The Role of Inflammation and Myeloperoxidase-Related Oxidative Stress in the Pathogenesis of Genetically Triggered Thoracic Aortic Aneurysms


{kind=link}
Abstract
1. Introduction
2. Pathogenesis of Thoracic Aortic Aneurysms
2.1. Structure of the Thoracic Aorta and Its Extracellular Matrix Components
2.2. Histopathology—Medial Degeneration
2.3. Molecular Mechanisms of Pathogenesis
3. Genetically Triggered Thoracic Aortic Aneurysms
3.1. Marfan Syndrome
3.2. Loeys–Dietz Syndrome
3.3. Vascular Ehlers–Danlos Syndrome
3.4. Bicuspid Aortic Valve
3.5. Familial Thoracic Aortic Aneurysm and Dissection
3.6. Sporadic and Degenerative Thoracic Aortic Aneurysms
4. Evidence of Inflammation in Thoracic Aortic Aneurysms
4.1. Monocytic/Macrophage Infiltration
4.2. T-cells
4.3. Interleukins
4.4. Inflammation and Disease Progression
5. Inflammation and Oxidative Stress: Myeloperoxidase and Reactive Oxygen Species
5.1. Myeloperoxidase
5.2. MPO-Derived Oxidants
5.3. MPO in Disease
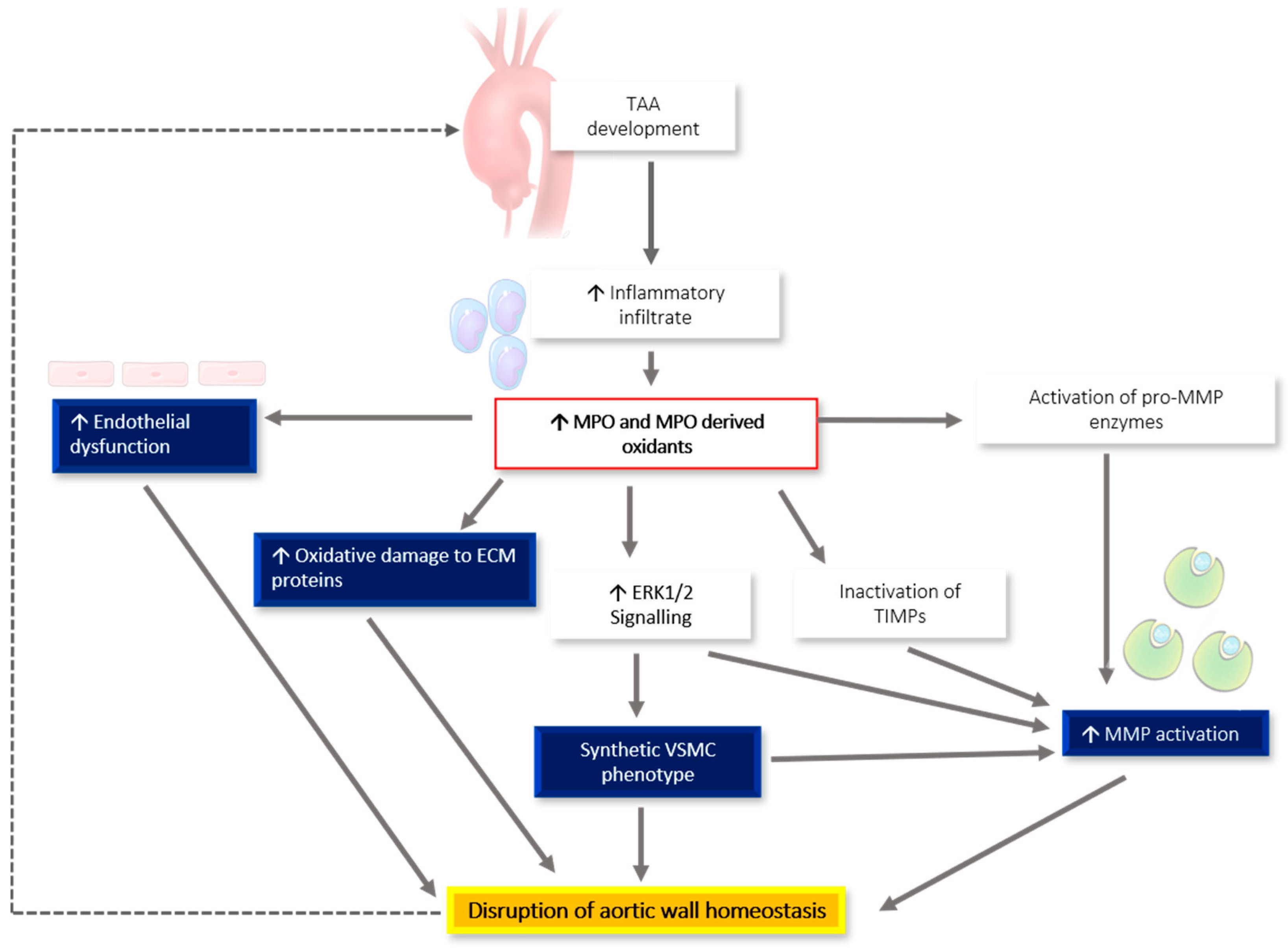
6. MPO-Associated Oxidative Stress and Links to TAA Pathogenesis
6.1. The Presence of MPO in TAA Samples and Other Aneurysmal Tissue
6.2. MPO and the Extracellular Matrix
6.3. MPO and Matrix Metalloproteinase Activation
6.4. MPO and Activation of the ERK1/2 Signaling Pathway
6.5. MPO, Nitric Oxide Availability, and Endothelial Dysfunction
6.6. MPO Utilizing Vascular NADPH Oxidase-Derived H2O2
6.7. MPO and Modifications to DNA
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AAA | Abdominal aortic aneurysms |
| AngII | Angiotensin II |
| BAV | Bicuspid aortic valve |
| ECM | Extracellular matrix |
| ECs | Endothelial cells |
| fTAAD | Familial thoracic aortic aneurysm and dissection |
| H2O2 | Hydrogen peroxide |
| HOBr | Hypobromous acid |
| HOCL | Hypochlorous acid |
| HOSCN | Hypothiocyanous acid |
| LDS | Loeys-Dietz syndrome |
| MCP-1 | Monocyte chemotactic protein-1 |
| M-CSF | Macrophage-colony stimulating factor |
| MFS | Marfan syndrome |
| MMPs | Matrix metalloproteinases |
| MPO | Myeloperoxidase |
| NADPH oxidases | NOX |
| NO | Nitric oxide |
| NO2. | Nitrogen dioxide |
| O2− | Superoxide |
| PTPN22 | protein tyrosine phosphatase, non-receptor type 22 |
| ROS | Reactive oxygen species |
| TAA | Thoracic aortic aneurysms |
| TIMPs | Tissue inhibitors of metalloproteinases |
| vEDS | Vascular Ehlers-Danlos syndrome |
| VSMC | Vascular smooth muscle cell |
References
- Morisaki, T.; Morisaki, H. Genetics of hereditary large vessel diseases. J. Hum. Genet. 2016, 61, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Elefteriades, J.A.; Farkas, E.A. Thoracic aortic aneurysm: Clinically pertinent controversies and uncertainties. J. Am. Coll. Cardiol. 2010, 55, 841–857. [Google Scholar] [CrossRef]
- Pinard, A.; Jones, G.T.; Milewicz, D.M. Genetics of Thoracic and Abdominal Aortic Diseases: Aneurysms, Dissections, and Ruptures. Circ. Res. 2019, 124, 588–606. [Google Scholar] [CrossRef] [PubMed]
- Maleszewski, J.J. Inflammatory ascending aortic disease: Perspectives from pathology. J. Thorac. Cardiovasc. Surg. 2015, 149, S176–S183. [Google Scholar] [CrossRef] [PubMed]
- Robertson, E.; Dilworth, C.; Lu, Y.; Hambly, B.; Jeremy, R. Molecular mechanisms of inherited thoracic aortic disease—From gene variant to surgical aneurysm. Biophys. Rev. 2015, 7, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Milewicz, D.M.; Guo, D.-C.; Tran-Fadulu, V.; Lafont, A.L.; Papke, C.L.; Inamoto, S.; Kwartler, C.S.; Pannu, H. Genetic basis of thoracic aortic aneurysms and dissections: Focus on smooth muscle cell contractile dysfunction. Annu. Rev. Genom. Hum. Genet. 2008, 9, 283–302. [Google Scholar] [CrossRef]
- Melvinsdottir, I.H.; Lund, S.H.; Agnarsson, B.A.; Sigvaldason, K.; Gudbjartsson, T.; Geirsson, A. The incidence and mortality of acute thoracic aortic dissection: Results from a whole nation study. Eur. J. Cardiothorac. Surg. 2016, 50, 1111–1117. [Google Scholar] [CrossRef]
- Hiratzka, L.F.; Bakris, G.L.; Beckman, J.A.; Bersin, R.M.; Carr, V.F.; Casey, D.E.; Eagle, K.A.; Hermann, L.K.; Isselbacher, E.M.; Kazerooni, E.A. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with thoracic aortic disease. J. Am. Coll. Cardiol. 2010, 55, e27–e129. [Google Scholar] [CrossRef] [PubMed]
- Pape, L.; Tsai, T.; Isselbacher, E.; Oh, J.; O’gara, P.; Evangelista, A.; Fattori, R.; Meinhardt, G.; Trimarchi, S.; Bossone, E. International Registry of Acute Aortic Dissection (IRAD) Investigators Aortic diameter > or = 5.5 cm is not a good predictor of type A aortic dissection: Observations from the International Registry of Acute Aortic Dissection (IRAD). Circulation 2007, 116, 1120–1127. [Google Scholar] [CrossRef]
- Goldfinger, J.Z.; Halperin, J.L.; Marin, M.L.; Stewart, A.S.; Eagle, K.A.; Fuster, V. Thoracic aortic aneurysm and dissection. J. Am. Coll. Cardiol. 2014, 64, 1725–1739. [Google Scholar] [CrossRef]
- Pannu, H.; Tran-Fadulu, V.; Milewicz, D.M. Genetic basis of thoracic aortic aneurysms and aortic dissections. Am. J. Med. Genet. C Semin. Med. Genet. 2005, 139C, 10–16. [Google Scholar] [CrossRef] [PubMed]
- De Backer, J.; Loeys, B.; Leroy, B.; Coucke, P.; Dietz, H.; De Paepe, A. Utility of molecular analyses in the exploration of extreme intrafamilial variability in the Marfan syndrome. Clin. Genet. 2007, 72, 188–198. [Google Scholar] [CrossRef]
