Dissecting Molecular Features of Gliomas: Genetic Loci and Validated Biomarkers

 , ,
, , 
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Germline Features and Loci Influencing the Risk of Glioma
3. Somatic Molecular Features for Glioma Classification
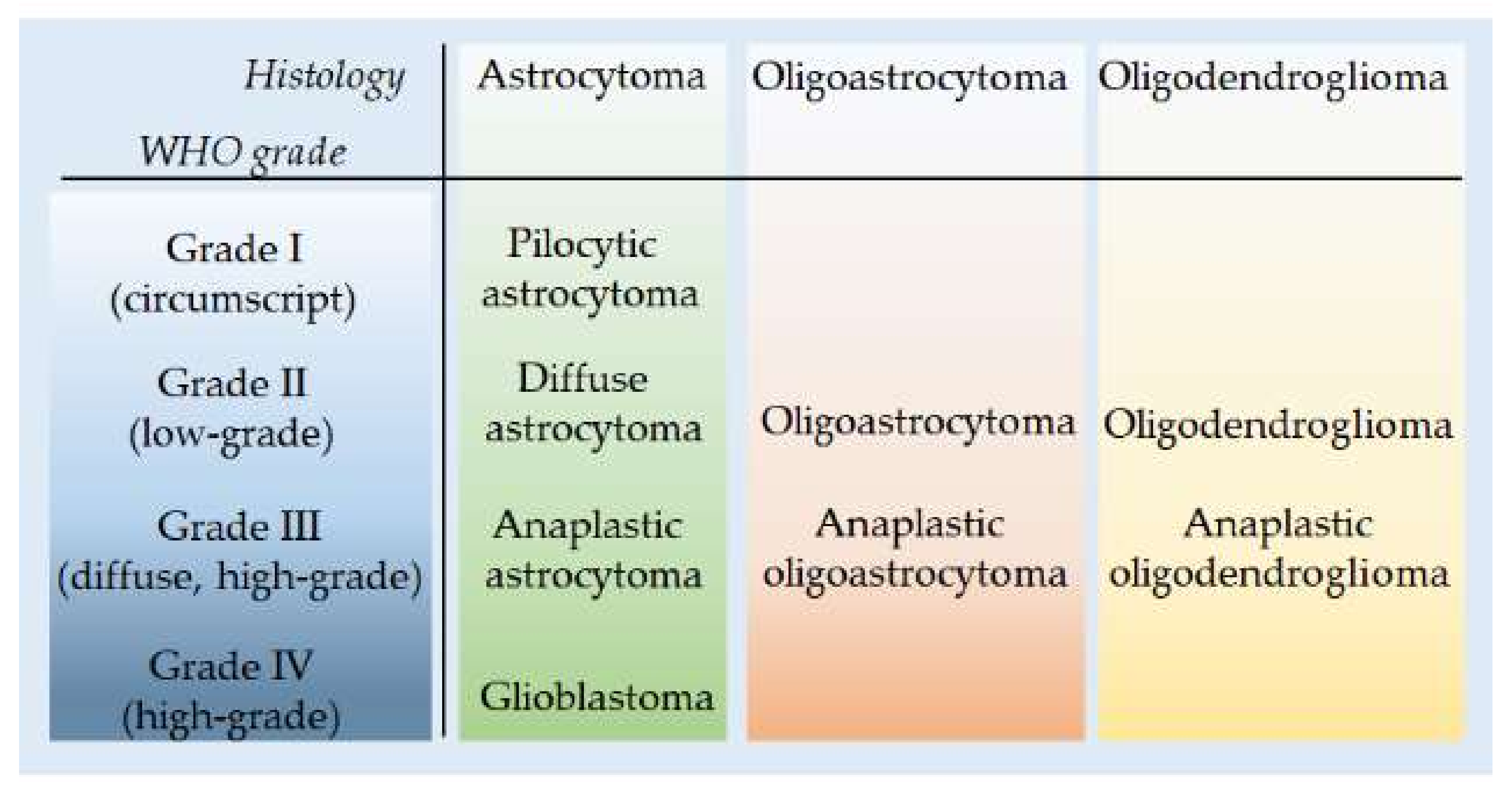
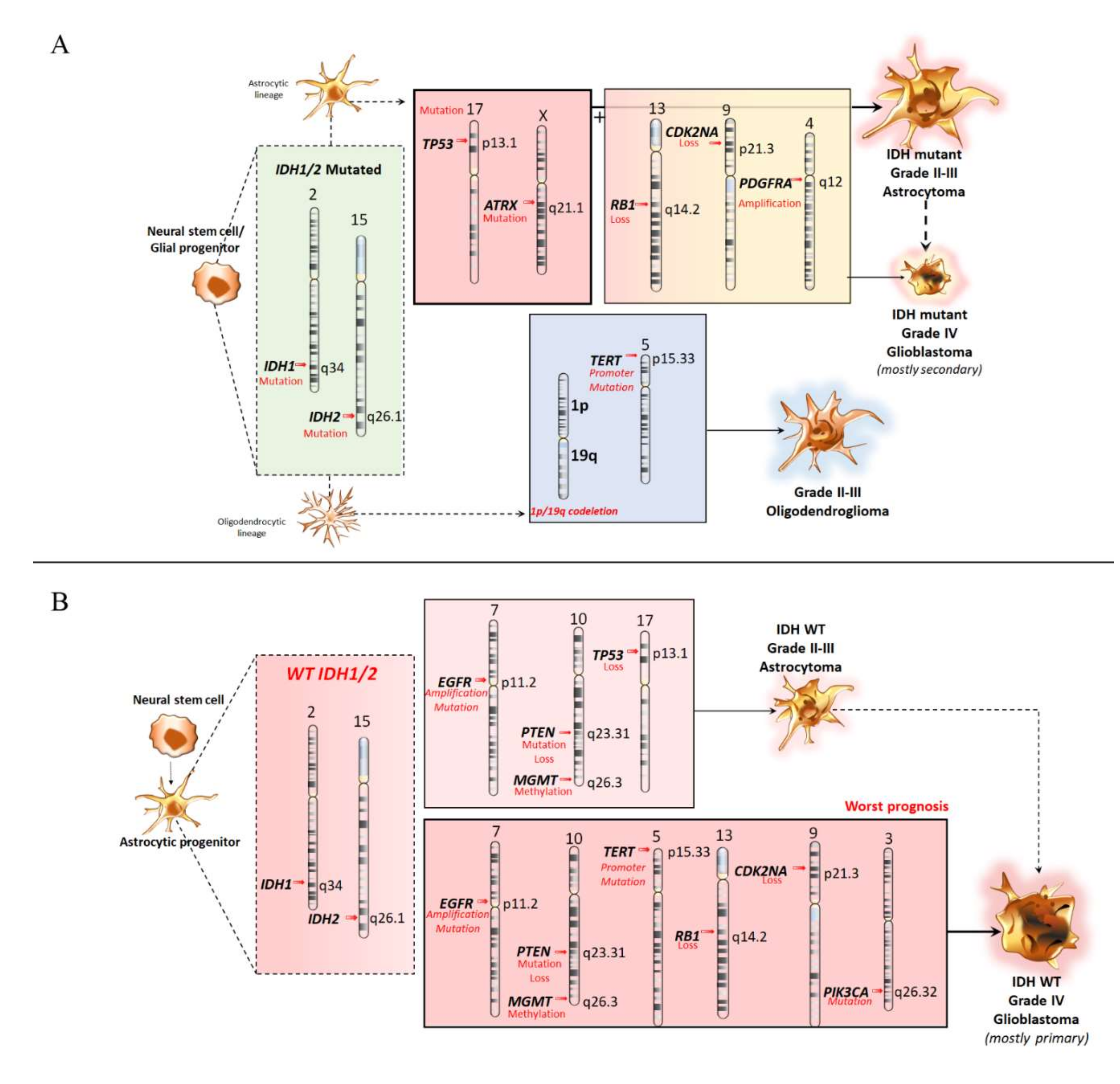
3.1. Molecular Features of Astrocytoma and Oligodendroglioma
3.2. Molecular Features of Primary Glioblastomas
3.3. Glioma Epigenetics
4. Somatic Molecular Features and Copy Number Variation
5. Diagnostic Approaches and NGS Application
6. Liquid Biopsies in Patients With Diffuse Glioma
7. Merging Genomics and Immunology: A Glance at Future Perspectives
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| aCGH | Array Comparative Genomic Hybridization |
| ACVR1 | Activin Receptor Type 1 |
| ALT | Alternative Lengthening Of Telomeres |
| ATRX α- | Thalassemia Mental Retardation X-Linked |
| BBB | Blood–Brain Barrier |
| CDKN2A, CDKN2B | Cyclin-Dependent Kinase Inhibitor 2A And B |
| CIC | Capicua Transcriptional Repressor |
| CIN | Chromosomal Instability |
| CNS | Central Nervous System |
| CNVs | Copy Number Variations |
| CSF | Cerebrospinal Fluid |
| ctDNSA | Circulating Tumor DNA |
| ddPCR | Droplet-Based Digital PCR |
| EGFR | Epidermal Growth Factor Receptor |
| EVs | Circulating Extracellular Vesicles |
| FFPE | Formalin-Fixed Paraffin-Embedded |
| FISH | Fluorescence In Situ Hybridization |
| GWAS | Genome-Wide Association Studies |
| IDH1 | I Isocitrate-Dehydrogenase Enzyme 1 |
| IDH2 | Isocitrate-Dehydrogenase Enzyme 2 |
| IH | Immunohistochemistry |
| LGGs | Low-Grade Gliomas |
| MGMT DNA | Repair Gene O6-Alkylguanine DNA Alkyltransferase |
| MIN | Microsatellite Instability |
| NADPH | Nicotinamide Adenine Dinucleotide Phosphate |
| NGS | Next Generation Sequencing |
| OPN | Osteopontin |
| PHLDB1 | Pleckstrin Homology-Like Domain Family B Member 1 |
| PIN | Point Mutation Instability |
| PTEN | Phosphatase And Tensin Homolog |
| RTEL1 | Regulator of Telomere Elongation Helicase 1 |
| SNVs | Single Nucleotide Variations |
| TERT | Telomerase Reverse Transcriptase |
| TP53 | Tumor Protein P53 |
| TP534 | Tumor Protein P53 |
| TSC1 | Tuberous Sclerosis 1/Hamartin |
| TSC2 | Tuberous Sclerosis 2/Tuberin |
| VEGF | Vascular Endothelial Growth Factor |
| WHO | World Health Organization |
References
- Vigneswaran, K.; Neill, S.; Hadjipanayis, C.G. Beyond the World Health Organization grading of infiltrating gliomas: Advances in the molecular genetics of glioma classification. Ann. Transl. Med. 2015, 3, 95. [Google Scholar]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K. (Eds.) WHO Classification of Tumours of the Central Nervous System, 4th ed.; IARC: Lyon, France; WHO Regional Office Europe: Copenhagen, Denmark, 2007; pp. 33–49. [Google Scholar]
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Kleihues, P. The definition of primary and secondary glioblastoma. Clin. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research. Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef] [PubMed]
