Binding Mode Exploration of B1 Receptor Antagonists’ by the Use of Molecular Dynamics and Docking Simulation—How Different Target Engagement Can Determine Different Biological Effects

 ,
,  ,
, 

Abstract
1. Introduction
2. Results
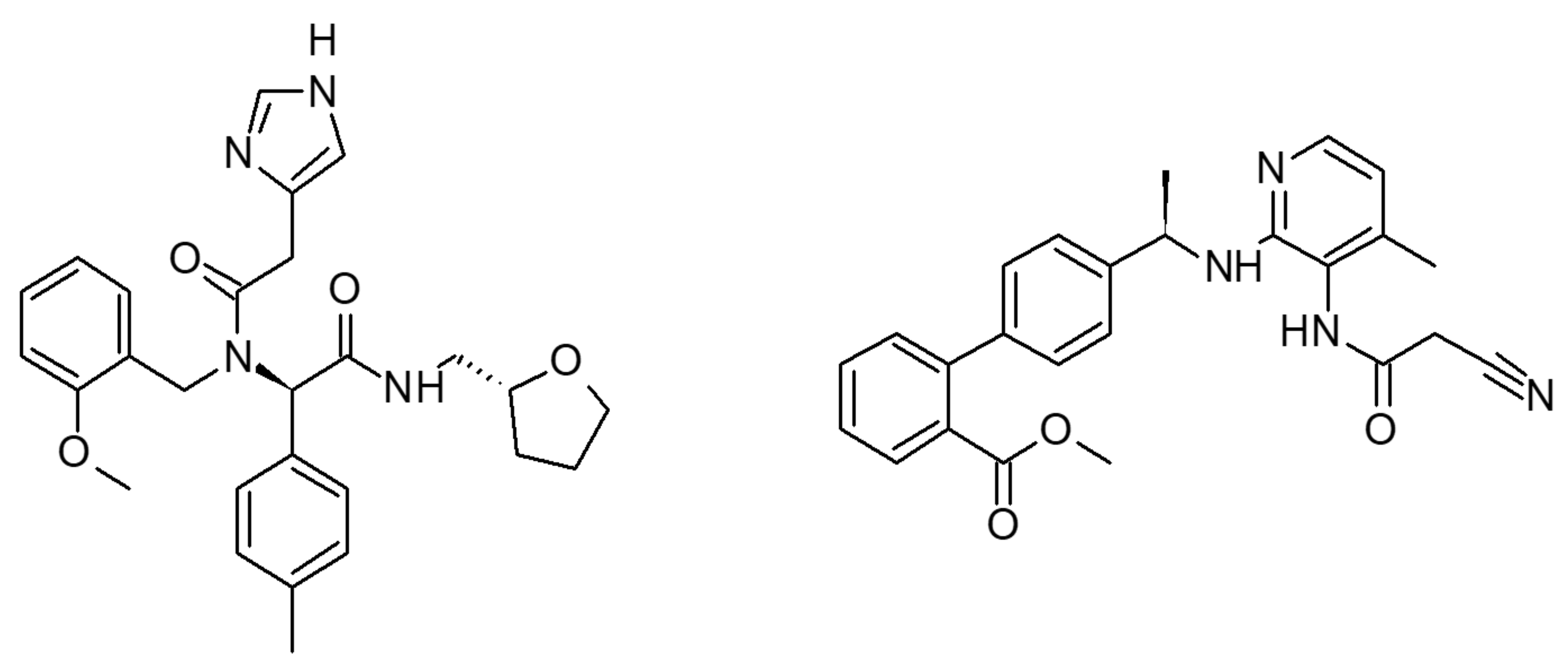
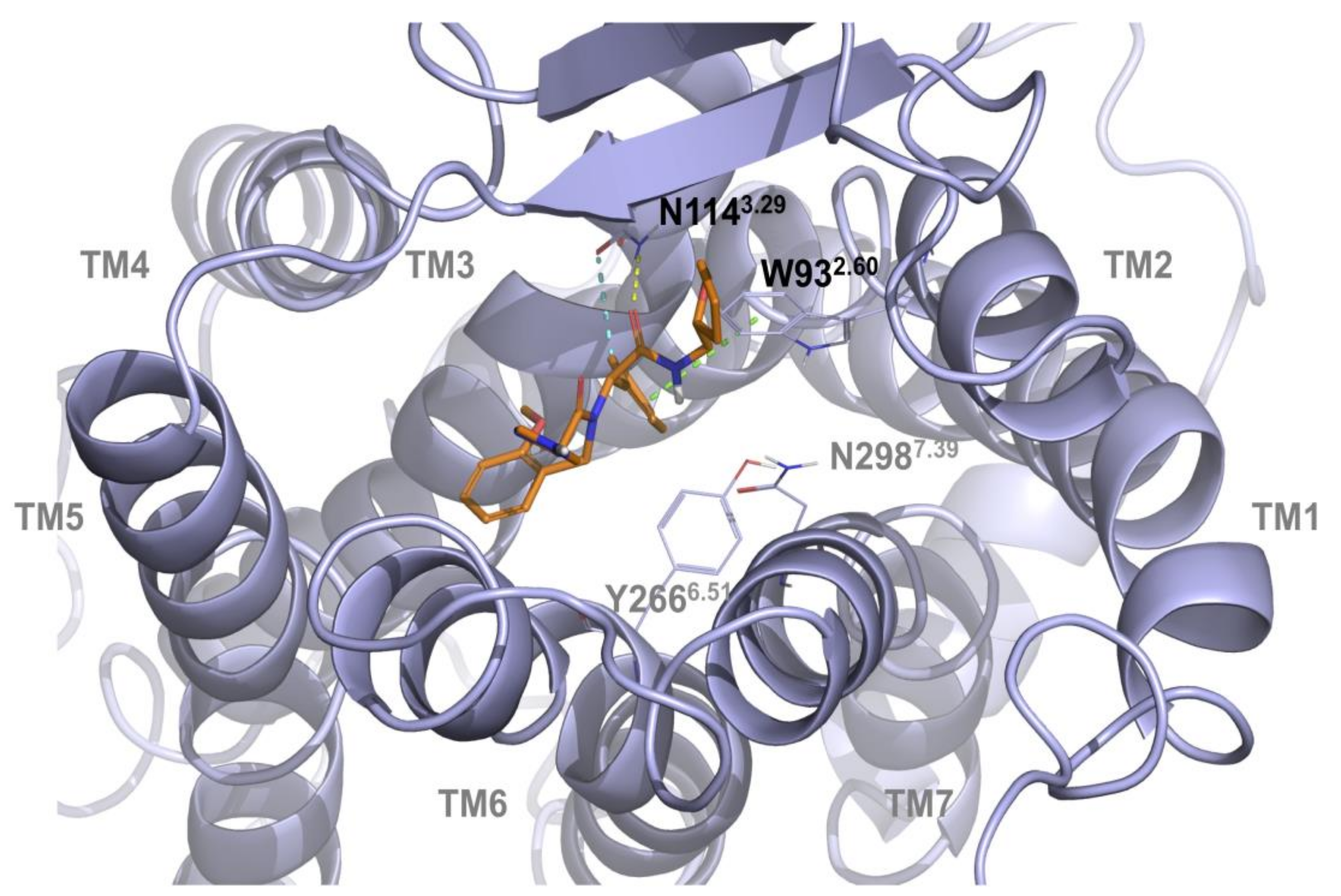
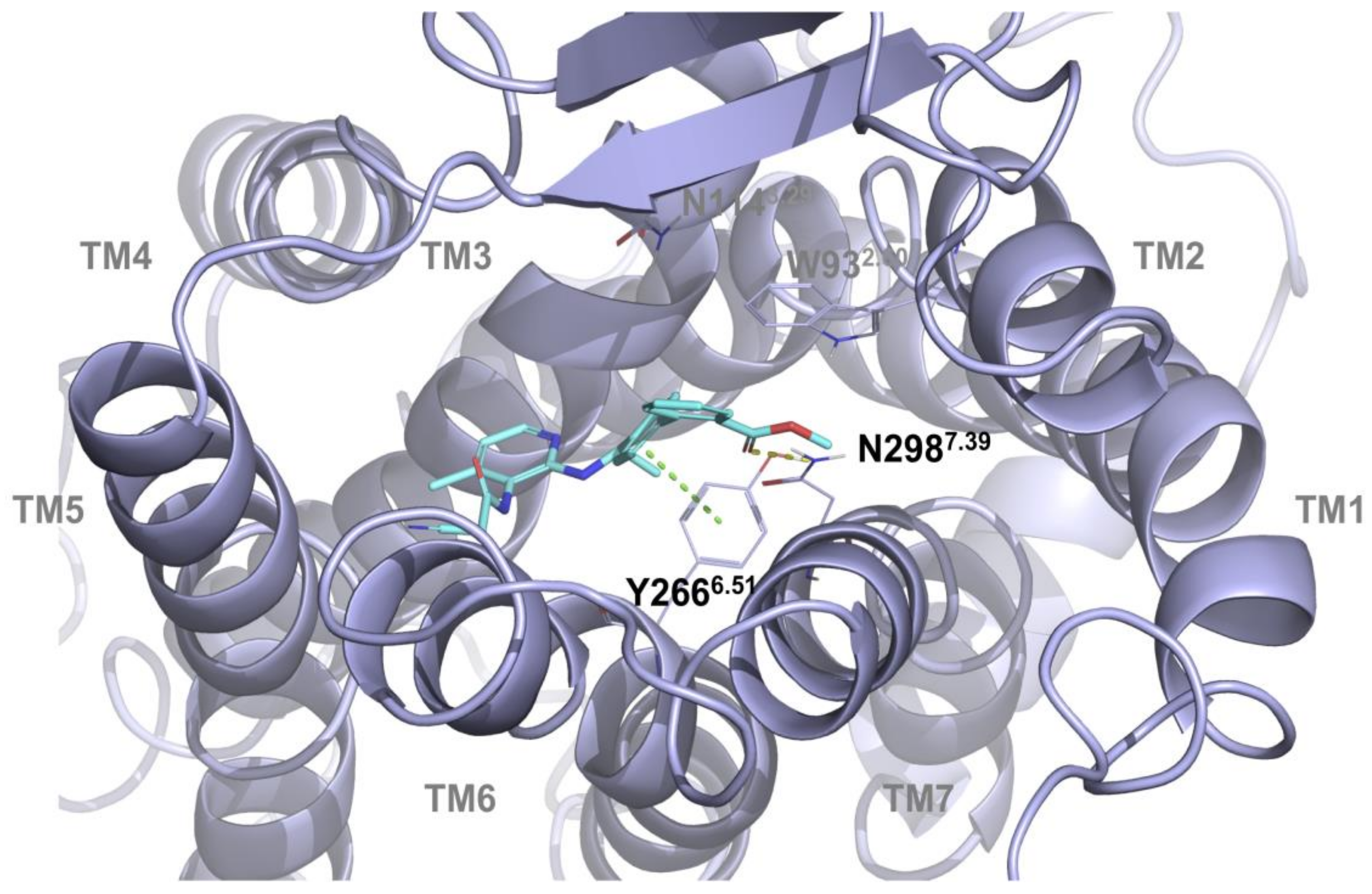
2.1. Multiple Receptor Conformations Molecular Docking Experiments
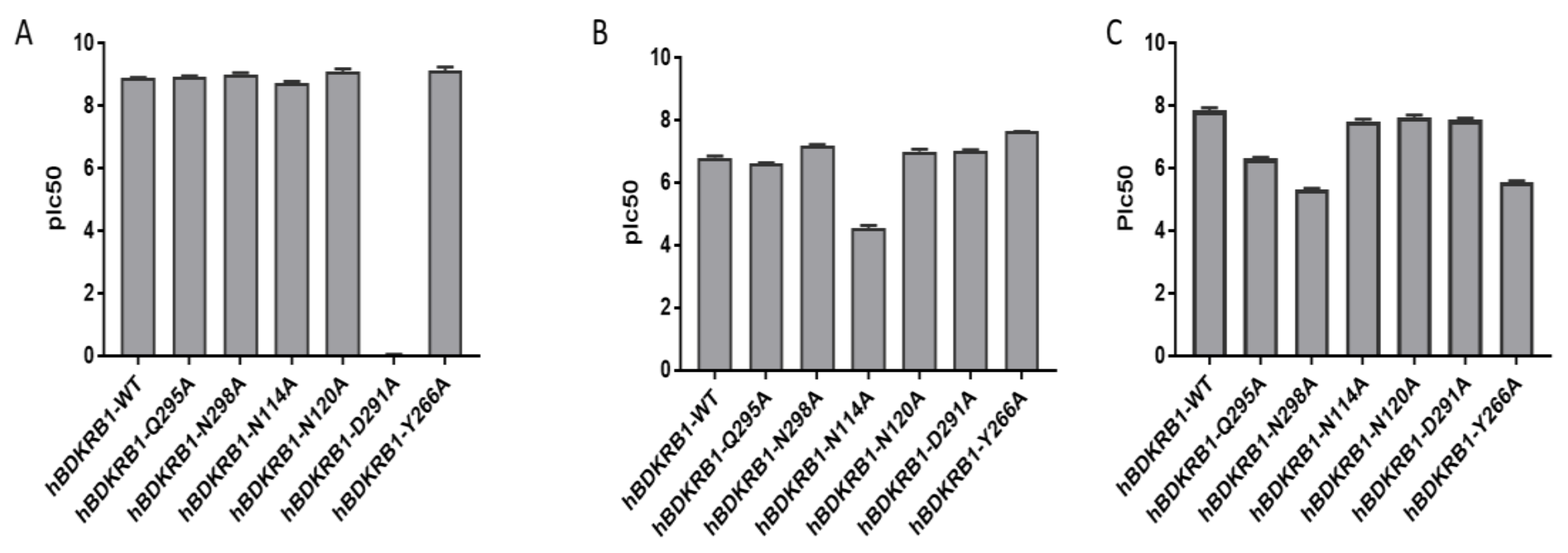
2.2. Validation of the Binding Site and Binding Mode through Single Point Mutagenesis and Radioligand Binding Experiments
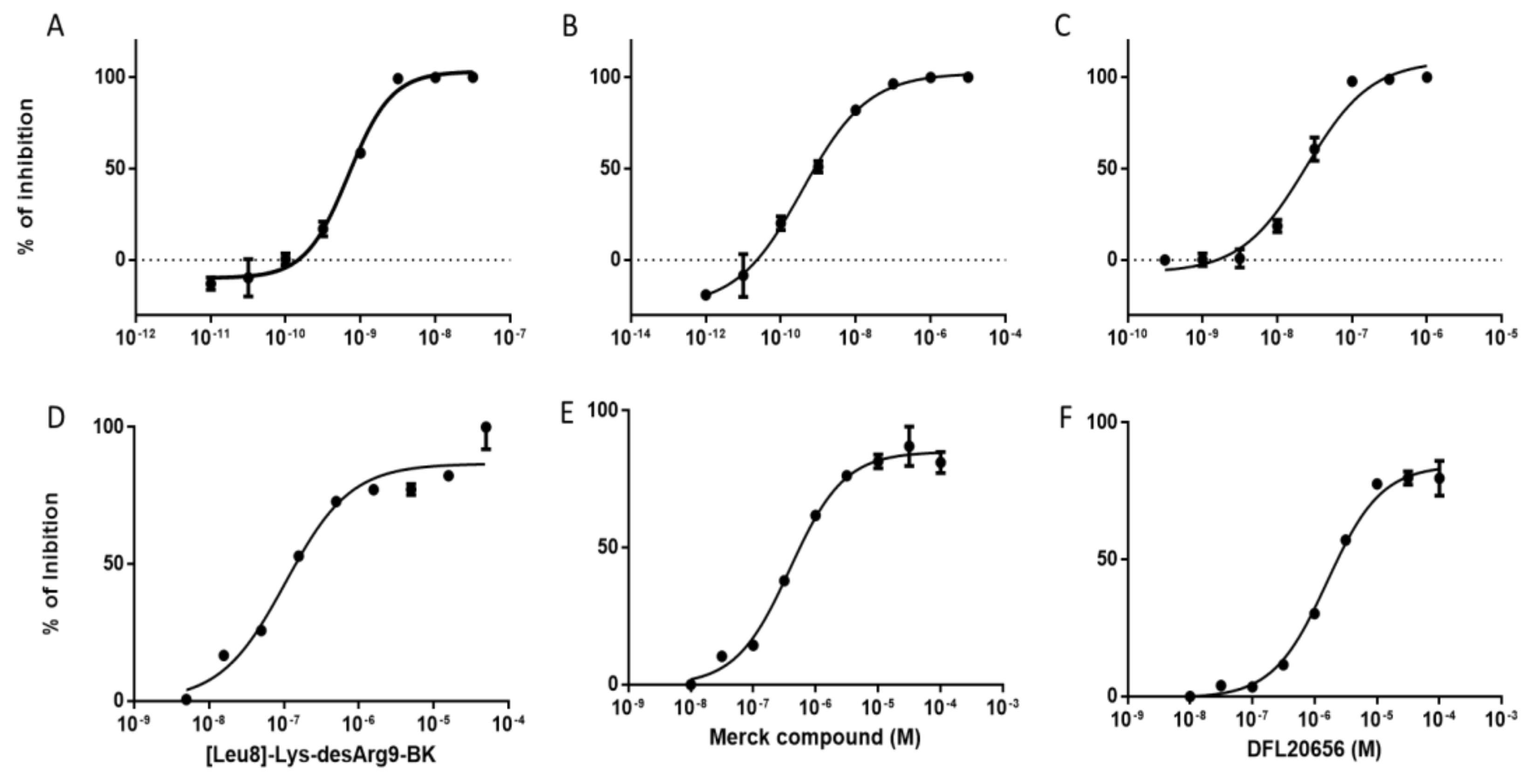
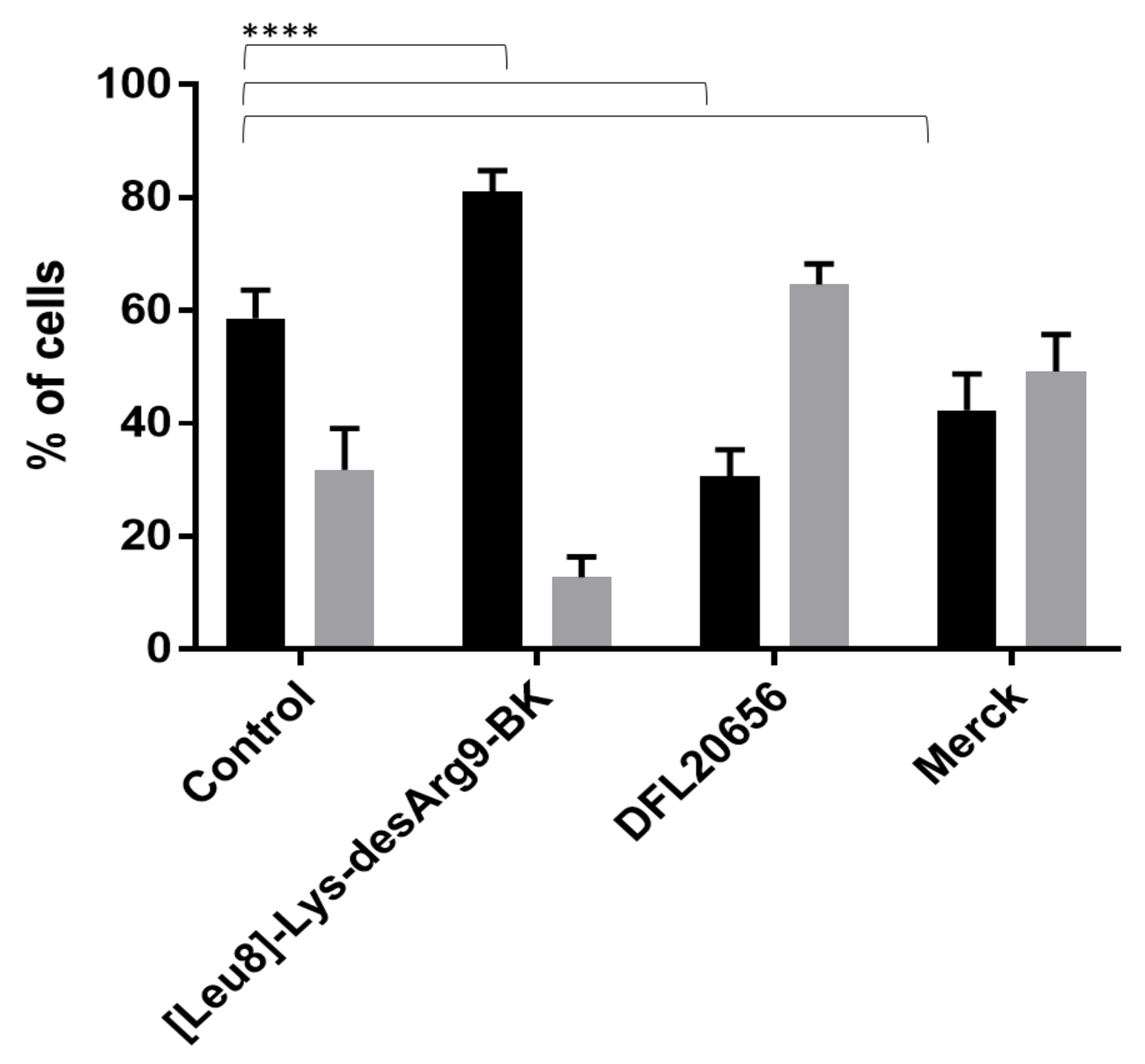
2.3. In Vitro Activity and Internalization
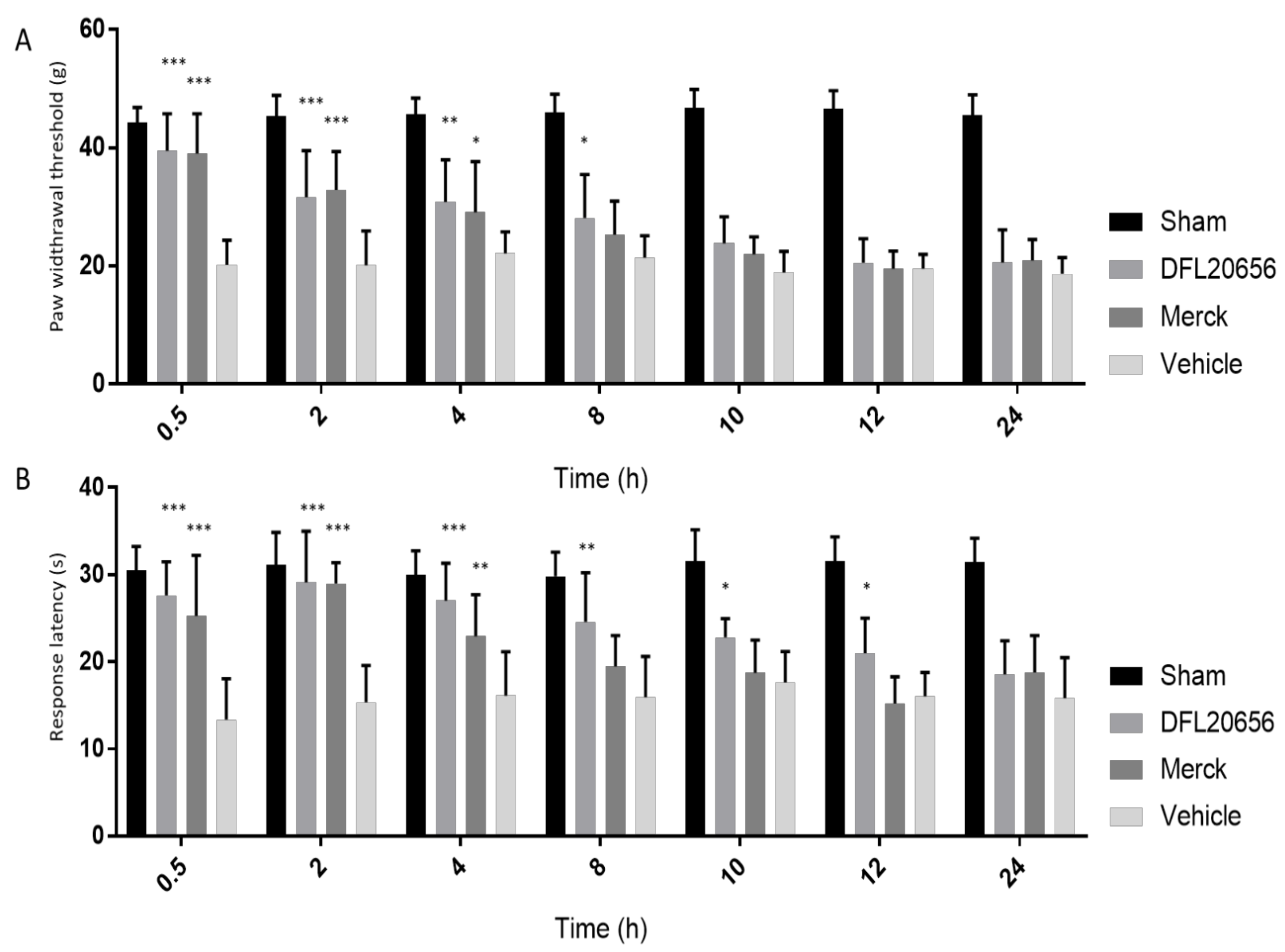
2.4. In Vivo Assays
3. Discussion and Conclusions
4. Materials and Methods
4.1. B1 Homology Model Generation and In Silico Molecular Docking Experiments
4.2. Chemical Compounds Tested
4.3. Radioligand Binding Assays
4.4. Cell Culture
4.5. Wild-Type and Mutant B1 Constructs and Transfection
4.6. Calcium Mobilization Assays
4.7. Flow Cytometry of IMR-90 Cells
4.8. In Vivo Compound Activity Evaluation
4.8.1. Animals
4.8.2. Experimental Groups and Procedures
4.8.3. Drug Treatment
4.8.4. Chronic Constriction Injury (CCI) Model of Neuropathic Pain
4.8.5. Mechanical Allodynia (Von Frey Test)
4.8.6. Thermal Hyperalgesia (Plantar Test, Hargreaves Apparatus)
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| TM | Transmembrane |
| GPCRs | G-protein-coupled receptors |
| CXCR1 | C-X-C Motif Chemokine Receptor 1 |
| CXCR2 | C-X-C Motif Chemokine Receptor 2 |
| C5a | Complement component 5a |
| I-TASSER | Iterative Threading ASSEmbly Refinement |
| MD | Molecular Dynamics |
| RMSD | Root mean square deviation |
| CHO | Chinese hamster ovary cells |
| CCI | Chronic constriction injury |
| HAE | Hereditary angioedema |
