evaluation of nicotinamide as an anti-inflammatory and anti-angiogenic agent in uveal melanoma cell lines. Arq. Bras. Oftalmol. 2017, 80, 74–77. [Google Scholar] [CrossRef]
- Godin, A.M.; Ferreira, W.C.; Rocha, L.T.; Seniuk, J.G.T.; Paiva, A.L.L.; Merlo, L.A.; Nascimento, E.B., Jr.; Bastos, L.F.S.; Coelho, M.M. Antinociceptive and anti-inflammatory activities of nicotinamide and its isomers in different experimental models. Pharmacol. Biochem. Behav. 2011, 99, 782–788. [Google Scholar] [CrossRef]
- Fricker, R.A.; Green, E.L.; Jenkins, S.I.; Griffin, S.M. The influence of nicotinamide on health and disease in the central nervous system. Int. J. Tryptophan Res. 2018, 11, 1178646918776658. [Google Scholar] [CrossRef]
- Siber, T.; Bušić, V.; Zobundžija, D.; Roca, S.; Vikić-Topić, D.; Vrandečić, K.; Gašo-Sokač, D. An improved method for the quaternization of nicotinamide and antifungal activities of its derivatives. Molecules 2019, 24, 1001. [Google Scholar] [CrossRef] [PubMed]
- Bušić, V.; Roca, S.; Vikić-Topić, D.; Vrandečić, K.; Ćosić, J.; Molnar, M.; Gašo-Sokač, D. Eco-friendly quaternization of nicotinamide and 2-bromoacetophenones in deep eutectic solvents. Antifungal activity of the products. Environ. Chem. Lett. 2020, 18, 889–894. [Google Scholar] [CrossRef]
- Pajouhesh, H.; Lenz, G.R. Medicinal chemical properties of successful central nervous system drugs. J. Am. Soc. Exp. Neurother 2005, 2, 541–553. [Google Scholar] [CrossRef]
- Simeon-Rudolf, V.; Šinko, G.; Štuglin, A.; Reiner, E. Inhibition of human blood acetylcholinesterase and butyrylcholinesterase by ethopropazine. Croat. Chem. Acta 2001, 74, 173–182. [Google Scholar]
- Reiner, E.; Radić, Z. Mechanism of action of cholinesterase inhibitors. In Cholinesterase and Cholinesterase Inhibitors; Giacobini, E., Ed.; Martin Dunitz Ltd.: London, UK, 2000; pp. 103–120. [Google Scholar]
- Saxena, A.; Fedorko, J.M.; Vinayaka, C.R.; Medhekar, R.; Radić, Z.; Taylor, P.; Lockridge, O.; Doctor, B.P. Aromatic amino-acid residues at the active and peripheral anionic sites control the binding of E2020 (Aricept) to cholinesterases. Eur. J. Biochem. 2003, 270, 4447–4458. [Google Scholar] [CrossRef]
- Giacobini, E. Cholinesterase inhibitors: From the Calabar bean to Alzheimer therapy. In Cholinestarse and Cholinesterase Inhibitors; Giacobini, E., Ed.; Martin Dunitz Ltd.: London, UK, 2000; pp. 181–227. [Google Scholar]
- Bourne, Y.; Kolb, H.C.; Radić, Z.; Sharpless, K.B.; Taylor, P.; Marchot, P. Freeze-frame inhibitor captures acetylcholinesterase in a unique conformation. Proc. Natl. Acad. Sci. USA 2004, 101, 1449–1454. [Google Scholar] [CrossRef]
- Bourne, Y.; Sharpless, K.B.; Taylor, P.; Marchot, P. Steric and dynamic parameters influencing in situ cycloadditions to form triazole inhibitors with crystalline acetylcholinesterase. J. Am. Chem. Soc. 2016, 138, 1611–1621. [Google Scholar] [CrossRef]
- Huang, N.; Shoichet, B.K. Exploiting ordered waters in molecular docking. J. Med. Chem. 2008, 51, 4862–4865. [Google Scholar] [CrossRef]