- Nickel, G.C.; Barnholtz-Sloan, J.; Gould, M.P.; McMahon, S.; Cohen, A.; Adams, M.D.; Guda, K.; Cohen, M.; Sloan, A.E.; LaFramboise, T. Characterizing mutational heterogeneity in a glioblastoma patient with double recurrence. PLoS ONE 2013, 7, e35262. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Ciriello, G.; Miller, M.L.; Aksoy, B.A.; Senbabaoglu, Y.; Schultz, N.; Sander, C. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 2013, 45, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Liao, P.; Rouse, C.; Chen, Y.; Dowling, J.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro Oncol. 2014, 16 (Suppl 4), iv1–iv63. [Google Scholar] [CrossRef]
- Wood, M.D.; Halfpenny, A.M.; Moore, S.R. Applications of molecular neuro-oncology-a review of diffuse glioma integrated diagnosis and emerging molecular entities. Diagn. Pathol. 2019, 14, 29. [Google Scholar] [CrossRef]
- Nikiforova, M.N.; Hamilton, R.L. Molecular diagnostics of gliomas. Arch. Pathol. Lab. Med. 2011, 135, 558–568. [Google Scholar]
- Ducray, F.; El Hallani, S.; Idbaih, A. Diagnostic and prognostic markers in gliomas. Curr. Opin. Oncol. 2009, 21, 537–542. [Google Scholar] [CrossRef]
- Riemenschneider, M.J.; Jeuken, J.W.; Wesseling, P.; Reifenberger, G. Molecular diagnostics of gliomas: State of the art. Acta Neuropathol. 2010, 120, 567–584. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef]
- Houillier, C.; Wang, X.; Kaloshi, G.; Mokhtari, K.; Guillevin, R.; Laffaire, J.; Paris, S.; Boisselier, B.; Idbaih, A.; Laigle-Donadey, F.; et al. IDH1 or IDH2 mutations predict longer survival and response to temozolomide in low-grade gliomas. Neurology 2010, 75, 1560–1566. [Google Scholar] [CrossRef]
- Hemminki, K.; Li, X. Familial risks in nervous system tumors. Cancer Epidemiol. Biomark. Prev. 2003, 12, 1137–1142. [Google Scholar]
- Kinnersley, B.; Labussiere, M.; Holroyd, A.; Di Stefano, A.L.; Broderick, P.; Vijayakrishnan, J.; Mokhtari, K.; Delattre, J.Y.; Gousias, K.; Schramm, J.; et al. Genome-wide association study identifies multiple susceptibility loci for glioma. Nat. Commun. 2015, 6, 8559. [Google Scholar] [CrossRef] [PubMed]
- Melin, B.S.; Barnholtz-Sloan, J.S.; Wrensch, M.R.; Johansen, C.; Il’Yasova, D.; Kinnersley, B.; Ostrom, Q.T.; Labreche, K.; Chen, Y.; Armstrong, G.; et al. Genome-wide association study of glioma subtypes identifies specific differences in genetic susceptibility to glioblastoma and non-glioblastoma tumors. Nat. Genet. 2017, 49, 789. [Google Scholar] [CrossRef] [PubMed]
- Rajaraman, P.; Melin, B.S.; Wang, Z.; McKean-Cowdin, R.; Michaud, D.S.; Wang, S.S.; Bondy, M.; Houlston, R.; Jenkins, R.B.; Wrensch, M.; et al. Genome-wide association study of glioma and meta-analysis. Hum. Genet. 2012, 131, 1877–1888. [Google Scholar] [CrossRef] [PubMed]
- Sanson, M.; Hosking, F.J.; Shete, S.; Zelenika, D.; Dobbins, S.E.; Ma, Y.; Enciso-Mora, V.; Idbaih, A.; Delattre, J.Y.; Hoang-Xuan, K.; et al. Chromosome 7p11.2 (EGFR) variation influences glioma risk. Hum. Mol. Genet. 2011, 20, 2897–2904. [Google Scholar] [CrossRef]