| OPM | Orientations of Proteins in Membranes |
| PME | Particle Mesh Ewald |
| RFU | Relative Fluorescence Units |
| PASS | Putative active site with spheres |
References
- Figueroa, C.D.; Ehrenfeld, P.; Bhoola, K.D. Kinin receptors as targets for cancer therapy. Expert Opin. Targets 2012, 16, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Regoli, D.; Barabé, J. Pharmacology of bradykinin and related kinins. Pharm. Rev. 1980, 32, 1–46. [Google Scholar] [PubMed]
- Regoli, D.; Gobeil, F. Kinins and peptide receptors. Biol. Chem. 2016, 397, 297. [Google Scholar] [CrossRef] [PubMed]
- Regoli, D.; Pheng, L.H.; Nsa Allogho, S.; Nguyen-Le, X.K.; Gobeil, F. Receptors for kinins: From classical pharmacology to molecular biology. Immunopharmacology 1996, 33, 116–122. [Google Scholar] [CrossRef]
- Rhaleb, N.; Drapeau, G.; Dion, S.; Jukic, D.; Rouissi, N.; Regoli, D. Structure-activity studies on bradykinin and related peptides: Agonists. Br. J. Pharm. 1990, 99, 445. [Google Scholar] [CrossRef]
- Regoli, D.; Allogho, S.N.; Rizzi, A.; Gobeil, F.J. Bradykinin receptors and their antagonists. Eur. J. Pharm. 1998, 348, 1–10. [Google Scholar] [CrossRef]
- Leeb-Lundberg, L.M.; Kang, D.S.; Lamb, M.E.; Fathy, D.B. The human B1 bradykinin receptor exhibits high ligand-independent, constitutive activity roles residues fourth intracell and third transmembrane Domains. J. Biol. Chem. 2001, 276, 8785. [Google Scholar] [CrossRef]
- Bock, M.G.; Longmore, J. Bradykinin antagonists: New opportunities. Curr. Opin. Chem. Biol. 2000, 4, 401–406. [Google Scholar] [CrossRef]
- Enquist, J.; Skröder, C.; Whistler, J.L.; Leeb-Lundberg, L.M.F. Kinins promote B2 receptor endocytosis and delay constitutive B1 receptor endocytosis. Mol. Pharm. 2007, 71, 494–507. [Google Scholar] [CrossRef]