- Li, Y.; Xu, J.; Chen, M.; Du, B.; Li, Q.; Xing, Q.; Zhang, Y. A FBN1 mutation association with different phenotypes of Marfan syndrome in a Chinese family. Clin. Chim. Acta 2016, 460, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Golledge, J. Abdominal aortic aneurysm: Update on pathogenesis and medical treatments. Nat. Rev. Cardiol. 2019, 16, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.J.; Dev, V.; Strauss, B.H.; Fedak, P.W.; Butany, J. Variation in the histopathological features of patients with ascending aortic aneurysms: A study of 111 surgically excised cases. J. Clin. Pathol. 2008, 61, 519–523. [Google Scholar] [CrossRef]
- Humphrey, J.D.; Schwartz, M.A.; Tellides, G.; Milewicz, D.M. Role of mechanotransduction in vascular biology: Focus on thoracic aortic aneurysms and dissections. Circ. Res. 2015, 116, 1448–1461. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, A.; Flachskampf, F.A.; Erbel, R.; Antonini-Canterin, F.; Vlachopoulos, C.; Rocchi, G.; Sicari, R.; Nihoyannopoulos, P.; Zamorano, J.; Pepi, M. Echocardiography in aortic diseases: EAE recommendations for clinical practice. Eur. J. Echocardiogr. 2010, 11, 645–658. [Google Scholar] [CrossRef]
- Emmott, A.; Garcia, J.; Chung, J.; Lachapelle, K.; El-Hamamsy, I.; Mongrain, R.; Cartier, R.; Leask, R.L. Biomechanics of the ascending thoracic aorta: A clinical perspective on engineering data. Can. J. Cardiol. 2016, 32, 35–47. [Google Scholar] [CrossRef]
- Chiesa, R.; Melissano, G.; Zangrillo, A. Thoraco-Abdominal Aorta: Surgical and Anesthetic Management; Springer Science & Business Media: Milan, Italy, 2011. [Google Scholar]
- Humphrey, J.D. Possible Mechanical Roles of Glycosaminoglycans in Thoracic Aortic Dissection and Associations with Dysregulated Transforming Growth Factor-β. J. Vasc. Res. 2013, 50, 1–10. [Google Scholar] [CrossRef]
- Karimi, A.; Milewicz, D.M. Structure of the elastin-contractile units in the thoracic aorta and how genes that cause thoracic aortic aneurysms and dissections disrupt this structure. Can. J. Cardiol. 2016, 32, 26–34. [Google Scholar] [CrossRef]
- Humphrey, J.D.; Milewicz, D.M.; Tellides, G.; Schwartz, M.A. Dysfunctional mechanosensing in aneurysms. Science 2014, 344, 477–479. [Google Scholar] [CrossRef]
- Jeremy, R.W.; Robertson, E.; Lu, Y.; Hambly, B.D. Perturbations of mechanotransduction and aneurysm formation in heritable aortopathies. Int. J. Cardiol. 2013, 169, 7–16. [Google Scholar] [CrossRef]
- Ammash, N.M.; Sundt, T.M.; Connolly, H.M. Marfan Syndrome—Diagnosis and Management. Curr. Probl. Cardiol. 2008, 33, 7–39. [Google Scholar] [CrossRef]
- Detaint, D.; Faivre, L.; Collod-Beroud, G.; Child, A.H.; Loeys, B.L.; Binquet, C.; Gautier, E.; Arbustini, E.; Mayer, K.; Arslan-Kirchner, M.; et al. Cardiovascular manifestations in men and women carrying a FBN1 mutation. Eur. Heart J. 2010, 31, 2223–2229. [Google Scholar] [CrossRef] [PubMed]
- Stuart, A.G.; Williams, A. Marfan’s syndrome and the heart. Arch. Dis. Child. 2007, 92, 351–356. [Google Scholar] [CrossRef]
- Chaudhry, S.S.; Cain, S.A.; Morgan, A.; Dallas, S.L.; Shuttleworth, C.A.; Kielty, C.M. Fibrillin-1 regulates the bioavailability of TGFβ1. J. Cell Biol. 2007, 176, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Meester, J.A.N.; Verstraeten, A.; Schepers, D.; Alaerts, M.; Van Laer, L.; Loeys, B.L. Differences in manifestations of Marfan syndrome, Ehlers-Danlos syndrome, and Loeys-Dietz syndrome. Ann. Cardiothorac. Surg. 2017, 6, 582–594. [Google Scholar] [CrossRef]
- Schepers, D.; Tortora, G.; Morisaki, H.; MacCarrick, G.; Lindsay, M.; Liang, D.; Mehta, S.G.; Hague, J.; Verhagen, J.; van de Laar, I. A mutation update on the LDS-associated genes TGFB2/3 and SMAD2/3. Hum. Mutat. 2018, 39, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P. Ehlers-Danlos syndrome type IV. Orphanet J. Rare Dis. 2007, 2, 32. [Google Scholar] [CrossRef]
- Beighton, P.; Paepe, A.D.; Steinmann, B.; Tsipouras, P.; Wenstrup, R. Ehlers-Danlos syndromes: Revised nosology, Villefranche, 1997. Am. J. Med. Genet. 1998, 77, 31–37. [Google Scholar] [CrossRef]
- Debiec, R.; Sall, H.; Samani, N.J.; Bolger, A. Genetic insights into bicuspid aortic valve disease. Cardiol. Rev. 2017, 25, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Tadros, T.M.; Klein, M.D.; Shapira, O.M. Ascending aortic dilatation associated with bicuspid aortic valve: Pathophysiology, molecular biology, and clinical implications. Circulation 2009, 119, 880–890. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, A. Bicuspid aortic valve and aortic root disease. Curr. Cardiol. Rep. 2011, 13, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Roman, M.J.; Pugh, N.L.; Devereux, R.B.; Eagle, K.A.; Holmes, K.; LeMaire, S.A.; Milewski, R.K.; Morris, S.A.; Prakash, S.K.; Pyeritz, R.E.; et al. Aortic dilatation associated with bicuspid aortic valve: Relation to sex, hemodynamics, and valve morphology (the national heart lung and blood institute-sponsored national registry of genetically triggered thoracic aortic aneurysms and cardiovascular conditions). Am. J. Cardiol. 2017, 120, 1171–1175. [Google Scholar] [CrossRef] [PubMed]
