Regulation of Platelet Production and Life Span: Role of Bcl-xL and Potential Implications for Human Platelet Diseases


Abstract
1. Platelets
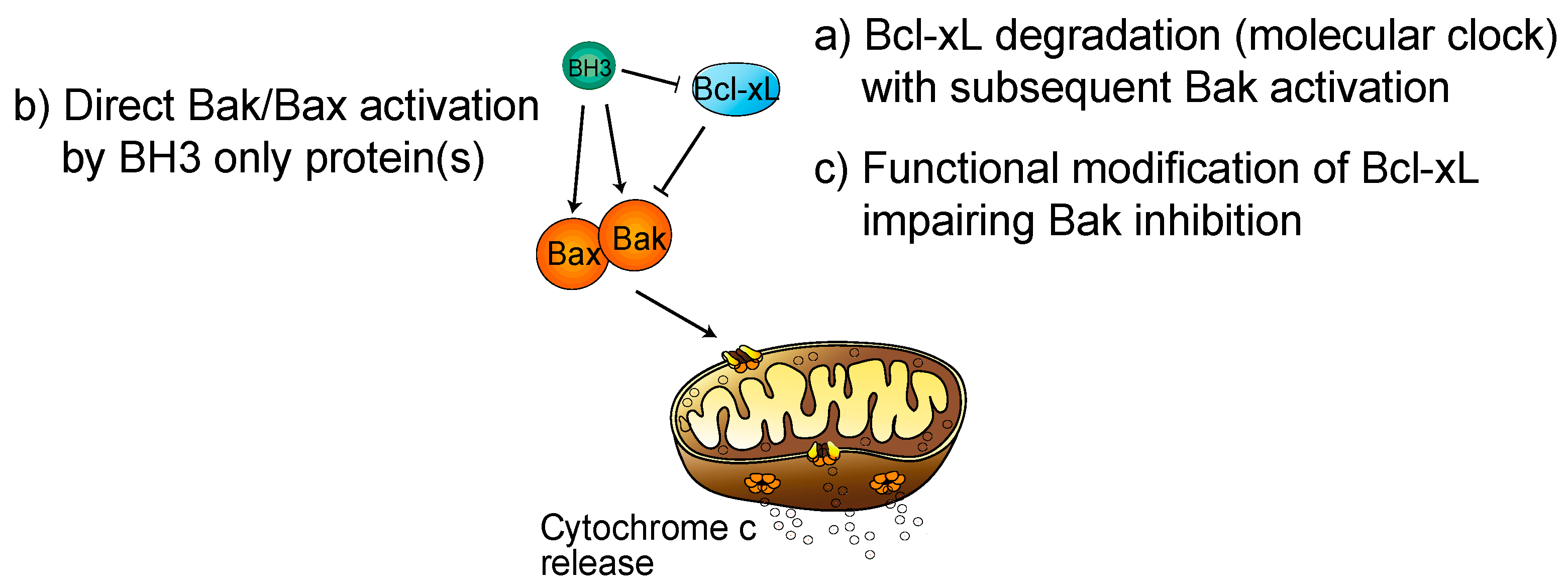
2. Regulation of Platelet Life Span by Apoptosis: Role of Bcl-xL
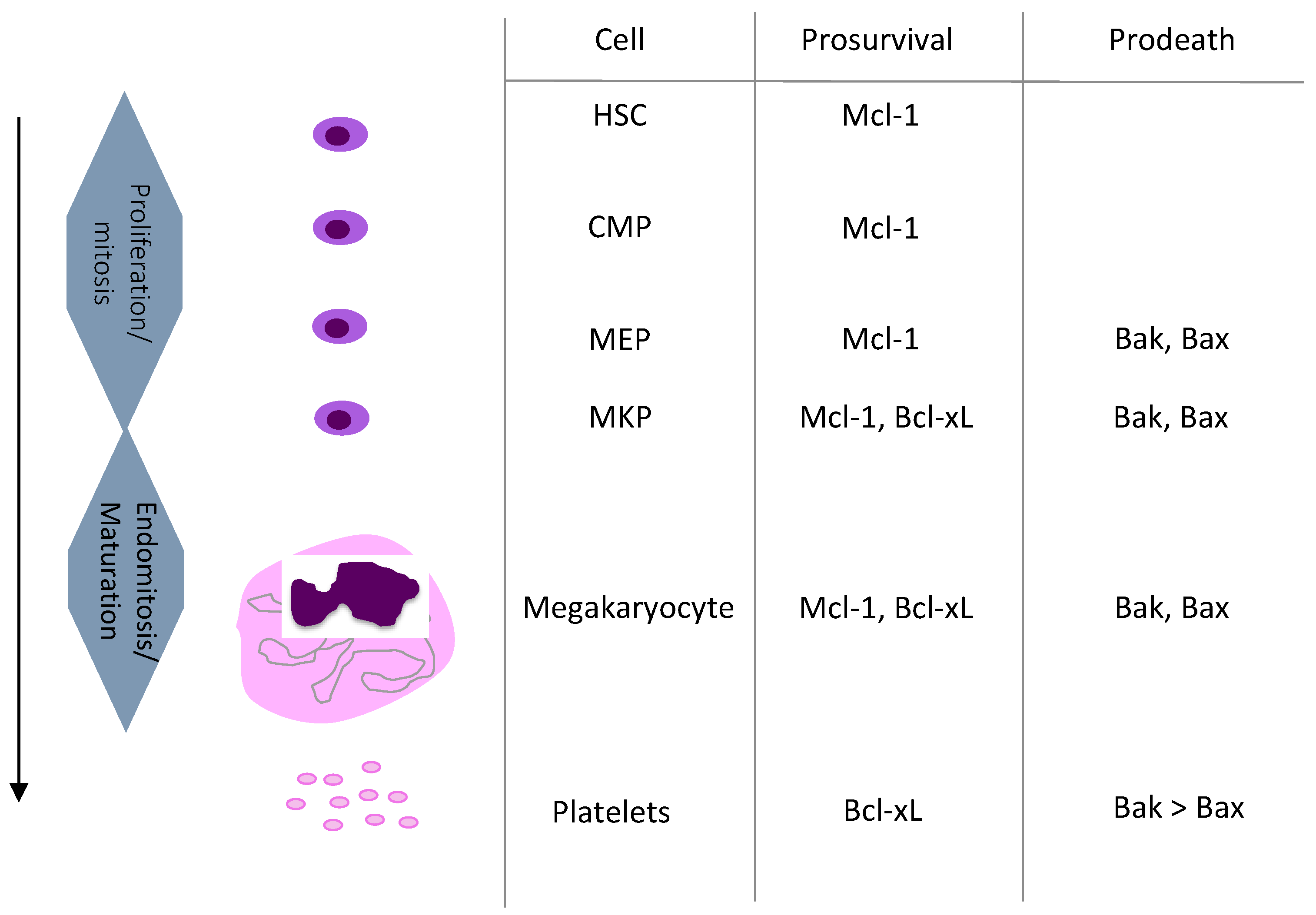
3. Megakaryopoiesis and Its Regulation
4. Diseases of Platelet Numbers in Humans
4.1. Thrombocytosis (Platelet Count over 400 × 106/mL)
4.2. Thrombocytopenia (Platelet Count Less Than 150 × 106/mL)
4.2.1. Thrombocytopenia with Abnormal Pooling
4.2.2. Thrombocytopenia due to Defective Platelet Production
4.2.3. Thrombocytopenia due to Increased Platelet Destruction or Consumption
5. Role of Bcl-xL in Megakaryocyte Survival
6. Conclusions
Funding
Conflicts of Interest
References
- Engelmann, B.; Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 2013, 13, 34–45. [Google Scholar] [CrossRef]
- Yeaman, M.R. Platelets in defense against bacterial pathogens. Cell Mol. Life Sci. 2010, 67, 525–544. [Google Scholar] [CrossRef]
- Gay, L.J.; Felding-Habermann, B. Contribution of platelets to tumour metastasis. Nat. Rev. Cancer 2011, 11, 123–134. [Google Scholar] [CrossRef]
- Goubran, H.A.; Stakiw, J.; Radosevic, M.; Burnouf, T. Platelets effects on tumor growth. Semin. Oncol. 2014, 41, 359–369. [Google Scholar] [CrossRef]
- Suzuki-Inoue, K. Essential in vivo roles of the platelet activation receptor CLEC-2 in tumour metastasis, lymphangiogenesis and thrombus formation. J. Biochem. 2011, 150, 127–132. [Google Scholar] [CrossRef]
- Sabrkhany, S.; Griffioen, A.W.; Oude Egbrink, M.G. The role of blood platelets in tumor angiogenesis. Biochim. Biophys. Acta 2011, 1815, 189–196. [Google Scholar] [CrossRef]
- Hess, P.R.; Rawnsley, D.R.; Jakus, Z.; Yang, Y.; Sweet, D.T.; Fu, J.; Herzog, B.; Lu, M.; Nieswandt, B.; Oliver, G.; et al. Platelets mediate lymphovenous hemostasis to maintain blood-lymphatic separation throughout life. J. Clin. Investig. 2014, 124, 273–284. [Google Scholar] [CrossRef]
- Boilard, E.; Blanco, P.; Nigrovic, P.A. Platelets: Active players in the pathogenesis of arthritis and SLE. Nat. Rev. Rheumatol. 2012, 8, 534–542. [Google Scholar] [CrossRef]
- Caudrillier, A.; Kessenbrock, K.; Gilliss, B.M.; Nguyen, J.X.; Marques, M.B.; Monestier, M.; Toy, P.; Werb, Z.; Looney, M.R. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J. Clin. Investig. 2012, 122, 2661–2671. [Google Scholar] [CrossRef]
- Danese, S.; Scaldaferri, F.; Papa, A.; Pola, R.; Sans, M.; Gasbarrini, G.; Pola, P.; Gasbarrini, A. Platelets: New players in the mucosal scenario of inflammatory bowel disease. Eur. Rev. Med. Pharm. Sci. 2004, 8, 193–198. [Google Scholar]