- Sandén, C.; Enquist, J.; Bengtson, S.H.; Herwald, H.; Leeb-Lundberg, L.M.F. Kinin B2 receptor-mediated bradykinin internalization and metalloendopeptidase EP24. 15-dependent intracellular bradykinin degradation. J. Pharm. Exp. 2008, 326, 24–32. [Google Scholar]
- Enquist, J.; Sandén, C.; Skröder, C.; Mathis, S.A.; Leeb-Lundberg, L.M.F. Kinin-stimulated B1 receptor signaling depends on receptor endocytosis whereas B2 receptor signaling does not. Neurochem. Res. 2014, 39, 1037–1047. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, A.; Perretti, M. B1 receptors as a new inflammatory target. Could this B the 1? Trends Pharm. Sci. 1999, 20, 100–104. [Google Scholar] [CrossRef]
- Kuduk, S.D.; Bock, M.G. Bradykinin B1 receptor antagonists as novel analgesics: A retrospective of selected medicinal chemistry developments. Curr. Top. Med. Chem. 2008, 8, 1420–1430. [Google Scholar] [CrossRef]
- Baron, R. Neuropathic pain: A clinical perspective. Handb. Exp. Pharm. 2009, 3. [Google Scholar] [CrossRef]
- Howl, J.; Payne, S.J. Bradykinin receptors as a therapeutic target. Expert Opin. Targets 2003, 7, 277–285. [Google Scholar] [CrossRef]
- Dias, J.P.; Ismael, M.A.; Pilon, M.; De Champlain, J.; Ferrari, B.; Carayon, P.; Couture, R. The kinin B1 receptor antagonist SSR240612 reverses tactile and cold allodynia in an experimental rat model of insulin resistance. Br. J. Pharm. 2007, 152, 280–287. [Google Scholar] [CrossRef]
- Gougat, J.; Ferrari, B.; Sarran, L.; Planchenault, C.; Poncelet, M.; Maruani, J.; Alonso, R.; Cudennec, A.; Croci, T.; Guagnini, F. SSR240612 [(2R)-2-[((3R)-3-(1, 3-benzodioxol-5-yl)-3-[[(6-methoxy-2-naphthyl) sulfonyl] amino] propanoyl) amino]-3-(4-[[2R, 6S)-2, 6-dimethylpiperidinyl] methyl] phenyl)-N-isopropyl-N-methylpropanamide hydrochloride], a new nonpeptide antagonist of the br. J. Pharm. Exp. 2004, 309, 661. [Google Scholar] [CrossRef]