- Andelfinger, G.; Loeys, B.; Dietz, H. A decade of discovery in the genetic understanding of thoracic aortic disease. Can. J. Cardiol. 2016, 32, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Biner, S.; Rafique, A.M.; Ray, I.; Cuk, O.; Siegel, R.J.; Tolstrup, K. Aortopathy is prevalent in relatives of bicuspid aortic valve patients. J. Am. Coll. Cardiol. 2009, 53, 2288–2295. [Google Scholar] [CrossRef] [PubMed]
- Albornoz, G.; Coady, M.A.; Roberts, M.; Davies, R.R.; Tranquilli, M.; Rizzo, J.A.; Elefteriades, J.A. Familial Thoracic Aortic Aneurysms and Dissections—Incidence, Modes of Inheritance, and Phenotypic Patterns. Ann. Thorac. Surg. 2006, 82, 1400–1406. [Google Scholar] [CrossRef]
- Robertson, E.N.; Jeremy, R.W. Uncovering the Burden of Familial Thoracic Aortic Aneurysm Disease. Circulation 2013, 128, A10144. [Google Scholar]
- Wu, D.; Shen, Y.H.; Russell, L.; Coselli, J.S.; LeMaire, S.A. Molecular mechanisms of thoracic aortic dissection. J. Surg. Res. 2013, 184, 907–924. [Google Scholar] [CrossRef]
- Pyeritz, R.E. Heritable thoracic aortic disorders. Curr. Opin. Cardiol. 2014, 29, 97–102. [Google Scholar] [CrossRef]
- Ostberg, N.P.; Zafar, M.A.; Ziganshin, B.A.; Elefteriades, J.A. The genetics of thoracic aortic aneurysms and dissection: A clinical perspective. Biomolecules 2020, 10, 182. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.-C.; Hostetler, E.M.; Fan, Y.; Kulmacz, R.J.; Zhang, D.; Nickerson, D.A.; Leal, S.M.; LeMaire, S.A.; Regalado, E.S.; Milewicz, D.M. Heritable thoracic aortic disease genes in sporadic aortic dissection. J. Am. Coll. Cardiol. 2017, 70, 2728–2730. [Google Scholar] [CrossRef] [PubMed]
- LeMaire, S.A.; McDonald, M.L.; Guo, D.C.; Russell, L.; Miller, C.C., III; Johnson, R.J.; Bekheirnia, M.R.; Franco, L.M.; Nguyen, M.; Pyeritz, R.E.; et al. Genome-wide association study identifies a susceptibility locus for thoracic aortic aneurysms and aortic dissections spanning FBN1 at 15q21.1. Nat. Genet. 2011, 43, 996–1000. [Google Scholar] [CrossRef] [PubMed]
- Sulkava, M.; Raitoharju, E.; Mennander, A.; Levula, M.; Seppälä, I.; Lyytikäinen, L.-P.; Järvinen, O.; Illig, T.; Klopp, N.; Mononen, N. Differentially expressed genes and canonical pathways in the ascending thoracic aortic aneurysm–The Tampere Vascular Study. Sci. Rep. 2017, 7, 1–8. [Google Scholar] [CrossRef]
- Malecki, C.; Robertson, E.N.; Liddy, K.A.; Sahagian, A.; Gover, C.; Lu, Y.; Kekic, M.; Lai, D.; Hambly, B.D.; Jeremy, R. Differential DNA Methylation in Marfan Syndrome is Associated With Inflammation and Cardiac Contraction. Circulation 2019, 140, A14231. [Google Scholar]
- Tang, P.C.; Yakimov, A.O.; Teesdale, M.A.; Coady, M.A.; Dardik, A.; Elefteriades, J.A.; Tellides, G. Transmural inflammation by interferon-γ-producing T cells correlates with outward vascular remodeling and intimal expansion of ascending thoracic aortic aneurysms. FASEB J. 2005, 19, 1528–1530. [Google Scholar] [CrossRef]
- Pereira, L.; Lee, S.Y.; Gayraud, B.; Andrikopoulos, K.; Shapiro, S.D.; Bunton, T.; Biery, N.J.; Dietz, H.C.; Sakai, L.Y.; Ramirez, F. Pathogenetic sequence for aneurysm revealed in mice underexpressing fibrillin-1. Proc. Natl. Acad. Sci. USA 1999, 96, 3819–3823. [Google Scholar] [CrossRef]
- Guo, G.; Booms, P.; Halushka, M.; Dietz, H.C.; Ney, A.; Stricker, S.; Hecht, J.; Mundlos, S.; Robinson, P.N. Induction of macrophage chemotaxis by aortic extracts of the mgR Marfan mouse model and a GxxPG-containing fibrillin-1 fragment. Circulation 2006, 114, 1855–1862. [Google Scholar] [CrossRef]
- Ju, X.; Ijaz, T.; Sun, H.; Lejeune, W.; Vargas, G.; Shilagard, T.; Recinos, A., 3rd; Milewicz, D.M.; Brasier, A.R.; Tilton, R.G. IL-6 regulates extracellular matrix remodeling associated with aortic dilation in a fibrillin-1 hypomorphic mgR/mgR mouse model of severe Marfan syndrome. J. Am. Heart Assoc. 2014, 3, e000476. [Google Scholar] [CrossRef]
- Hibender, S.; Wanga, S.; van der Made, I.; Vos, M.; Mulder, B.J.; Balm, R.; de Vries, C.J.; de Waard, V. Renal cystic disease in the Fbn1C1039G/+ Marfan mouse is associated with enhanced aortic aneurysm formation. Cardiovasc. Pathol. 2019, 38, 1–6. [Google Scholar] [CrossRef]
- Guo, G.; Gehle, P.; Doelken, S.; Martin-Ventura, J.L.; Von Kodolitsch, Y.; Hetzer, R.; Robinson, P.N. Induction of macrophage chemotaxis by aortic extracts from patients with Marfan syndrome is related to elastin binding protein. PLoS ONE 2011, 6, e20138. [Google Scholar] [CrossRef] [PubMed]
- Heald, B.; Rigelsky, C.; Moran, R.; LaGuardia, L.; O’Malley, M.; Burke, C.A.; Zahka, K. Prevalence of thoracic aortopathy in patients with juvenile Polyposis Syndrome-Hereditary Hemorrhagic Telangiectasia due to SMAD4. Am. J. Med. Genet. A 2015, 167, 1758–1762. [Google Scholar] [CrossRef] [PubMed]
- Holm, T.M.; Habashi, J.P.; Doyle, J.J.; Bedja, D.; Chen, Y.; van Erp, C.; Lindsay, M.E.; Kim, D.; Schoenhoff, F.; Cohn, R.D.; et al. Noncanonical TGFbeta signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science 2011, 332, 358–361. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, H.-Y.; Bian, G.-L.; Yu, Y.-S.; Ye, W.-X.; Hua, F.; Chen, Y.-H.; Shen, Z.-Y. A functional variant of SMAD4 enhances thoracic aortic aneurysm and dissection risk through promoting smooth muscle cell apoptosis and proteoglycan degradation. EBioMedicine 2017, 21, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yin, P.; Chen, Y.-H.; Yu, Y.-S.; Ye, W.-X.; Huang, H.-Y.; Ji, Z.-C.; Shen, Z.-Y. A functional variant of SMAD4 enhances macrophage recruitment and inflammatory response via TGF-β signal activation in Thoracic aortic aneurysm and dissection. Aging 2018, 10, 3683–3701. [Google Scholar] [CrossRef]