- Shete, S.; Hosking, F.J.; Robertson, L.B.; Dobbins, S.E.; Sanson, M.; Malmer, B.; Simon, M.; Marie, Y.; Boisselier, B.; Delattre, J.Y.; et al. Genome-wide association study identifies five susceptibility loci for glioma. Nat. Genet. 2009, 41, 899–904. [Google Scholar] [CrossRef]
- Wrensch, M.; Jenkins, R.B.; Chang, J.S.; Yeh, R.F.; Xiao, Y.; Decker, P.A.; Ballman, K.V.; Berger, M.; Buckner, J.C.; Chang, S.; et al. Variants in the CDKN2B and RTEL1 regions are associated with high-grade glioma susceptibility. Nat. Genet. 2009, 41, 905–908. [Google Scholar] [CrossRef]
- Enciso-Mora, V.; Hosking, F.J.; Di Stefano, A.L.; Zelenika, D.; Shete, S.; Broderick, P.; Idbaih, A.; Delattre, J.Y.; Hoang-Xuan, K.; Marie, Y.; et al. Low penetrance susceptibility to glioma is caused by the TP53 variant rs78378222. Br. J. Cancer 2013, 108, 2178–21785. [Google Scholar] [CrossRef]
- Jenkins, R.B.; Xiao, Y.; Sicotte, H.; Decker, P.A.; Kollmeyer, T.M.; Hansen, H.M.; Kosel, M.L.; Zheng, S.; Walsh, K.M.; Rice, T.; et al. A low-frequency variant at 8q24.21 is strongly associated with risk of oligodendroglial tumors and astrocytomas with IDH1 or IDH2 mutation. Nat. Genet. 2012, 44, 1122–1125. [Google Scholar] [CrossRef]
- Enciso-Mora, V.; Hosking, F.J.; Kinnersley, B.; Wang, Y.; Shete, S.; Zelenika, D.; Broderick, P.; Idbaih, A.; Delattre, J.Y.; Hoang-Xuan, K.; et al. Deciphering the 8q24.21 association for glioma. Hum. Mol. Genet. 2013, 22, 2293–2302. [Google Scholar] [CrossRef]
- Walsh, K.M.; Codd, V.; Smirnov, I.V.; Rice, T.; Decker, P.A.; Hansen, H.M.; Kollmeyer, T.; Kosel, M.L.; Molinaro, A.M.; McCoy, L.S.; et al. Variants near TERT and TERC influencing telomere length are associated with high-grade glioma risk. Nat. Genet. 2014, 46, 731–735. [Google Scholar] [CrossRef] [PubMed]
- Malmer, B.; Iselius, L.; Holmberg, E.; Collins, A.; Henriksson, R.; Gronberg, H. Genetic epidemiology of glioma. Br. J. Cancer 2001, 84, 429–434. [Google Scholar] [CrossRef] [PubMed][Green Version]
- De Andrade, M.; Barnholtz, J.S.; Amos, C.I.; Adatto, P.; Spencer, C.; Bondy, M.L. Segregation analysis of cancer in families of glioma patients. Genet. Epidemiol. 2001, 20, 258–270. [Google Scholar] [CrossRef]
- Stacey, S.N.; Sulem, P.; Jonasdottir, A.; Masson, G.; Gudmundsson, J.; Gudbjartsson, D.F.; Magnusson, O.T.; Gudjonsson, S.A.; Sigurgeirsson, B.; Thorisdottir, K.; et al. A germline variant in the TP53 polyadenylation signal confers cancer susceptibility. Nat. Genet. 2011, 43, 1098–1103. [Google Scholar] [CrossRef]
- Schwartzbaum, J.A.; Xiao, Y.; Liu, Y.; Tsavachidis, S.; Berger, M.S.; Bondy, M.L.; Chang, J.S.; Chang, S.M.; Decker, P.A.; Ding, B.; et al. Inherited variation in immune genes and pathways and glioblastoma risk. Carcinogenesis 2010, 31, 1770–1777. [Google Scholar] [CrossRef]
- Andersson, U.; Schwartzbaum, J.; Wiklund, F.; Sjostrom, S.; Liu, Y.; Tsavachidis, S.; Ahlbom, A.; Auvinen, A.; Collatz-Laier, H.; Feychting, M.; et al. A comprehensive study of the association between the EGFR and ERBB2 genes and glioma risk. Acta Oncol. 2010, 49, 767–775. [Google Scholar] [CrossRef]