- Golebiewska, E.M.; Poole, A.W. Platelet secretion: From haemostasis to wound healing and beyond. Blood Rev. 2015, 29, 153–162. [Google Scholar] [CrossRef]
- Kirschbaum, M.; Jenne, C.N.; Veldhuis, Z.J.; Sjollema, K.A.; Lenting, P.J.; Giepmans, B.N.G.; Porte, R.J.; Kubes, P.; Denis, C.V.; Lisman, T. Transient von Willebrand factor-mediated platelet influx stimulates liver regeneration after partial hepatectomy in mice. Liver Int. 2017, 37, 1731–1737. [Google Scholar] [CrossRef]
- Jarre, A.; Gowert, N.S.; Donner, L.; Münzer, P.; Klier, M.; Borst, O.; Schaller, M.; Lang, F.; Korth, C.; Elvers, M. Pre-activated blood platelets and a pro-thrombotic phenotype in APP23 mice modeling Alzheimer’s disease. Cell. Signal. 2014, 26, 2040–2050. [Google Scholar] [CrossRef]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef]
- Kuwana, T.; Bouchier-Hayes, L.; Chipuk, J.E.; Bonzon, C.; Sullivan, B.A.; Green, D.R.; Newmeyer, D.D. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol. Cell 2005, 17, 525–535. [Google Scholar] [CrossRef]
- Ren, D.; Tu, H.C.; Kim, H.; Wang, G.X.; Bean, G.R.; Takeuchi, O.; Jeffers, J.R.; Zambetti, G.P.; Hsieh, J.J.; Cheng, E.H. BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program. Science 2010, 330, 1390–1393. [Google Scholar] [CrossRef]
- Villunger, A.; Labi, V.; Bouillet, P.; Adams, J.; Strasser, A. Can the analysis of BH3-only protein knockout mice clarify the issue of ‘direct versus indirect’ activation of Bax and Bak? Cell Death Differ. 2011, 18, 1545–1546. [Google Scholar] [CrossRef][Green Version]
- Willis, S.N.; Fletcher, J.I.; Kaufmann, T.; van Delft, M.F.; Chen, L.; Czabotar, P.E.; Ierino, H.; Lee, E.F.; Fairlie, W.D.; Bouillet, P.; et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 2007, 315, 856–859. [Google Scholar] [CrossRef]
- Mason, K.D.; Carpinelli, M.R.; Fletcher, J.I.; Collinge, J.E.; Hilton, A.A.; Ellis, S.; Kelly, P.N.; Ekert, P.G.; Metcalf, D.; Roberts, A.W.; et al. Programmed anuclear cell death delimits platelet life span. Cell 2007, 128, 1173–1186. [Google Scholar] [CrossRef]
- Zhang, H.; Nimmer, P.M.; Tahir, S.K.; Chen, J.; Fryer, R.M.; Hahn, K.R.; Iciek, L.A.; Morgan, S.J.; Nasarre, M.C.; Nelson, R.; et al. Bcl-2 family proteins are essential for platelet survival. Cell Death Differ. 2007, 14, 943–951. [Google Scholar] [CrossRef]
- Lebois, M.; Josefsson, E.C. Regulation of platelet lifespan by apoptosis. Platelets 2016, 27, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Josefsson, E.C.; James, C.; Henley, K.J.; Debrincat, M.A.; Rogers, K.L.; Dowling, M.R.; White, M.J.; Kruse, E.A.; Lane, R.M.; Ellis, S.; et al. Megakaryocytes possess a functional intrinsic apoptosis pathway that must be restrained to survive and produce platelets. J. Exp. Med. 2011, 208, 2017–2031. [Google Scholar] [CrossRef] [PubMed]