- Zhang, P.; Hou, S.; Chen, J.; Zhang, J.; Lin, F.; Ju, R.; Cheng, X.; Ma, X.; Song, Y.; Zhang, Y. Smad4 deficiency in smooth muscle cells initiates the formation of aortic aneurysm. Circ. Res. 2016, 118, 388–399. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Guo, D.-C.; Estrera, A.L.; Safi, H.J.; Huynh, T.T.; Yin, Z.; Cao, S.-N.; Lin, J.; Kurian, T.; Buja, L.M. Characterization of the inflammatory and apoptotic cells in the aortas of patients with ascending thoracic aortic aneurysms and dissections. J. Thorac. Cardiovasc. Surg. 2006, 131, 671–678.e2. [Google Scholar] [CrossRef]
- He, R.; Guo, D.-C.; Sun, W.; Papke, C.L.; Duraisamy, S.; Estrera, A.L.; Safi, H.J.; Ahn, C.; Maximilian Buja, L.; Arnett, F.C.; et al. Characterization of the inflammatory cells in ascending thoracic aortic aneurysms in patients with Marfan syndrome, familial thoracic aortic aneurysms, and sporadic aneurysms. J. Thorac. Cardiovasc. Surg. 2008, 136, 922–929.e1. [Google Scholar] [CrossRef]
- Radonic, T.; de Witte, P.; Groenink, M.; de Waard, V.; Lutter, R.; van Eijk, M.; Jansen, M.; Timmermans, J.; Kempers, M.; Scholte, A.J.; et al. Inflammation aggravates disease severity in Marfan syndrome patients. PLoS ONE 2012, 7, e32963. [Google Scholar] [CrossRef]
- Zhou, H.-f.; Yan, H.; Cannon, J.L.; Springer, L.E.; Green, J.M.; Pham, C.T. CD43-mediated IFN-γ production by CD8+ T cells promotes abdominal aortic aneurysm in mice. J. Immunol. 2013, 190, 5078–5085. [Google Scholar] [CrossRef]
- Shah, A.A.; Gregory, S.G.; Krupp, D.; Feng, S.; Dorogi, A.; Haynes, C.; Grass, E.; Lin, S.S.; Hauser, E.R.; Kraus, W.E.; et al. Epigenetic profiling identifies novel genes for ascending aortic aneurysm formation with bicuspid aortic valves. Heart Surg. Forum 2015, 18, E134–E139. [Google Scholar] [CrossRef]
- Didion, S.P. Cellular and Oxidative Mechanisms Associated with Interleukin-6 Signaling in the Vasculature. Int. J. Mol. Sci. 2017, 18, 2563. [Google Scholar] [CrossRef] [PubMed]
- Pope, N.H.; Salmon, M.; Johnston, W.F.; Lu, G.; Lau, C.L.; Upchurch, G.R., Jr.; Ailawadi, G. Interleukin-6 receptor inhibition prevents descending thoracic aortic aneurysm formation. Ann. Thorac. Surg. 2015, 100, 1620–1626. [Google Scholar] [CrossRef]
- Dawson, J.; Cockerill, G.W.; Choke, E.; Belli, A.-M.; Loftus, I.; Thompson, M.M. Aortic aneurysms secrete interleukin-6 into the circulation. J. Vasc. Surg. 2007, 45, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Fujita, D.; Preiss, L.; Aizawa, K.; Asch, F.; Eagle, K.; Suzuki, T.; Investigators, G.R. Circulating interleukin-6 (IL-6) levels are associated with aortic dimensions in genetic aortic conditions. PLoS ONE 2019, 14, e0214084. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Runge, M.S.; Brasier, A.R. Angiotensin II induces interleukin-6 transcription in vascular smooth muscle cells through pleiotropic activation of nuclear factor-κB transcription factors. Circ. Res. 1999, 84, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Isselbacher, E.M. Losartan for the Treatment of Marfan Syndrome: Hope Fades. J. Am. Coll. Cardiol. 2018. [Google Scholar] [CrossRef]
- Ouyang, W.; O’Garra, A. IL-10 family cytokines IL-10 and IL-22: From basic science to clinical translation. Immunity 2019, 50, 871–891. [Google Scholar] [CrossRef]
- Black, R.L.; Roos, C.M.; Hagler, M.A.; Thalji, N.M.; Zhang, B.; Verzosa, G.C.; Miller, J.D. Potential Tissue-specific Interactions Between Inflammation and Canonical Tgfβ Signaling in Marfan Syndrome. Arterioscler. Thromb. Vasc. Biol. 2017, 37, A249. [Google Scholar]
- Wang, Y.; Barbacioru, C.C.; Shiffman, D.; Balasubramanian, S.; Iakoubova, O.; Tranquilli, M.; Albornoz, G.; Blake, J.; Mehmet, N.N.; Ngadimo, D.; et al. Gene expression signature in peripheral blood detects thoracic aortic aneurysm. PLoS ONE 2007, 2, e1050. [Google Scholar] [CrossRef]
- Pinderski Oslund, L.J.; Hedrick, C.C.; Olvera, T.; Hagenbaugh, A.; Territo, M.; Berliner, J.A.; Fyfe, A.I. Interleukin-10 blocks atherosclerotic events in vitro and in vivo. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2847–2853. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef] [PubMed]
- Johnston, W.F.; Salmon, M.; Pope, N.H.; Meher, A.; Su, G.; Stone, M.L.; Lu, G.; Owens, G.K.; Upchurch, G.R., Jr.; Ailawadi, G. Inhibition of interleukin-1β decreases aneurysm formation and progression in a novel model of thoracic aortic aneurysms. Circulation 2014, 130, S51–S59. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.F.; Guo, L.L.; Zhang, L.W.; Chu, Y.X.; Zhu, G.L.; Lu, Y.; Zhang, L.; Lu, Q.S.; Jing, Z.P. Local upregulation of interleukin-1 beta in aortic dissecting aneurysm: Correlation with matrix metalloproteinase-2, 9 expression and biomechanical decrease. Interact. Cardiovasc. Thorac. Surg. 2019, 28, 344–352. [Google Scholar] [CrossRef]
- Le, S.; Zhang, H.; Huang, X.; Chen, S.; Wu, J.; Chen, S.; Ding, X.; Chen, S.; Zhao, J.; Xu, H.; et al. PKM2 Activator TEPP-46 Attenuates Thoracic Aortic Aneurysm and Dissection by Inhibiting NLRP3 Inflammasome-Mediated IL-1β Secretion. J. Cardiovasc. Pharmacol. Ther. 2020, 25, 364–376. [Google Scholar] [CrossRef]