- Liu, Y.; Shete, S.; Hosking, F.J.; Robertson, L.B.; Bondy, M.L.; Houlston, R.S. New insights into susceptibility to glioma. Arch. Neurol. 2010, 67, 275–278. [Google Scholar] [CrossRef]
- Deng, L.; Xiong, P.; Luo, Y.; Bu, X.; Qian, S.; Zhong, W.; Lv, S. Association between IDH1/2 mutations and brain glioma grade. Oncol. Lett. 2018, 1, 5405–5409. [Google Scholar] [CrossRef]
- Molenaar, R.J.; Verbaan, D.; Lamba, S.; Zanon, C.; Jeuken, J.W.; Boots-Sprenger, S.H.; Wesseling, P.; Hulsebos, T.J.; Troost, D.; van Tilborg, A.A.; et al. The combination of IDH1 mutations and MGMT methylation status predicts survival in glioblastoma better than either IDH1 or MGMT alone. Neuro Oncol. 2014, 16, 1263–1273. [Google Scholar] [CrossRef]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Balss, J.; Meyer, J.; Mueller, W.; Korshunov, A.; Hartmann, C.; von Deimling, A. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 2008, 116, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, K.; Pearson, D.M.; Kocialkowski, S.; Bäcklund, L.M.; Chan, R.; Jones, D.T.; Collins, V.P. IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro Oncol. 2009, 11, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Horbinski, C. What do we know about IDH1/2 mutations so far, and how do we use it? Acta Neuropathol. 2013, 125, 621–636. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.P.; Wen, J.; Bang, S.; Park, S.; Song, S.Y. CD44-positive cells are responsible for gemcitabine resistance in pancreatic cancer cells. Int. J. Cancer 2009, 125, 2323–2331. [Google Scholar] [CrossRef] [PubMed]
- Hagmann, W.; Jesnowski, R.; Faissner, R.; Guo, C.; Lohr, J.M. ATPbinding cassette C transporters in human pancreatic carcinoma cell lines. Upregulation in 5-fluorouracil-resistant cells. Pancreatology 2009, 9, 136–144. [Google Scholar] [PubMed]
- Cui, Y.; Ko¨nig, J.; Buchholz, J.K.; Spring, H.; Leier, I.; Keppler, D. Drug resistance and ATP-dependent conjugate transport mediated by the apical multidrug resistance protein, MRP2, permanently expressed in human and canine cells. Mol. Pharmacol. 1999, 55, 929–937. [Google Scholar]
- Oguri, T.; Bessho, Y.; Achiwa, H.; Ozasa, H.; Maeno, K.; Maeda, H.; Sato, S.; Ueda, R. MRP8/ABCC11 directly confers resistance to 5-fluorouracil. Mol. Cancer Ther. 2007, 6, 122–127. [Google Scholar] [CrossRef]
- Pratt, S.; Shepard, R.L.; Kandasamy, R.A.; Johnston, P.A.; Perry, W.; Dantzig, A.H. The multidrug resistance protein 5 (ABCC5) confers resistance to 5-fluorouracil and transports its monophosphorylated metabolites. Mol. Cancer Ther. 2005, 4, 855–863. [Google Scholar] [CrossRef]
- Kieran, M.W.; Roberts, C.W.; Chi, S.N.; Ligon, K.L.; Rich, B.E.; Macconaill, L.E.; Garraway, L.A.; Biegel, J.A. Absence of oncogenic canonical pathway mutations in aggressive pediatric rhabdoid tumors. Pediatr. Blood Cancer 2012, 59, 1155–1157. [Google Scholar] [CrossRef]