- Karlsson, D.; Fallarero, A.; Brunhofer, G.; Mayer, C.; Prakash, O.; Mohan, C.G.; Vuorela, P.; Erker, T. The exploration of thienothiazines as selective butyrylcholinesterase inhibitors. Eur. J. Pharm. Sci. 2012, 47, 190–205. [Google Scholar] [CrossRef]
- Li, J.C.; Zhang, J.; Rodrigues, M.C.; Ding, D.J.; Longo, J.P.F.; Azevedo, R.B.; Muehlmann, L.A.; Jiang, C.S. Synthesis and evaluation of novel 1,2,3-triazole-based acetylcholinesterase inhibitors with neuroprotective activity. Bioorg. Med. Chem. Lett. 2016, 26, 3881–3885. [Google Scholar] [CrossRef]
- Bosak, A.; Knežević, A.; Gazić Smilović, I.; Šinko, G.; Kovarik, Z. Resorcinol-, catechol- and saligenin-based bronchodilating β2-agonists as inhibitors of human cholinesterase activity. J. Enzyme Inhib. Med. Chem. 2017, 32, 789–797. [Google Scholar] [CrossRef]
- Bosak, A.; Ramić, A.; Šmidlehner, T.; Hrenar, T.; Primožič, I.; Kovarik, Z. Design and evaluation of selective butyrylcholinesterase inhibitors based on Cinchona alkaloid scaffold. PLoS ONE 2018, 13, e0205193. [Google Scholar] [CrossRef]
- Yang, J.; Yun, Y.; Miao, Y.; Sun, J.; Wang, X. Synthesis and biological evaluation of 3-arylbenzofuranone derivatives as potential anti-Alzheimer’s disease agents. J. Enzyme Inhib. Med. Chem. 2020, 35, 805–814. [Google Scholar] [CrossRef]
- Manning, B.D.; Toker, A. AKT/PKB signaling: Navigating the network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef]
- Evans, A.M.; Hardie, D.G. AMPK and the need to breathe and feed: What’s the matter with oxygen? Int. J. Mol. Sci. 2020, 21, 3518. [Google Scholar] [CrossRef]
- Zhuo, L.; Fu, B.; Bai, X.; Zhang, B.; Wu, L.; Cui, J.; Cui, S.; Wei, R.; Chen, X.; Cai, G. NAD blocks high glucose induced mesangial hypertrophy via activation of the sirtuins-AMPK-mTOR pathway. Cell Physiol. Biochem. 2011, 27, 681–690. [Google Scholar] [CrossRef]
- Wilson, J.W.; Shakir, D.; Batie, M.; Frost, M.; Rocha, S. Oxygen-sensing mechanisms in cells. FEBS J. 2020. [Google Scholar] [CrossRef]
- Ye, Y.H.; Ma, L.; Dai, Z.C.; Xiao, Y.; Zhang, Y.Y.; Li, D.D.; Wang, J.X.; Zhu, H.L. Synthesis and antifungal activity of nicotinamide derivatives as succinate dehydrogenase inhibitors. J. Agric. Food. Chem. 2014, 62, 4063–4071. [Google Scholar] [CrossRef]
- Lv, X.H.; Ren, Z.L.; Liu, P.; Li, B.X.; Li, Q.S.; Chu, M.J.; Cao, H.Q. Design, synthesis and biological evaluation of novel nicotinamide derivatives bearing a substituted pyrazole moiety as potential SDH inhibitors. Pest. Manag. Sci. 2017, 73, 1585–1592. [Google Scholar] [CrossRef]
- Yang, Z.; Guo, L.; Zhou, C.; Wang, X.; Yu, M.; Xul, M.; Yang, K. Synthesis and biological evaluation of nicotinamide derivatives with a diarylamine-modified scaffold as succinate dehydrogenase inhibitors. J. Pestic Sci. 2020, 45, 39–44. [Google Scholar] [CrossRef]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef]
- SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [CrossRef]