- Zhang, L.; Liao, M.F.; Tian, L.; Zou, S.L.; Lu, Q.S.; Bao, J.M.; Pei, Y.F.; Jing, Z.P. Overexpression of interleukin-1β and interferon-γ in type I thoracic aortic dissections and ascending thoracic aortic aneurysms: Possible correlation with matrix metalloproteinase-9 expression and apoptosis of aortic media cells. Eur. J. Cardio-Thorac. Surg. 2011, 40, 17–22. [Google Scholar] [CrossRef]
- Xu, Y.; Ye, J.; Wang, M.; Wang, Y.; Ji, Q.; Huang, Y.; Zeng, T.; Wang, Z.; Ye, D.; Jiang, H.; et al. Increased interleukin-11 levels in thoracic aorta and plasma from patients with acute thoracic aortic dissection. Clin. Chim. Acta Int. J. Clin. Chem. 2018, 481, 193–199. [Google Scholar] [CrossRef]
- Ye, J.; Wang, M.; Jiang, H.; Ji, Q.; Huang, Y.; Liu, J.; Zeng, T.; Xu, Y.; Wang, Z.; Lin, Y.; et al. Increased levels of interleukin-22 in thoracic aorta and plasma from patients with acute thoracic aortic dissection. Clin. Chim. Acta Int. J. Clin. Chem. 2018, 486, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhang, C.; Jia, L.; Chen, B.; Liu, L.; Sun, J.; Zhang, W.; You, B.; Li, Y.; Li, P.; et al. Interleukin-3 stimulates matrix metalloproteinase 12 production from macrophages promoting thoracic aortic aneurysm/dissection. Clin. Sci. 2018, 132, 655–668. [Google Scholar] [CrossRef]
- Niki, E. Oxidant-specific biomarkers of oxidative stress. Association with atherosclerosis and implication for antioxidant effects. Free Radic. Biol. Med. 2018, 120, 425–440. [Google Scholar] [CrossRef]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Wang, Y.; Branicky, R.; Noë, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef] [PubMed]
- Aratani, Y. Myeloperoxidase: Its role for host defense, inflammation, and neutrophil function. Arch. Biochem. Biophys. 2018, 640, 47–52. [Google Scholar] [CrossRef]
- Segal, A.W. How neutrophils kill microbes. Annu. Rev. Immunol. 2005, 23, 197–223. [Google Scholar] [CrossRef]
- Inazawa, J.; Inoue, K.; Nishigaki, H.; Tsuda, S.; Taniwaki, M.; Misawa, S.; Abe, T. Assignment of the human myeloperoxidase gene (MPO) to bands q21. 3→ q23 of chromosome 17. Cytogenet. Genome Res. 1989, 50, 135–136. [Google Scholar] [CrossRef]
- Van der Veen, B.S.; de Winther, M.P.; Heeringa, P. Myeloperoxidase: Molecular mechanisms of action and their relevance to human health and disease. Antioxid. Redox Signal. 2009, 11, 2899–2937. [Google Scholar] [CrossRef] [PubMed]
- Hansson, M.; Olsson, I.; Nauseef, W.M. Biosynthesis, processing, and sorting of human myeloperoxidase. Arch. Biochem. Biophys. 2006, 445, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Fenna, R.E. X-ray crystal structure of canine myeloperoxidase at 3 A resolution. J. Mol. Biol. 1992, 226, 185–207. [Google Scholar] [CrossRef]
- Furtmüller, P.G.; Zederbauer, M.; Jantschko, W.; Helm, J.; Bogner, M.; Jakopitsch, C.; Obinger, C. Active site structure and catalytic mechanisms of human peroxidases. Arch. Biochem. Biophys. 2006, 445, 199–213. [Google Scholar] [CrossRef]
- Khan, A.A.; Alsahli, M.A.; Rahmani, A.H. Myeloperoxidase as an active disease biomarker: Recent biochemical and pathological perspectives. Med Sci. 2018, 6, 33. [Google Scholar] [CrossRef] [PubMed]
- Harrison, J.E.; Araiso, T.; Palcic, M.M.; Dunford, H.B. Compound I of myeloperoxidase. Biochem. Biophys. Res. Commun. 1980, 94, 34–40. [Google Scholar] [CrossRef]
- Senthilmohan, R.; Kettle, A.J. Bromination and chlorination reactions of myeloperoxidase at physiological concentrations of bromide and chloride. Arch. Biochem. Biophys. 2006, 445, 235–244. [Google Scholar] [CrossRef]
- Rees, M.D.; McNiven, T.N.; Davies, M. Degradation of extracellular matrix and its components by hypobromous acid. Biochem. J. 2007, 401, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Bartesaghi, S.; Radi, R. Fundamentals on the biochemistry of peroxynitrite and protein tyrosine nitration. Redox Biol. 2018, 14, 618–625. [Google Scholar] [CrossRef]
- Davies, M.J. Myeloperoxidase-derived oxidation: Mechanisms of biological damage and its prevention. J. Clin. Biochem. Nutr. 2011, 48, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Meuwese, M.C.; Stroes, E.S.; Hazen, S.L.; van Miert, J.N.; Kuivenhoven, J.A.; Schaub, R.G.; Wareham, N.J.; Luben, R.; Kastelein, J.J.; Khaw, K.-T. Serum myeloperoxidase levels are associated with the future risk of coronary artery disease in apparently healthy individuals: The EPIC-Norfolk Prospective Population Study. J. Am. Coll. Cardiol. 2007, 50, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Nagra, R.M.; Becher, B.; Tourtellotte, W.W.; Antel, J.P.; Gold, D.; Paladino, T.; Smith, R.A.; Nelson, J.R.; Reynolds, W.F. Immunohistochemical and genetic evidence of myeloperoxidase involvement in multiple sclerosis. J. Neuroimmunol. 1997, 78, 97–107. [Google Scholar] [CrossRef]
- Gray, E.; Thomas, T.L.; Betmouni, S.; Scolding, N.; Love, S. Elevated activity and microglial expression of myeloperoxidase in demyelinated cerebral cortex in multiple sclerosis. Brain Pathol. 2008, 18, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Malle, E.; Waeg, G.; Schreiber, R.; Gröne, E.F.; Sattler, W.; Gröne, H.J. Immunohistochemical evidence for the myeloperoxidase/H2O2/halide system in human atherosclerotic lesions: Colocalization of myeloperoxidase and hypochlorite-modified proteins. Eur. J. Biochem. 2000, 267, 4495–4503. [Google Scholar] [CrossRef]