- Rosenkilde, M.M.; Benned-Jensen, T.; Frimurer, T.M.; Schwartz, T.W. The minor binding pocket: A major player in 7TM receptor activation. Trends Pharm. Sci. 2010, 31, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Allegretti, M.; Bertini, R.; Bizzarri, C.; Beccari, A.; Mantovani, A.; Locati, M. Allosteric inhibitors of chemoattractant receptors: Opportunities and pitfalls. Trends Pharm. Sci. 2008, 29, 280–286. [Google Scholar] [CrossRef]
- Moriconi, A.; Cunha, T.M.; Souza, G.R.; Lopes, A.H.; Cunha, F.Q.; Carneiro, V.L.; Pinto, L.G.; Brandolini, L.; Aramini, A.; Bizzarri, C. Targeting the minor pocket of C5aR for the rational design of an oral allosteric inhibitor for inflammatory and neuropathic pain relief. Proc. Natl. Acad. Sci. USA 2014, 111, 16937–16942. [Google Scholar] [CrossRef]
- Lupala, C.S.; Rasaeifar, B.; Gomez-Gutierrez, P.; Perez, J.J. Using molecular dynamics for the refinement of atomistic models of GPCRs by homology modeling. J. Biomol. Struct. Dyn. 2018, 36, 2436–2448. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.-M.; Wai, J.M.; Kuduk, S.D.; Ng, C.; Murphy, K.L.; Ransom, R.W.; Reiss, D.; Chang, R.S.L.; Harrell, C.M.; MacNeil, T.; et al. 2,3-Diaminopyridine as a platform for designing structurally unique nonpeptide bradykinin B1 receptor antagonists. Bioorg. Med. Chem. Lett. 2005, 15, 2385–2388. [Google Scholar] [CrossRef] [PubMed]
- Lupala, C.S.; Gomez-Gutierrez, P.; Perez, J.J. New insights into the stereochemical requirements of the bradykinin B1 receptor antagonists binding. J. Mol. Graph. Model. 2016, 68, 184–196. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.N.; Hey, P.J.; Ransom, R.W.; Bock, M.G.; Su, D.-S.; Murphy, K.L.; Chang, R.; Chen, T.-B.; Pettibone, D.; Hess, J.F. Identification of the critical residues of bradykinin receptor B1 for interaction with the kinins guided by site-directed mutagenesis and molecular modeling. Biochemistry 2006, 45, 14355–14361. [Google Scholar] [CrossRef]