- Egan, W.J.; Lauri, G. Prediction of intestinal permeability. Adv. Drug Deliv. Rev. 2002, 54, 273–289. [Google Scholar] [CrossRef]
- Simeon-Rudolf, V.; Evans, R.T. Interlaboratory study into the proficiency of attribution of human serum butyrylcholinesterase phenotypes: Reference values of activities and inhibitor numbers. Acta Pharm. 2001, 51, 289–296. [Google Scholar]
- Reiner, E.; Bosak, A.; Simeon-Rudolf, V. Activity of cholinesterases in human whole blood measured with acetylthiocholine as substrate and ethopropazine as selective inhibitor of plasma butyrylcholinesterase. Arh. Hig. Rada Toksikol. 2004, 55, 1–4. [Google Scholar]
- Lucić Vrdoljak, A.; Čalić, M.; Radić, B.; Berend, S.; Jun, D.; Kuca, K.; Kovarik, Z. Pretreatment with pyridinium oximes improves antidotal therapy against tabun poisoning. Toxicology 2006, 228, 41–50. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Featherstone, R.M. New and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Šinko, G.; Katalinić, M.; Kovarik, Z. para- and ortho-pyridinium aldoximes in reaction with acetylthiocholine. FEBS Lett. 2006, 580, 3167–3172. [Google Scholar] [CrossRef]
- Hunter, A.; Downs, C.E. The inhibition of arginase by amino acids. J. Biol. Chem. 1945, 157, 427–446. [Google Scholar]
- Čalić, M.; Lucić Vrdoljak, A.; Radić, B.; Jelić, D.; Jun, D.; Kuča, K.; Kovarik, Z. In vitro and in vivo evaluation of pyridinium oximes: Mode of interaction with acetylcholinesterase, effect on tabun- and soman-poisoned mice and their cytotoxicity. Toxicology 2006, 219, 85–96. [Google Scholar] [CrossRef]
- Koska, J.; Spassov, V.Z.; Maynard, A.J.; Yan, L.; Austin, N.; Flook, P.K.; Venkatachalam, C.M. Fully automated molecular mechanics based induced fit protein-ligand docking method. J. Chem. Inf. Model. 2008, 48, 1965–1973. [Google Scholar] [CrossRef]
- Protein Data Bank—4PQE Crystal Structure of Human Acetylcholinesterase. Available online: https://www.rcsb.org/structure/4PQE (accessed on 27 April 2020).
- Ngamelue, M.N.; Homma, K.; Lockridge, O.; Asojo, O.A. Crystallization and X-ray structure of full-length recombinant human butyrylcholinesterase. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2007, 63, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Colletier, J.P.; Weik, M.; Jiang, H.; Moult, J.; Silman, I.; Sussman, J.K. Flexibility of aromatic residues in the active-site gorge of acetylcholinesterase: X-ray versus molecular dynamics. Biophys. J. 2008, 95, 2500–2511. [Google Scholar] [CrossRef] [PubMed]
- Rosenberry, T.L.; Brazzolotto, X.; Macdonald, I.R.; Wandhammer, M.; Trovaslet-Leroy, M.; Darvesh, S.; Nachon, F. Comparison of the binding of reversible inhibitors to human butyrylcholinesterase and acetylcholinesterase: A crystallographic, kinetic and calorimetric study. Molecules 2017, 22, 2098. [Google Scholar] [CrossRef]