- Karakas, M.; Koenig, W. Myeloperoxidase production by macrophage and risk of atherosclerosis. Curr. Atheroscler. Rep. 2012, 14, 277–283. [Google Scholar] [CrossRef] [PubMed]
- La Rocca, G.; Di Stefano, A.; Eleuteri, E.; Anzalone, R.; Magno, F.; Corrao, S.; Loria, T.; Martorana, A.; Di Gangi, C.; Colombo, M. Oxidative stress induces myeloperoxidase expression in endocardial endothelial cells from patients with chronic heart failure. Basic Res. Cardiol. 2009, 104, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, S.; Kugiyama, K.; Aikawa, M.; Nakamura, S.; Ogawa, H.; Libby, P. Hypochlorous acid, a macrophage product, induces endothelial apoptosis and tissue factor expression: Involvement of myeloperoxidase-mediated oxidant in plaque erosion and thrombogenesis. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1309–1314. [Google Scholar] [CrossRef]
- Sugiyama, S.; Okada, Y.; Sukhova, G.K.; Virmani, R.; Heinecke, J.W.; Libby, P. Macrophage myeloperoxidase regulation by granulocyte macrophage colony-stimulating factor in human atherosclerosis and implications in acute coronary syndromes. Am. J. Pathol. 2001, 158, 879–891. [Google Scholar] [CrossRef]
- Arslan, S.; Berkan, Ö.; Bayyurt, B.; Beton, O.; Şahin, N.l.Ö.l.; Aydemir, E.I. Effects of MPO-463G/A and-129G/A polymorphisms on coronary artery disease risk and patient survival in a Turkish population. Biomed. Rep. 2017, 7, 547–552. [Google Scholar] [CrossRef]
- Khalil, A.; Medfai, H.; Poelvoorde, P.; Kazan, M.F.; Delporte, C.; Van Antwerpen, P.; El-Makhour, Y.; Biston, P.; Delrée, P.; Badran, B.; et al. Myeloperoxidase promotes tube formation, triggers ERK1/2 and Akt pathways and is expressed endogenously in endothelial cells. Arch. Biochem. Biophys. 2018, 654, 55–69. [Google Scholar] [CrossRef]
- Green, P.S.; Mendez, A.J.; Jacob, J.S.; Crowley, J.R.; Growdon, W.; Hyman, B.T.; Heinecke, J.W. Neuronal expression of myeloperoxidase is increased in Alzheimer’s disease. J. Neurochem. 2004, 90, 724–733. [Google Scholar] [CrossRef]
- Maki, R.A.; Holzer, M.; Motamedchaboki, K.; Malle, E.; Masliah, E.; Marsche, G.; Reynolds, W.F. Human myeloperoxidase (hMPO) is expressed in neurons in the substantia nigra in Parkinson’s disease and in the hMPO-α-synuclein-A53T mouse model, correlating with increased nitration and aggregation of α-synuclein and exacerbation of motor impairment. Free Radic. Biol. Med. 2019, 141, 115–140. [Google Scholar] [CrossRef]
- Roumeguère, T.; Delree, P.; Van Antwerpen, P.; Rorive, S.; Vanhamme, L.; de Ryhove, L.d.l.K.; Serteyn, D.; Wespes, E.; Vanhaerverbeek, M.; Boudjeltia, K.Z. Intriguing location of myeloperoxidase in the prostate: A preliminary immunohistochemical study. Prostate 2012, 72, 507–513. [Google Scholar] [CrossRef]
- Maki, R.A.; Tyurin, V.A.; Lyon, R.C.; Hamilton, R.L.; DeKosky, S.T.; Kagan, V.E.; Reynolds, W.F. Aberrant expression of myeloperoxidase in astrocytes promotes phospholipid oxidation and memory deficits in a mouse model of Alzheimer disease. J. Biol. Chem. 2009, 284, 3158–3169. [Google Scholar] [CrossRef]
- Chu, Y.; Wilson, K.; Gu, H.; Wegman-Points, L.; Dooley, S.A.; Pierce, G.L.; Cheng, G.; Pena Silva, R.A.; Heistad, D.D.; Hasan, D. Myeloperoxidase is increased in human cerebral aneurysms and increases formation and rupture of cerebral aneurysms in mice. Stroke 2015, 46, 1651–1656. [Google Scholar] [CrossRef] [PubMed]
- Ollikainen, E.; Tulamo, R.; Lehti, S.; Hernesniemi, J.; Niemelä, M.; Kovanen, P.T.; Frösen, J. Myeloperoxidase associates with degenerative remodeling and rupture of the saccular intracranial aneurysm wall. J. Neuropathol. Exp. Neurol. 2018, 77, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Milewicz, D.M.; Østergaard, J.R.; Ala-Kokko, L.M.; Khan, N.; Grange, D.K.; Mendoza-Londono, R.; Bradley, T.J.; Olney, A.H.; Adès, L.; Maher, J.F.; et al. De novo ACTA2 mutation causes a novel syndrome of multisystemic smooth muscle dysfunction. Am. J. Med. Genet. A 2010, 152a, 2437–2443. [Google Scholar] [CrossRef]
- Kim, H.W.; Blomkalns, A.L.; Ogbi, M.; Thomas, M.; Gavrila, D.; Neltner, B.S.; Cassis, L.A.; Thompson, R.W.; Weiss, R.M.; Lindower, P.D.; et al. Role of myeloperoxidase in abdominal aortic aneurysm formation: Mitigation by taurine. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H1168–H1179. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Mozo, P.; Madrigal-Matute, J.; Martinez-Pinna, R.; Blanco-Colio, L.M.; Lopez, J.A.; Camafeita, E.; Meilhac, O.; Michel, J.-B.; Aparicio, C.; De Ceniga, M.V. Proteomic analysis of polymorphonuclear neutrophils identifies catalase as a novel biomarker of abdominal aortic aneurysm: Potential implication of oxidative stress in abdominal aortic aneurysm progression. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 3011–3019. [Google Scholar] [CrossRef]
- Memon, A.A.; Zarrouk, M.; Ågren-Witteschus, S.; Sundquist, J.; Gottsäter, A.; Sundquist, K. Identification of novel diagnostic and prognostic biomarkers for abdominal aortic aneurysm. Eur. J. Prev. Cardiol. 2020, 27, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Zagrapan, B.; Eilenberg, W.; Prausmueller, S.; Nawrozi, P.; Muench, K.; Hetzer, S.; Elleder, V.; Rajic, R.; Juster, F.; Martelanz, L.; et al. A Novel Diagnostic and Prognostic Score for Abdominal Aortic Aneurysms Based on D-Dimer and a Comprehensive Analysis of Myeloid Cell Parameters. Thromb. Haemost. 2019, 119, 807–820. [Google Scholar] [CrossRef]