- Pleines, I.; Lebois, M.; Gangatirkar, P.; Au, A.E.; Lane, R.M.; Henley, K.J.; Kauppi, M.; Corbin, J.; Cannon, P.; Bernardini, J.; et al. Intrinsic apoptosis circumvents the functional decline of circulating platelets but does not cause the storage lesion. Blood 2018, 132, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Schoenwaelder, S.M.; Yuan, Y.; Josefsson, E.C.; White, M.J.; Yao, Y.; Mason, K.D.; O’Reilly, L.A.; Henley, K.J.; Ono, A.; Hsiao, S.; et al. Two distinct pathways regulate platelet phosphatidylserine exposure and procoagulant function. Blood 2009, 114, 663–666. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.W.; Seymour, J.F.; Brown, J.R.; Wierda, W.G.; Kipps, T.J.; Khaw, S.L.; Carney, D.A.; He, S.Z.; Huang, D.C.; Xiong, H.; et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: Results of a phase I study of navitoclax in patients with relapsed or refractory disease. J. Clin. Oncol. 2012, 30, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Bertino, A.M.; Qi, X.Q.; Li, J.; Xia, Y.; Kuter, D.J. Apoptotic markers are increased in platelets stored at 37 degrees C. Transfusion 2003, 43, 857–866. [Google Scholar] [CrossRef]
- Debrincat, M.A.; Pleines, I.; Lebois, M.; Lane, R.M.; Holmes, M.L.; Corbin, J.; Vandenberg, C.J.; Alexander, W.S.; Ng, A.P.; Strasser, A.; et al. BCL-2 is dispensable for thrombopoiesis and platelet survival. Cell Death Dis. 2015, 6, e1721. [Google Scholar] [CrossRef]
- Kodama, T.; Takehara, T.; Hikita, H.; Shimizu, S.; Shigekawa, M.; Li, W.; Miyagi, T.; Hosui, A.; Tatsumi, T.; Ishida, H.; et al. BH3-only activator proteins Bid and Bim are dispensable for Bak/Bax-dependent thrombocyte apoptosis induced by Bcl-xL deficiency: Molecular requisites for the mitochondrial pathway to apoptosis in platelets. J. Biol. Chem. 2011, 286, 13905–13913. [Google Scholar] [CrossRef]
- Leytin, V.; Allen, D.J.; Mykhaylov, S.; Lyubimov, E.; Freedman, J. Thrombin-triggered platelet apoptosis. J. Thromb. Haemost. JTH 2006, 4, 2656–2663. [Google Scholar] [CrossRef]
- Li, S.; Wang, Z.; Liao, Y.; Zhang, W.; Shi, Q.; Yan, R.; Ruan, C.; Dai, K. The glycoprotein Ibalpha-von Willebrand factor interaction induces platelet apoptosis. J. Thromb. Haemost. JTH 2010, 8, 341–350. [Google Scholar] [CrossRef]
- Liu, Z.J.; Hoffmeister, K.M.; Hu, Z.; Mager, D.E.; Ait-Oudhia, S.; Debrincat, M.A.; Pleines, I.; Josefsson, E.C.; Kile, B.T.; Italiano, J., Jr.; et al. Expansion of the neonatal platelet mass is achieved via an extension of platelet lifespan. Blood 2014, 123, 3381–3389. [Google Scholar] [CrossRef] [PubMed]