- Jiao, Y.; Killela, P.J.; Reitman, Z.J.; Rasheed, A.B.; Heaphy, C.M.; de Wilde, R.F.; Rodriguez, F.J.; Rosemberg, S.; Oba-Shinjo, S.M.; Nagahashi Marie, S.K.; et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget 2012, 3, 709–722. [Google Scholar] [CrossRef]
- Kannan, K.; Inagaki, A.; Silber, J.; Gorovets, D.; Zhang, J.; Kastenhuber, E.R.; Heguy, A.; Petrini, J.H.; Chan, T.A.; Huse, J.T.; et al. Whole exome sequencing identified ATRX mutation as a key molecular determinant in lower-grade glioma. Oncotarget 2012, 3, 1194–1203. [Google Scholar] [CrossRef] [PubMed]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tönjes, M.; et al. Driver mutations in histone H3·3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Brat, D.J.; Verhaak, R.G.; Aldape, K.D.; Yung, W.K.; Salama, S.R.; Cooper, L.A.; Rheinbay, E.; Miller, C.R.; Vitucci, M.; et al.; Cancer Genome Atlas Research Network Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [Google Scholar] [PubMed]
- Clynes, D.; Jelinska, C.; Xella, B.; Ayyub, H.; Scott, C.; Mitson, M.; Taylor, S.; Higgs, D.R.; Gibbons, R.J. Suppression of the alternative lengthening of telomere pathway by the chromatin remodelling factor ATRX. Nat. Commun. 2015, 6, 753. [Google Scholar] [CrossRef]
- Heaphy, C.M.; de Wilde, R.F.; Jiao, Y.; Klein, A.P.; Edil, B.H.; Shi, C.; Bettegowda, C. Altered telomeres in tumors with ATRX and DAXX mutations. Science 2011, 333, 425. [Google Scholar] [CrossRef]
- Suzuki, H.; Aoki, K.; Chiba, K.; Sato, Y.; Shiozawa, Y.; Shiraishi, Y.; Shimamura, T.; Niida, A.; Motomura, K.; Ohka, F.; et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nat. Genet. 2015, 47, 458–468. [Google Scholar] [CrossRef]
- Liu, T.; Yuan, X.; Xu, D. Cancer-Specific Telomerase Reverse Transcriptase (TERT) Promoter Mutations: Biological and Clinical Implications. Genes 2016, 7, 3. [Google Scholar] [CrossRef]
- Killela, P.J.; Reitman, Z.J.; Jiao, Y.; Bettegowda, C.; Agrawal, N.; Diaz, L.A., Jr.; Friedman, A.H.; Friedman, H.; Gallia, G.L.; Giovanella, B.C.; et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. USA 2013, 110, 6021–6026. [Google Scholar] [CrossRef]
- Andersson, U.; Osterman, P.; Sjostrom, S.; Johansen, C.; Henriksson, R.; Brannstrom, T.; Broholm, H.; Christensen, H.C.; Ahlbom, A.; Auvinen, A.; et al. MNS16A minisatellite genotypes in relation to risk of glioma and meningioma and to glioblastoma outcome. Int. J. Cancer 2009, 125, 968–972. [Google Scholar] [CrossRef]
- Chiba, K.; Johnson, J.Z.; Vogan, J.M.; Wagner, T.; Boyle, J.M.; Hockemeyer, D. Cancer-associated TERT promoter mutations abrogate telomerase silencing. Elife 2015, 4, e07918. [Google Scholar] [CrossRef]
- Guo, C.; Pirozzi, C.J.; Lopez, G.Y.; Yan, H. Isocitrate dehydrogenase mutations in gliomas: Mechanisms, biomarkers and therapeutic target. Curr. Opin. Neurol. 2011, 24, 648–652. [Google Scholar] [CrossRef] [PubMed]
- Arteaga, C.L.; Engelman, J.A. ERBB receptors: From oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell 2014, 25, 282–303. [Google Scholar] [CrossRef] [PubMed]