- Sherrah, A.; Wilson, M.; Thomas, S.; Chan, E.; Yeung, A.; Robertson, E.; Jeremy, R.; Hambly, B.; Qasabian, R.; Vallely, M. Haem-Enzymes Predictive of Coronary Artery Disease Are Present in Thoracic and Abdominal Aortic Aneurysm. Heart Lung Circ. 2017, 26, S405. [Google Scholar] [CrossRef][Green Version]
- Ali, O.A.; Chapman, M.; Nguyen, T.H.; Chirkov, Y.Y.; Heresztyn, T.; Mundisugih, J.; Horowitz, J.D. Interactions between inflammatory activation and endothelial dysfunction selectively modulate valve disease progression in patients with bicuspid aortic valve. Heart 2014, 100, 800–805. [Google Scholar] [CrossRef]
- Mehrkens, D.; Dohr, J.; Mollenhauer, M.; Kochen, M.; Silva, A.; Sengle, G.; Rudolph, V.; Klinke, A.; Adam, M.; Baldus, S. P4551 Myeloperoxidase activity aggravates aortic wall remodeling and participates in aneurysm development in Marfan Syndrome. Eur. Heart J. 2018, 39, ehy563.P4551. [Google Scholar] [CrossRef]
- Nataatmadja, M.; West, M.; West, J.; Summers, K.; Walker, P.; Nagata, M.; Watanabe, T. Abnormal extracellular matrix protein transport associated with increased apoptosis of vascular smooth muscle cells in marfan syndrome and bicuspid aortic valve thoracic aortic aneurysm. Circulation 2003, 108 (Suppl. S1), II329–II334. [Google Scholar] [CrossRef]
- Nybo, T.; Cai, H.; Chuang, C.Y.; Gamon, L.F.; Rogowska-Wrzesinska, A.; Davies, M. Chlorination and oxidation of human plasma fibronectin by myeloperoxidase-derived oxidants, and its consequences for smooth muscle cell function. Redox Biol. 2018, 19, 388–400. [Google Scholar] [CrossRef]
- Nybo, T.; Dieterich, S.; Gamon, L.F.; Chuang, C.Y.; Hammer, A.; Hoefler, G.; Malle, E.; Rogowska-Wrzesinska, A.; Davies, M. Chlorination and oxidation of the extracellular matrix protein laminin and basement membrane extracts by hypochlorous acid and myeloperoxidase. Redox Biol. 2019, 20, 496–513. [Google Scholar] [CrossRef]
- Cai, H.; Chuang, C.Y.; Vanichkitrungruang, S.; Hawkins, C.L.; Davies, M. Hypochlorous acid-modified extracellular matrix contributes to the behavioral switching of human coronary artery smooth muscle cells. Free Radic. Biol. Med. 2019, 134, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Löffek, S.; Schilling, O.; Franzke, C.W. Biological role of matrix metalloproteinases: A critical balance. Eur. Respir. J. 2011, 38, 191–208. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Aikawa, M. Many faces of matrix metalloproteinases in aortic aneurysms. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 752–754. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Kassim, S.Y.; Parks, W.C.; Heinecke, J.W. Hypochlorous acid oxygenates the cysteine switch domain of pro-matrilysin (MMP-7) A mechanism for matrix metalloproteinase activation and atherosclerotic plaque rupture by myeloperoxidase. J. Biol. Chem. 2001, 276, 41279–41287. [Google Scholar] [CrossRef]
- Fu, X.; Kassim, S.Y.; Parks, W.C.; Heinecke, J.W. Hypochlorous acid generated by myeloperoxidase modifies adjacent tryptophan and glycine residues in the catalytic domain of matrix Metalloproteinase-7 (Matrilysin) an oxidative mechanism for restraining proteolytic activity during inflammation. J. Biol. Chem. 2003, 278, 28403–28409. [Google Scholar] [CrossRef]
- Meli, D.N.; Christen, S.; Leib, S.L. Matrix metalloproteinase-9 in pneumococcal meningitis: Activation via an oxidative pathway. J. Infect. Dis. 2003, 187, 1411–1415. [Google Scholar] [CrossRef]
- Michaelis, J.; Vissers, M.C.; Winterbourn, C.C. Different effects of hypochlorous acid on human neutrophil metalloproteinases: Activation of collagenase and inactivation of collagenase and gelatinase. Arch. Biochem. Biophys. 1992, 292, 555–562. [Google Scholar] [CrossRef]
- Peppin, G.J.; Weiss, S.J. Activation of the endogenous metalloproteinase, gelatinase, by triggered human neutrophils. Proc. Natl. Acad. Sci. USA 1986, 83, 4322–4326. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.J.; Peppin, G.; Ortiz, X.; Ragsdale, C.; Test, S.T. Oxidative autoactivation of latent collagenase by human neutrophils. Science 1985, 227, 747–749. [Google Scholar] [CrossRef]
- Zhang, Y.; Dong, H.; Seeburg, D.P.; Wojtkiewicz, G.R.; Waterman, P.; Pulli, B.; Forghani, R.; Ali, M.; Iwamoto, Y.; Swirski, F.K. Multimodal molecular imaging demonstrates myeloperoxidase regulation of matrix metalloproteinase activity in neuroinflammation. Mol. Neurobiol. 2019, 56, 954–962. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Rosen, H.; Madtes, D.K.; Shao, B.; Martin, T.R.; Heinecke, J.W.; Fu, X. Myeloperoxidase inactivates TIMP-1 by oxidizing its N-terminal cysteine residue an oxidative mechanism for regulating proteolysis during inflammation. J. Biol. Chem. 2007, 282, 31826–31834. [Google Scholar] [CrossRef] [PubMed]
- Son, Y.; Cheong, Y.K.; Kim, N.H.; Chung, H.T.; Kang, D.G.; Pae, H.O. Mitogen-activated protein kinases and reactive oxygen species: How can ROS activate MAPK pathways? J. Signal. Transduct. 2011, 2011, 792639. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Zhang, K.; Liu, Z.; Xu, Q.; You, B.; Li, C.; Cao, J.; Zhou, H.; Li, X.; Chen, J. VPO1 modulates vascular smooth muscle cell phenotypic switch by activating extracellular signal-regulated kinase 1/2 (ERK 1/2) in abdominal aortic aneurysms. J. Am. Heart Assoc. 2018, 7, e010069. [Google Scholar] [CrossRef]