- Vanags, D.M.; Orrenius, S.; Aguilar-Santelises, M. Alterations in Bcl-2/Bax protein levels in platelets form part of an ionomycin-induced process that resembles apoptosis. Br. J. Haematol. 1997, 99, 824–831. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.W.; Davids, M.S.; Pagel, J.M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.; et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Print, C.G.; Loveland, K.L.; Gibson, L.; Meehan, T.; Stylianou, A.; Wreford, N.; de Kretser, D.; Metcalf, D.; Kontgen, F.; Adams, J.M.; et al. Apoptosis regulator bcl-w is essential for spermatogenesis but appears otherwise redundant. Proc. Natl. Acad. Sci. USA 1998, 95, 12424–12431. [Google Scholar] [CrossRef]
- Ross, A.J.; Waymire, K.G.; Moss, J.E.; Parlow, A.F.; Skinner, M.K.; Russell, L.D.; MacGregor, G.R. Testicular degeneration in Bclw-deficient mice. Nat. Genet. 1998, 18, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xia, Y.; Bertino, A.M.; Coburn, J.P.; Kuter, D.J. The mechanism of apoptosis in human platelets during storage. Transfusion 2000, 40, 1320–1329. [Google Scholar] [CrossRef]
- Delbridge, A.R.; Chappaz, S.; Ritchie, M.E.; Kile, B.T.; Strasser, A.; Grabow, S. Loss of PUMA (BBC3) does not prevent thrombocytopenia caused by the loss of BCL-XL (BCL2L1). Br. J. Haematol. 2016, 174, 962–969. [Google Scholar] [CrossRef]
- Hamasaki, A.; Sendo, F.; Nakayama, K.; Ishida, N.; Negishi, I.; Nakayama, K.; Hatakeyama, S. Accelerated neutrophil apoptosis in mice lacking A1-a, a subtype of the bcl-2-related A1 gene. J. Exp. Med. 1998, 188, 1985–1992. [Google Scholar] [CrossRef]
- Ottina, E.; Grespi, F.; Tischner, D.; Soratroi, C.; Geley, S.; Ploner, A.; Reichardt, H.M.; Villunger, A.; Herold, M.J. Targeting antiapoptotic A1/Bfl-1 by in vivo RNAi reveals multiple roles in leukocyte development in mice. Blood 2012, 119, 6032–6042. [Google Scholar] [CrossRef]
- Josefsson, E.C.; Kile, B.T. Cell death in the hematopoietic system. In Essentials of Apoptosis, 2nd ed.; Yin, X.-M., Dong, Z., Eds.; Springer: New York, NY, USA, 2009; pp. 443–459. [Google Scholar] [CrossRef]
- Debrincat, M.A.; Josefsson, E.C.; James, C.; Henley, K.J.; Ellis, S.; Lebois, M.; Betterman, K.L.; Lane, R.M.; Rogers, K.L.; White, M.J.; et al. Mcl-1 and Bcl-x(L) coordinately regulate megakaryocyte survival. Blood 2012, 119, 5850–5858. [Google Scholar] [CrossRef] [PubMed]