- Šinko, G. Assessment of scoring functions and in silico parameters for AChE-ligand interactions as a tool for predicting inhibition potency. Chem. Biol. Interact. 2019, 308, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Katalinić, M.; Zandona, A.; Ramić, A.; Zorbaz, T.; Primožič, I.; Kovarik, Z. New Cinchona oximes evaluated as reactivators of acetylcholinesterase and butyrylcholinesterase inhibited by organophosphorus compounds. Molecules 2017, 22, 1234. [Google Scholar] [CrossRef]
- Zhang, T.; Faraggi, E.; Zhou, Y. Fluctuations of backbone torsion angles obtained from NMR-determined structures and their prediction. Proteins 2010, 78, 3353–3362. [Google Scholar] [CrossRef] [PubMed]





| Compound | −R | logBB * | Level ** |
|---|---|---|---|
| 1 | 4-H | −0.885 | 3 |
| 2 | 4-F | −0.821 | 3 |
| 3 | 4-Cl | −0.679 | 3 |
| 4 | 4-Br | −0.654 | 3 |
| 5 | 4-NO2 | −1.595 | 3 |
| 6 | 4-CH3 | −0.735 | 3 |
| 7 | 4-OCH3 | −1.031 | 3 |
| 8 | 4-Ph | −0.416 | 2 |
| 9 | 2-OCH3 | −1.031 | 3 |
| Compound | −R | Ki ± SE (µM) | Ki (AChE)/Ki (BChE) | |
|---|---|---|---|---|
| AChE | BChE | |||
| 1 | 4-H | 79 ± 11 | 232 ± 20 | 0.34 |
| 2 | 4-F | 85 ± 15 | 595 ± 73 | 0.14 |
| 3 | 4-Cl | 33 ± 5 | 224 ± 23 | 0.15 |
| 4 | 4-Br | 11 ± 3 | 47 ± 15 | 0.23 |
| 5 | 4-NO2 | 39 ± 5 | 145 ± 24 | 0.27 |
| 6 | 4-CH3 | 3 ± 1 | 242 ± 22 | 0.01 |
| 7 | 4-OCH3 | 19 ± 2 | 216 ± 34 | 0.08 |
| 8 | 4-Ph | 4 ± 0.2 | 8 ± 1 | 0.50 |
| 9 | 2-OCH3 | 26 ± 4 | 180 ± 18 | 0.14 |
| Ethopropazine 1 | - | 161 | 0.16 | 1010 |
| Donepezil 2 | - | 0.0043 | 2.3 | 0.0019 |
| Tacrin 3 | - | 0.190 | 0.047 | 4.04 |
| Huperizine A 3 | - | 0.082 | 74.4 | 0.0011 |
| Compound | −R | IC50 (μM) | |
|---|---|---|---|
| SH-SY5Y | HEK293 | ||
| 1 | 4-H | ≥800 | ≤800 1 |
| 2 | 4-F | ≥800 | ≥800 |
| 3 | 4-Cl | ≤800 1 | 85 ± 1 |
| 4 | 4-Br | 501 ± 2 | 83 ± 1 |
| 5 | 4-NO2 | 214 ± 5 | 417 ± 1 |
| 6 | 4-CH3 | ≥800 | ≤8001 |
| 7 | 4-OCH3 | ≥800 | ≥800 |
| 8 | 4-Ph | 257 ± 1 | 155 ± 1 |
| 9 | 2-OCH3 | ≥800 | ≥800 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zandona, A.; Lihtar, G.; Maraković, N.; Miš, K.; Bušić, V.; Gašo-Sokač, D.; Pirkmajer, S.; Katalinić, M. Vitamin B3-Based Biologically Active Compounds as Inhibitors of Human Cholinesterases. Int. J. Mol. Sci. 2020, 21, 8088. https://doi.org/10.3390/ijms21218088
Zandona A, Lihtar G, Maraković N, Miš K, Bušić V, Gašo-Sokač D, Pirkmajer S, Katalinić M. Vitamin B3-Based Biologically Active Compounds as Inhibitors of Human Cholinesterases. International Journal of Molecular Sciences. 2020; 21(21):8088. https://doi.org/10.3390/ijms21218088
Chicago/Turabian StyleZandona, Antonio, Gabriela Lihtar, Nikola Maraković, Katarina Miš, Valentina Bušić, Dajana Gašo-Sokač, Sergej Pirkmajer, and Maja Katalinić. 2020. "Vitamin B3-Based Biologically Active Compounds as Inhibitors of Human Cholinesterases" International Journal of Molecular Sciences 21, no. 21: 8088. https://doi.org/10.3390/ijms21218088
APA StyleZandona, A., Lihtar, G., Maraković, N., Miš, K., Bušić, V., Gašo-Sokač, D., Pirkmajer, S., & Katalinić, M. (2020). Vitamin B3-Based Biologically Active Compounds as Inhibitors of Human Cholinesterases. International Journal of Molecular Sciences, 21(21), 8088. https://doi.org/10.3390/ijms21218088