- Mu, H.; Wang, X.; Lin, P.; Yao, Q.; Chen, C. Nitrotyrosine promotes human aortic smooth muscle cell migration through oxidative stress and ERK1/2 activation. Biochim. Biophys. Acta Mol. Cell Res. 2008, 1783, 1576–1584. [Google Scholar] [CrossRef][Green Version]
- Mu, H.; Wang, X.; Lin, P.H.; Yao, Q.; Chen, C. Chlorotyrosine promotes human aortic smooth muscle cell migration through increasing superoxide anion production and ERK1/2 activation. Atherosclerosis 2008, 201, 67–75. [Google Scholar] [CrossRef]
- Petsophonsakul, P.; Furmanik, M.; Forsythe, R.; Dweck, M.; Schurink, G.W.; Natour, E.; Reutelingsperger, C.; Jacobs, M.; Mees, B.; Schurgers, L. Role of Vascular Smooth Muscle Cell Phenotypic Switching and Calcification in Aortic Aneurysm Formation: Involvement of Vitamin K-Dependent Processes. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1351–1368. [Google Scholar] [CrossRef]
- Landis, B.J.; Corvera, J.S.; Idrees, M.; Ware, S.M. COQ8B: A candidate genetic modifier of oxidative metabolism and thoracic aortic aneurysm severity. Circulation 2018, 138, A14841. [Google Scholar]
- Jimenez-Altayo, F.; Meirelles, T.; Crosas-Molist, E.; Sorolla, M.A.; Del Blanco, D.G.; Lopez-Luque, J.; Mas-Stachurska, A.; Siegert, A.M.; Bonorino, F.; Barbera, L.; et al. Redox stress in Marfan syndrome: Dissecting the role of the NADPH oxidase NOX4 in aortic aneurysm. Free Radic. Biol. Med. 2018, 118, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Cook, N.L.; Moeke, C.H.; Fantoni, L.I.; Pattison, D.I.; Davies, M.J. The myeloperoxidase-derived oxidant hypothiocyanous acid inhibits protein tyrosine phosphatases via oxidation of key cysteine residues. Free Radic. Biol. Med. 2016, 90, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Lane, A.E.; Tan, J.T.; Hawkins, C.L.; Heather, A.K.; Davies, M.J.J.B.J. The myeloperoxidase-derived oxidant HOSCN inhibits protein tyrosine phosphatases and modulates cell signalling via the mitogen-activated protein kinase (MAPK) pathway in macrophages. Biochem. J. 2010, 430, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Cyr, A.R.; Huckaby, L.V.; Shiva, S.S.; Zuckerbraun, B.S. Nitric Oxide and Endothelial Dysfunction. Crit. Care Clin. 2020, 36, 307–321. [Google Scholar] [CrossRef]
- Stocker, R.; Huang, A.; Jeranian, E.; Hou, J.Y.; Wu, T.T.; Thomas, S.R.; Keaney, J.F., Jr. Hypochlorous acid impairs endothelium-derived nitric oxide bioactivity through a superoxide-dependent mechanism. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2028–2033. [Google Scholar] [CrossRef]
- Eiserich, J.P.; Baldus, S.; Brennan, M.-L.; Ma, W.; Zhang, C.; Tousson, A.; Castro, L.; Lusis, A.J.; Nauseef, W.M.; White, C.R. Myeloperoxidase, a leukocyte-derived vascular NO oxidase. Science 2002, 296, 2391–2394. [Google Scholar] [CrossRef]
- Chung, A.; Au Yeung, K.; Cortes, S.; Sandor, G.; Judge, D.; Dietz, H.C.; Van Breemen, C. Endothelial dysfunction and compromised eNOS/Akt signaling in the thoracic aorta during the progression of Marfan syndrome. Br. J. Pharmacol. 2007, 150, 1075–1083. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, J.; Jacobs, J.D.; Jennings, L.K. Interaction of myeloperoxidase with vascular NAD (P) H oxidase-derived reactive oxygen species in vasculature: Implications for vascular diseases. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H2563–H2572. [Google Scholar] [CrossRef]
- Jenner, A.M.; Ruiz, J.E.; Dunster, C.; Halliwell, B.; Mann, G.E.; Siow, R.C. Vitamin C protects against hypochlorous acid–induced glutathione depletion and DNA base and protein damage in human vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 574–580. [Google Scholar] [CrossRef]
- Acilan, C.; Serhatli, M.; Kacar, O.; Adiguzel, Z.; Tuncer, A.; Hayran, M.; Baysal, K. Smooth muscle cells isolated from thoracic aortic aneurysms exhibit increased genomic damage, but similar tendency for apoptosis. DNA Cell Biol. 2012, 31, 1523–1534. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malecki, C.; Hambly, B.D.; Jeremy, R.W.; Robertson, E.N. The Role of Inflammation and Myeloperoxidase-Related Oxidative Stress in the Pathogenesis of Genetically Triggered Thoracic Aortic Aneurysms. Int. J. Mol. Sci. 2020, 21, 7678. https://doi.org/10.3390/ijms21207678
Malecki C, Hambly BD, Jeremy RW, Robertson EN. The Role of Inflammation and Myeloperoxidase-Related Oxidative Stress in the Pathogenesis of Genetically Triggered Thoracic Aortic Aneurysms. International Journal of Molecular Sciences. 2020; 21(20):7678. https://doi.org/10.3390/ijms21207678
Chicago/Turabian StyleMalecki, Cassandra, Brett D. Hambly, Richmond W. Jeremy, and Elizabeth N. Robertson. 2020. "The Role of Inflammation and Myeloperoxidase-Related Oxidative Stress in the Pathogenesis of Genetically Triggered Thoracic Aortic Aneurysms" International Journal of Molecular Sciences 21, no. 20: 7678. https://doi.org/10.3390/ijms21207678
APA StyleMalecki, C., Hambly, B. D., Jeremy, R. W., & Robertson, E. N. (2020). The Role of Inflammation and Myeloperoxidase-Related Oxidative Stress in the Pathogenesis of Genetically Triggered Thoracic Aortic Aneurysms. International Journal of Molecular Sciences, 21(20), 7678. https://doi.org/10.3390/ijms21207678