- Phagoo, S.B.; Poole, S.; Leeb-Lundberg, L.M.F. Autoregulation of bradykinin receptors: Agonists in the presence of interleukin-1β shift the repertoire of receptor subtypes from B2 to B1 in human lung fibroblasts. Mol. Pharm. 1999, 56, 325–333. [Google Scholar] [CrossRef]
- Schanstra, J.P.; Bataillé, E.; Castano, M.E.M.; Barascud, Y.; Hirtz, C.; Pesquero, J.B.; Pecher, C.; Gauthier, F.; Girolami, J.-P.; Bascands, J.-L. The B1-agonist [des-Arg10]-kallidin activates transcription factor NF-kappaB and induces homologous upregulation of the bradykinin B1-receptor in cultured human lung fibroblasts. J. Clin. Investig. 1998, 101, 2080–2091. [Google Scholar] [CrossRef]
- Huang, H.; Player, M.R. Bradykinin B1 receptor antagonists as potential therapeutic agents for pain. J. Med. Chem. 2010, 53, 5383–5399. [Google Scholar] [CrossRef]
- Marceau, F.; Regoli, D. Bradykinin receptor ligands: Therapeutic perspectives. Nat. Rev. Drug Discov. 2004, 3, 845–852. [Google Scholar] [CrossRef]
- Qadri, F.; Bader, M. Kinin B1 receptors as a therapeutic target for inflammation. Expert Opin. Targets 2018, 22, 31–44. [Google Scholar] [CrossRef]
- Farkas, H. Icatibant as acute treatment for hereditary angioedema in adults. Expert Rev. Clin. Pharm. 2016, 9, 779–788. [Google Scholar] [CrossRef]
- Copeland, R.A. The drug–target residence time model: A 10-year retrospective. Nat. Rev. Drug Discov. 2016, 15, 87. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Huang, H.; Wu, C.H. Protein Bioinformatics Databases and Resources. In Protein Bioinformatics. Methods in Molecular Biology; Wu, C., Arighi, C., Ross, K., Eds.; Humana Press: New York, NY, USA, 2017; Volume 1558, pp. 3–39. [Google Scholar]