- Kodama, T.; Hikita, H.; Kawaguchi, T.; Shigekawa, M.; Shimizu, S.; Hayashi, Y.; Li, W.; Miyagi, T.; Hosui, A.; Tatsumi, T.; et al. Mcl-1 and Bcl-xL regulate Bak/Bax-dependent apoptosis of the megakaryocytic lineage at multistages. Cell Death Differ. 2012, 19, 1856–1869. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, C.J.; Josefsson, E.C.; Campbell, K.J.; James, C.; Lawlor, K.E.; Kile, B.T.; Cory, S. Loss of Bak enhances lymphocytosis but does not ameliorate thrombocytopaenia in BCL-2 transgenic mice. Cell Death Differ. 2014, 21, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Koehler, M.F.; Bergeron, P.; Choo, E.F.; Lau, K.; Ndubaku, C.; Dudley, D.; Gibbons, P.; Sleebs, B.E.; Rye, C.S.; Nikolakopoulos, G.; et al. Structure-Guided Rescaffolding of Selective Antagonists of BCL-XL. ACS Med. Chem. Lett. 2014, 5, 662–667. [Google Scholar] [CrossRef]
- Lessene, G.; Czabotar, P.E.; Sleebs, B.E.; Zobel, K.; Lowes, K.N.; Adams, J.M.; Baell, J.B.; Colman, P.M.; Deshayes, K.; Fairbrother, W.J.; et al. Structure-guided design of a selective BCL-X(L) inhibitor. Nat. Chem. Biol. 2013, 9, 390–397. [Google Scholar] [CrossRef]
- Leverson, J.D.; Phillips, D.C.; Mitten, M.J.; Boghaert, E.R.; Diaz, D.; Tahir, S.K.; Belmont, L.D.; Nimmer, P.; Xiao, Y.; Ma, X.M.; et al. Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci. Transl. Med. 2015, 7, 279ra240. [Google Scholar] [CrossRef] [PubMed]
- Tao, Z.F.; Hasvold, L.; Wang, L.; Wang, X.; Petros, A.M.; Park, C.H.; Boghaert, E.R.; Catron, N.D.; Chen, J.; Colman, P.M.; et al. Discovery of a Potent and Selective BCL-XL Inhibitor with in Vivo Activity. ACS Med. Chem. Lett. 2014, 5, 1088–1093. [Google Scholar] [CrossRef]
- Josefsson, E.C.; Dowling, M.R.; Lebois, M.; Kile, B.T. The regulation of platelet life span. In Platelets, 3rd ed.; Michelson, A., Ed.; Elsevier Inc.: London, UK, 2013; pp. 51–65. [Google Scholar]
- Deppermann, C.; Kratofil, R.M.; Peiseler, M.; David, B.A.; Zindel, J.; Castanheira, F.; van der Wal, F.; Carestia, A.; Jenne, C.N.; Marth, J.D.; et al. Macrophage galactose lectin is critical for Kupffer cells to clear aged platelets. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Hoffmeister, K.M.; Falet, H. Platelet clearance by the hepatic Ashwell-Morrell receptor: Mechanisms and biological significance. Thromb. Res. 2016, 141 (Suppl. 2), S68–S72. [Google Scholar] [CrossRef]
- Kelly, P.N.; White, M.J.; Goschnick, M.W.; Fairfax, K.A.; Tarlinton, D.M.; Kinkel, S.A.; Bouillet, P.; Adams, J.M.; Kile, B.T.; Strasser, A. Individual and overlapping roles of BH3-only proteins Bim and Bad in apoptosis of lymphocytes and platelets and in suppression of thymic lymphoma development. Cell Death Differ. 2010, 17, 1655–1664. [Google Scholar] [CrossRef]
- Lefrançais, E.; Ortiz-Muñoz, G.; Caudrillier, A.; Mallavia, B.; Liu, F.; Sayah, D.M.; Thornton, E.E.; Headley, M.B.; David, T.; Coughlin, S.R.; et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature 2017, 544, 105–109. [Google Scholar] [CrossRef]