- Furnari, F.B.; Cloughesy, T.F.; Cavenee, W.K.; Mischel, P.S. Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat. Rev. Cancer 2015, 15, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Sugawa, N.; Ekstrand, A.J.; James, C.D.; Collins, V.P. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc. Natl. Acad. Sci. USA 1990, 87, 8602–8606. [Google Scholar] [CrossRef] [PubMed]
- Eskilsson, E.; Rosland, G.V.; Talasila, K.M.; Knappskog, S.; Keunen, O.; Sottoriva, A.; Foerster, S.; Solecki, G.; Taxt, T.; Jirik, R.; et al. EGFRvIII mutations can emerge as late and heterogenous events in glioblastoma development and promote angiogenesis through Src activation. Neuro Oncol. 2016, 18, 1644–1655. [Google Scholar] [CrossRef]
- Crespo, I.; Vital, A.L.; Nieto, A.B.; Rebelo, O.; Tão, H.; Lopes, M.C.; Oliveira, C.R.; French, P.J.; Orfao, A.; Tabernero, M.D. Detailed Characterization of Alterations of Chromosomes 7, 9, and 10 in Glioblastomas as Assessed by Single-Nucleotide Polymorphism Arrays. J. Mol. Diagn. 2011, 13, 634–647. [Google Scholar] [CrossRef]
- Kanwal, R.; Gupta, S. Epigenetic modifications in cancer. Clin. Genet. 2012, 81, 303–311. [Google Scholar] [CrossRef]
- Delpu, Y.; Cordelier, P.; Cho, W.C.; Torrisani, J. DNA methylation and cancer diagnosis. Int. J. Mol. Sci. 2013, 14, 15029–15058. [Google Scholar] [CrossRef]
- Malmstrom, A.; Gronberg, B.H.; Marosi, C.; Stupp, R.; Frappaz, D.; Schultz, H.; Abacioglu, U.; Tavelin, B.; Lhermitte, B.; Hegi, M.E.; et al. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: The Nordic randomised, phase 3 trial. Lancet Oncol. 2012, 13, 916–926. [Google Scholar] [CrossRef]
- Perry, J.R.; Laperriere, N.; O’Callaghan, C.J.; Brandes, A.A.; Menten, J.; Phillips, C.; Fay, M.; Nishikawa, R.; Cairncross, J.G.; Roa, W.; et al. Short-course radiation plus temozolomide in elderly patients with glioblastoma. N. Engl. J. Med. 2017, 376, 1027–1037. [Google Scholar] [CrossRef] [PubMed]
- Christmann, M.; Kaina, B. Epigenetic regulation of DNA repair genes and implications for tumor therapy. Mutat. Res. 2019, 780, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Bouras, E.; Karakioulaki, M.; Bougioukas, K.I.; Aivaliotis, M.; Tzimagiorgis, G.; Chourdakis, M. Gene promoter methylation and cancer: An umbrella review. Gene 2019, 710, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Donson, A.M.; Addo-Yobo, S.O.; Handler, M.H.; Gore, L.; Foreman, N.K. MGMT promoter methylation correlates with survival benefit and sensitivity to temozolomide inpediatric glioblastoma. Pediatr. Blood Cancer 2007, 48, 403–407. [Google Scholar] [CrossRef]
- Herrlinger, U.; Schafer, N.; Steinbach, J.P.; Weyerbrock, A.; Hau, P.; Goldbrunner, R.; Friedrich, F.; Rohde, V.; Ringel, F.; Schlegel, U.; et al. Bevacizumab plus irinotecan versus temozolomide in newly diagnosed O6-methylguanine-DNA methyltransferase nonmethylated glioblastoma: The randomized GLARIUS trial. J. Clin. Oncol. 2016, 34, 1611–1619. [Google Scholar] [CrossRef]