- Zhang, J.; Yang, J.; Jang, R.; Zhang, Y. GPCR-I-TASSER: A Hybrid Approach to G Protein-Coupled Receptor Structure Modeling and the Application to the Human Genome. Structure 2015, 23, 1538–1549. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols and influence on virtual screening enrichments. J. Comput. Aided. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the OPLS force field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, A.C.; Kumar, S.; Wei, D.Q.; Sahi, S. Structure based virtual screening studies to identify novel potential compounds for GPR142 and their relative dynamic analysis for study of type 2 diabetes. Front. Chem. 2018, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Seow, V.; Lim, J.; Cotterell, A.J.; Yau, M.-K.; Xu, W.; Lohman, R.-J.; Kok, W.M.; Stoermer, M.J.; Sweet, M.J.; Reid, R.C. Receptor residence time trumps drug-likeness and oral bioavailability in determining efficacy of complement C5a antagonists. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kaczor, A.A.; Jörg, M.; Capuano, B. The dopamine D 2 receptor dimer and its interaction with homobivalent antagonists: Homology modeling, docking and molecular dynamics. J. Mol. Model. 2016, 22, 203. [Google Scholar] [CrossRef]
- Ng, H.W.; Laughton, C.A.; Doughty, S.W. Molecular dynamics simulations of the adenosine A2a receptor: Structural stability, sampling and convergence. J. Chem. Inf. Model. 2013, 53, 1168–1178. [Google Scholar] [CrossRef]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM database and PPM web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 2011, 40, D370–D376. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all--atom protein loop prediction. Proteins Struct. Funct. Bioinform. 2004, 55, 351–367. [Google Scholar] [CrossRef]
- Lyman, E.; Higgs, C.; Kim, B.; Lupyan, D.; Shelley, J.C.; Farid, R.; Voth, G.A. A Role for a Specific Cholesterol Interaction in Stabilizing the Apo Configuration of the Human A2A Adenosine Receptor. Structure 2009, 17, 1660–1668. [Google Scholar] [CrossRef]