- Chao, M.P.; Seita, J.; Weissman, I.L. Establishment of a normal hematopoietic and leukemia stem cell hierarchy. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2008; Volume 73, pp. 439–449. [Google Scholar] [CrossRef]
- Gekas, C.; Graf, T. CD41 expression marks myeloid-biased adult hematopoietic stem cells and increases with age. Blood 2013, 121, 4463–4472. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Fraticelli, A.E.; Weinreb, C.; Wang, S.W.; Migueles, R.P.; Jankovic, M.; Usart, M.; Klein, A.M.; Lowell, S.; Camargo, F.D. Single-cell lineage tracing unveils a role for TCF15 in haematopoiesis. Nature 2020, 583, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Mazzi, S.; Lordier, L.; Debili, N.; Raslova, H.; Vainchenker, W. Megakaryocyte and polyploidization. Exp. Hematol. 2018, 57, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Junt, T.; Schulze, H.; Chen, Z.; Massberg, S.; Goerge, T.; Krueger, A.; Wagner, D.D.; Graf, T.; Italiano, J.E., Jr.; Shivdasani, R.A.; et al. Dynamic visualization of thrombopoiesis within bone marrow. Science 2007, 317, 1767–1770. [Google Scholar] [CrossRef]
- Thon, J.N.; Montalvo, A.; Patel-Hett, S.; Devine, M.T.; Richardson, J.L.; Ehrlicher, A.; Larson, M.K.; Hoffmeister, K.; Hartwig, J.H.; Italiano, J.E., Jr. Cytoskeletal mechanics of proplatelet maturation and platelet release. J. Cell Biol. 2010, 191, 861–874. [Google Scholar] [CrossRef]
- Potts, K.S.; Farley, A.; Dawson, C.A.; Rimes, J.; Biben, C.; de Graaf, C.; Potts, M.A.; Stonehouse, O.J.; Carmagnac, A.; Gangatirkar, P.; et al. Membrane budding is a major mechanism of in vivo platelet biogenesis. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Kaushansky, K. The molecular mechanisms that control thrombopoiesis. J. Clin. Investig. 2005, 115, 3339–3347. [Google Scholar] [CrossRef]
- Plo, I.; Bellanné-Chantelot, C.; Mosca, M.; Mazzi, S.; Marty, C.; Vainchenker, W. Genetic Alterations of the Thrombopoietin/MPL/JAK2 Axis Impacting Megakaryopoiesis. Front. Endocrinol. 2017, 8, 234. [Google Scholar] [CrossRef]
- Sanz, C.; Benet, I.; Richard, C.; Badia, B.; Andreu, E.J.; Prosper, F.; Fernández-Luna, J.L. Antiapoptotic protein Bcl-x(L) is up-regulated during megakaryocytic differentiation of CD34(+) progenitors but is absent from senescent megakaryocytes. Exp. Hematol. 2001, 29, 728–735. [Google Scholar] [CrossRef]
- Fielder, P.J.; Gurney, A.L.; Stefanich, E.; Marian, M.; Moore, M.W.; Carver-Moore, K.; de Sauvage, F.J. Regulation of thrombopoietin levels by c-mpl-mediated binding to platelets. Blood 1996, 87, 2154–2161. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.R.; Hartwig, J.H.; Italiano, J.E., Jr. The biogenesis of platelets from megakaryocyte proplatelets. J. Clin. Investig. 2005, 115, 3348–3354. [Google Scholar] [CrossRef] [PubMed]