- Minniti, G.; Scaringi, C.; Arcella, A.; Lanzetta, G.; Di Stefano, D.; Scarpino, S.; Bozzao, A.; Pace, A.; Villani, V.; Salvati, M.; et al. IDH1 mutation and MGMT methylation status predict survival in patients with anaplastic astrocytoma treated with temozolomide-based chemoradiotherapy. J. Neurooncol. 2014, 118, 377–383. [Google Scholar] [CrossRef]
- Baumert, B.G.; Hegi, M.E.; van den Bent, M.J.; von Deimling, A.; Gorlia, T.; Hoang-Xuan, K.; Brandes, A.A.; Kantor, G.; Taphoorn, M.J.B.; Hassel, M.B.; et al. Temozolomide chemotherapy versus radiotherapy in high-risk lowgrade glioma (EORTC 22033–26033): A randomised, open-label, phase 3 intergroup study. Lancet Oncol. 2016, 17, 1521–1532. [Google Scholar] [CrossRef]
- Bady, P.; Delorenzi, M.; Hegi, M.E. Sensitivity analysis of the MGMT-STP27 model and impact of genetic and epigenetic context to predict the MGMT methylation status in gliomas and other tumors. J. Mol. Diagn. 2016, 18, 350–361. [Google Scholar] [CrossRef]
- Vlassenbroeck, I.; Califice, S.; Diserens, A.C.; Migliavacca, E.; Straub, J.; Di Stefano, I.; Moreau, F.; Hamou, M.F.; Renard, I.; Delorenzi, M.; et al. Validation of real-time methylation-specific PCR to determine O6-methylguanine-DNA methyltransferase gene promoter methylation in glioma. J. Mol.Diagn. 2008, 10, 332–337. [Google Scholar] [CrossRef]
- Bady, P.; Diserens, A.C.; Castella, V.; Kalt, S.; Heinimann, K.; Hamou, M.F.; Delorenzi, M.; Hegi, M.E. DNA fingerprinting of glioma cell lines and considerations on similarity measurements. Neuro Oncol. 2012, 14, 701–711. [Google Scholar] [CrossRef]
- Van den Bent, J.; Erdem Eraslan, L.; Idbaih, A.; de Rooi, J.J.; Eilers, P.H.; Spliet, W.; den Dunnen, W.F.; Tijssen, C.; Wesseling, P.; Sillevis Smitt, P.A.; et al. MGMT-STP27 methylation status as predictive marker for response to PCVin anaplastic oligodendrogliomas and oligoastrocytomas. A report from EORTCstudy 26951. Clin. Cancer Res. 2013, 19, 5513–5522. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L.A. Mutator phenotype may be required for multistage carcinogenesis. Cancer Res. 1991, 51, 3075–3079. [Google Scholar] [PubMed]
- Bielas, J.H.; Loeb, K.R.; Rubin, B.P.; True, L.D.; Loeb, L.A. Human cancers express a mutator phenotype. Proc. Natl. Acad. Sci. USA 2006, 103, 18238–18242. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.J.; Cleveland, D.W. Boveri revisited: Chromosomal instability, aneuploidy and tumorigenesis. Nat. Rev. Mol. Cell Biol. 2009, 10, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Orr, B.; Godek, K.M.; Compton, D. Aneuploidy. Curr Biol. 2015, 25, R538–R542. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Endesfelder, D.; Rowan, A.J.; Walther, A.; Birkbak, N.J.; Futreal, A.P.; Downward, J.; Szallasi, Z.; Tomlinson, I.P.; Howell, M.; et al. Chromosomal instability confers intrinsic multidrug resistance. Cancer Res. 2011, 71, 1858–1870. [Google Scholar] [CrossRef] [PubMed]
- Woehrer, A.; Sander, P.; Haberler, C.; Kern, S.; Maier, H.; Preusser, M.; Hartmann, C.; Kros, J.M.; Hainfellner, J.A. FISH-based detection of 1p 19q codeletion in oligodendroglial tumors: Procedures and protocols for neuropathological practice—A publication under the auspices of the Research Committee of the European Confederation of Neuropathological Societies (Euro-CNS). Clin. Neuropathol. 2011, 30, 47–55. [Google Scholar] [CrossRef]