- Clark, T. G-Protein coupled receptors: Answers from simulations. Beilstein J. Org. Chem. 2017, 13, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Almerico, A.M.; Tutone, M.; Pantano, L.; Lauria, A. A3 adenosine receptor: Homology modeling and 3D-QSAR studies. J. Mol. Graph. Model. 2013, 42, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the SC ’06: Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; p. 43. [Google Scholar]
- Beato, C.; Beccari, A.R.; Cavazzoni, C.; Lorenzi, S.; Costantino, G. Use of Experimental Design to Optimize Docking Performance: The Case of LiGenDock, the Docking Module of Ligen, a New De Novo Design Program. J. Chem. Inf. Model. 2013, 53, 1503–1517. [Google Scholar] [CrossRef] [PubMed]
- Beccari, A.R.; Cavazzoni, C.; Beato, C.; Costantino, G. LiGen: A High Performance Workflow for Chemistry Driven de Novo Design. J. Chem. Inf. Model. 2013, 53, 1518–1527. [Google Scholar] [CrossRef] [PubMed]
- Brady, G.P.; Stouten, P.F.W. Fast prediction and visualization of protein binding pockets with PASS. J. Comput. Aided. Mol. Des. 2000, 14, 383–401. [Google Scholar] [CrossRef] [PubMed]
- Bennett, G.J.; Xie, Y.-K. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 1988, 33, 87–107. [Google Scholar] [CrossRef]
- Hargreaves, K.; Dubner, R.; Brown, F.; Flores, C.; Joris, J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 1988, 32, 77–88. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Koff(min−1) | Residence Time (min) | Kon (M − 1 min−1) | Ki[Kd](M) |
|---|---|---|---|---|
| Merck compound 14 | 6.08 × 10−2 ± 9.78 × 10−3 | 19.06 ± 3.41 | 3.92 × 107± 3.54 × 106 | 1.53 × 10−9 ± 1.89 × 10−10 |
| DFL20656 | 5.34 × 10−3 ± 5.66 × 10−4 | 198.14 ± 20.41 | 5.06 × 106 ± 2.57 × 105 | 1.05 × 10−9 ± 8.44 × 10−11 |