- Xavier-Ferrucio, J.; Scanlon, V.; Li, X.; Zhang, P.X.; Lozovatsky, L.; Ayala-Lopez, N.; Tebaldi, T.; Halene, S.; Cao, C.; Fleming, M.D.; et al. Low iron promotes megakaryocytic commitment of megakaryocytic-erythroid progenitors in humans and mice. Blood 2019, 134, 1547–1557. [Google Scholar] [CrossRef]
- Vainchenker, W.; Kralovics, R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood 2017, 129, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Malherbe, J.A.; Fuller, K.A.; Arshad, A.; Nangalia, J.; Romeo, G.; Hall, S.L.; Meehan, K.S.; Guo, B.; Howman, R.; Erber, W.N. Megakaryocytic hyperplasia in myeloproliferative neoplasms is driven by disordered proliferative, apoptotic and epigenetic mechanisms. J. Clin. Pathol. 2016, 69, 155–163. [Google Scholar] [CrossRef] [PubMed]
- López, J.A.; Berliner, N. Introduction to a series of reviews on clinical platelet disorders. Blood 2017, 129, 2821–2822. [Google Scholar] [CrossRef]
- Nurden, A.T.; Nurden, P. Inherited thrombocytopenias: History, advances and perspectives. Haematologica 2020. [Google Scholar] [CrossRef]
- Lambert, M.P.; Gernsheimer, T.B. Clinical updates in adult immune thrombocytopenia. Blood 2017, 129, 2829–2835. [Google Scholar] [CrossRef]
- Clarke, M.C.; Savill, J.; Jones, D.B.; Noble, B.S.; Brown, S.B. Compartmentalized megakaryocyte death generates functional platelets committed to caspase-independent death. J. Cell Biol. 2003, 160, 577–587. [Google Scholar] [CrossRef]
- De Botton, S.; Sabri, S.; Daugas, E.; Zermati, Y.; Guidotti, J.E.; Hermine, O.; Kroemer, G.; Vainchenker, W.; Debili, N. Platelet formation is the consequence of caspase activation within megakaryocytes. Blood 2002, 100, 1310–1317. [Google Scholar] [CrossRef]
- Morison, I.M.; Bordé, E.M.C.; Cheesman, E.J.; Cheong, P.L.; Holyoake, A.J.; Fichelson, S.; Weeks, R.J.; Lo, A.; Davies, S.M.; Wilbanks, S.M.; et al. A mutation of human cytochrome c enhances the intrinsic apoptotic pathway but causes only thrombocytopenia. Nat. Genet. 2008, 40, 387–389. [Google Scholar] [CrossRef] [PubMed]
- De Rocco, D.; Cerqua, C.; Goffrini, P.; Russo, G.; Pastore, A.; Meloni, F.; Nicchia, E.; Moraes, C.T.; Pecci, A.; Salviati, L.; et al. Mutations of cytochrome c identified in patients with thrombocytopenia THC4 affect both apoptosis and cellular bioenergetics. Biochim. Biophys. Acta 2014, 1842, 269–274. [Google Scholar] [CrossRef]
- Liptak, M.D.; Fagerlund, R.D.; Ledgerwood, E.C.; Wilbanks, S.M.; Bren, K.L. The proapoptotic G41S mutation to human cytochrome c alters the heme electronic structure and increases the electron self-exchange rate. J. Am. Chem. Soc. 2011, 133, 1153–1155. [Google Scholar] [CrossRef]