| [Leu8]-Lys-desArg9-BK | 5.83 × 10−3± 3.93 × 10−4 | 176.04 ± 13.08 | 4.87 × 107± 1.92 × 106 | 1.21 × 10−10 ± 9.57 × 10−12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gemei, M.; Talarico, C.; Brandolini, L.; Manelfi, C.; Za, L.; Bovolenta, S.; Liberati, C.; Del Vecchio, L.; Russo, R.; Cerchia, C.; et al. Binding Mode Exploration of B1 Receptor Antagonists’ by the Use of Molecular Dynamics and Docking Simulation—How Different Target Engagement Can Determine Different Biological Effects. Int. J. Mol. Sci. 2020, 21, 7677. https://doi.org/10.3390/ijms21207677
Gemei M, Talarico C, Brandolini L, Manelfi C, Za L, Bovolenta S, Liberati C, Del Vecchio L, Russo R, Cerchia C, et al. Binding Mode Exploration of B1 Receptor Antagonists’ by the Use of Molecular Dynamics and Docking Simulation—How Different Target Engagement Can Determine Different Biological Effects. International Journal of Molecular Sciences. 2020; 21(20):7677. https://doi.org/10.3390/ijms21207677
Chicago/Turabian StyleGemei, Marica, Carmine Talarico, Laura Brandolini, Candida Manelfi, Lorena Za, Silvia Bovolenta, Chiara Liberati, Luigi Del Vecchio, Roberto Russo, Carmen Cerchia, and et al. 2020. "Binding Mode Exploration of B1 Receptor Antagonists’ by the Use of Molecular Dynamics and Docking Simulation—How Different Target Engagement Can Determine Different Biological Effects" International Journal of Molecular Sciences 21, no. 20: 7677. https://doi.org/10.3390/ijms21207677
APA StyleGemei, M., Talarico, C., Brandolini, L., Manelfi, C., Za, L., Bovolenta, S., Liberati, C., Del Vecchio, L., Russo, R., Cerchia, C., Allegretti, M., & Beccari, A. R. (2020). Binding Mode Exploration of B1 Receptor Antagonists’ by the Use of Molecular Dynamics and Docking Simulation—How Different Target Engagement Can Determine Different Biological Effects. International Journal of Molecular Sciences, 21(20), 7677. https://doi.org/10.3390/ijms21207677