- Afreen, S.; Bohler, S.; Müller, A.; Demmerath, E.M.; Weiss, J.M.; Jutzi, J.S.; Schachtrup, K.; Kunze, M.; Erlacher, M. BCL-XL expression is essential for human erythropoiesis and engraftment of hematopoietic stem cells. Cell Death Dis. 2020, 11, 8. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |



{kind=link}
{kind=link}
| Primitive |
| Acquired: - Myeloproliferative Neoplasms (Essential thrombocytopenia, Polycythemia Vera, Primitive Myelofibrosis, Chronic Myeloid Leukemia) |
| - Rarely: Myelodysplastic syndromes (5q-) Constitutional (inherited) thrombocytosis |
| Reactive |
| Iron Deficiency |
| Inflammation/Cancer |
| Secondary to asplenism |
| Due to defective platelet production |
| Acquired: - Bone marrow infiltration (by malignant cells in most cases) |
| - Vitamin deficiency (B12, folates) - Defect in megakaryocyte differentiation (myelodysplastic syndromes) Constitutional (inherited) thrombocytopenia - Absence of megakaryocytes - Defects in megakaryocyte differentiation (mutations in transcription factors) - Defects in proplatelet formation |
| Due to increased platelet destruction |
| Immunological processes - Immune Thrombocytopenia Purpura - Drug-induced thrombocytopenia |
| Non-Immunological processes: platelet consumption - Microangiopathies (including thrombotic thrombocytopenia) - Large hemorrhages - Disseminated intravascular coagulation - Sepsis |
| Secondary to abnormal pooling (hypersplenism due to splenomegaly) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Josefsson, E.C.; Vainchenker, W.; James, C. Regulation of Platelet Production and Life Span: Role of Bcl-xL and Potential Implications for Human Platelet Diseases. Int. J. Mol. Sci. 2020, 21, 7591. https://doi.org/10.3390/ijms21207591
Josefsson EC, Vainchenker W, James C. Regulation of Platelet Production and Life Span: Role of Bcl-xL and Potential Implications for Human Platelet Diseases. International Journal of Molecular Sciences. 2020; 21(20):7591. https://doi.org/10.3390/ijms21207591
Chicago/Turabian StyleJosefsson, Emma C., William Vainchenker, and Chloe James. 2020. "Regulation of Platelet Production and Life Span: Role of Bcl-xL and Potential Implications for Human Platelet Diseases" International Journal of Molecular Sciences 21, no. 20: 7591. https://doi.org/10.3390/ijms21207591
APA StyleJosefsson, E. C., Vainchenker, W., & James, C. (2020). Regulation of Platelet Production and Life Span: Role of Bcl-xL and Potential Implications for Human Platelet Diseases. International Journal of Molecular Sciences, 21(20), 7591. https://doi.org/10.3390/ijms21207591