- Griffin, C.A.; Burger, P.; Morsberger, L.; Yonescu, R.; Swierczynski, S.; Weingart, J.D.; Murphy, K.M. Identification of der(1;19)(q10;p10) in five oligodendrogliomas suggests mechanism of concurrent 1p and 19q loss. J. Neuropathol. Exp. Neurol. 2006, 65, 988–994. [Google Scholar] [CrossRef]
- Jenkins, R.B.; Blair, H.; Ballman, K.V.; Giannini, C.; Arusell, R.M.; Law, M.; Flynn, H.; Passe, S.; Felten, S.; Brown, P.D.; et al. A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res. 2006, 66, 9852–9861. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Bauchet, L.; Davis, F.G.; Deltour, I.; Fisher, J.L.; Langer, C.E.; Pekmezci, M.; Schwartzbaum, J.A.; Turner, M.C.; Walsh, K.M.; et al. The epidemiology of glioma in adults: A “state of the science” review. Neuro Oncol. 2014, 16, 896–913. [Google Scholar] [CrossRef]
- Idbaih, A.; Marie, Y.; Pierron, G.; Brennetot, C.; Hoang-Xuan, K.; Kujas, M.; Mokhtari, K.; Sanson, M.; Lejeune, J.; Aurias, A.; et al. Two types of chromosome 1p losses with opposite significance in gliomas. Ann. Neurol. 2005, 58, 483–487. [Google Scholar] [CrossRef] [PubMed]
- Qu, M.; Olofsson, T.; Sigurdardottir, S.; You, C.; Kalimo, H.; Nistér, M.; Smits, A.; Ren, Z.P. Genetically distinct astrocytic and oligodendroglial components in oligoastrocytomas. Acta Neuropathol. 2007, 113, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Sim, J.; Nam, D.H.; Kim, Y.; Lee, I.H.; Choi, J.W.; Sa, J.K.; Suh, Y.L. Comparison of 1p and 19q status of glioblastoma by whole exome sequencing, array-comparative genomic hybridization, and fluorescence in situ hybridization. Med Oncol. 2018, 35, 60. [Google Scholar] [CrossRef] [PubMed]
- Labussiere, M.; Idbaih, A.; Wang, X.W.; Marie, Y.; Boisselier, B.; Falet, C.; Paris, S.; Laffaire, J.; Carpentier, C.; Crinière, E.; et al. All the 1p19q codeleted gliomas are mutated on IDH1 or IDH2. Neurology 2010, 74, 1886–1890. [Google Scholar] [CrossRef] [PubMed]
- Yip, S.; Butterfield, Y.S.; Morozova, O.; Chittaranjan, S.; Blough, M.D.; An, J.; Birol, I.; Chesnelong, C.; Chiu, R.; Chuah, E.; et al. Concurrent CIC mutations, IDH mutations, and 1p/19q loss distinguish oligodendrogliomas from other cancers. J. Pathol. 2012, 226, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Bettegowda, C.; Agrawal, N.; Jiao, Y.; Sausen, M.; Wood, L.D.; Hruban, R.H.; Rodriguez, F.J.; Cahill, D.P.; McLendon, R.; Riggins, G.; et al. Mutations in CIC and FUBP1 contribute to human oligodendroglioma. Science 2011, 333, 1453–1455. [Google Scholar] [CrossRef] [PubMed]
- Schaaf, C.P.; Wiszniewska, J.; Beaudet, A.L. Copy number and SNP arrays in clinical diagnostics. Annu. Rev. Genomics Hum. Genet. 2011, 12, 25–51. [Google Scholar] [CrossRef]
- Synhaeve, N.E.; van den Bent, M.J.; French, P.J.; Dinjens, W.N.M.; Atmodimedjo, P.N.; Kros, J.M.; Verdijk, R.; Dirven, C.M.F.; Dubbink, H.J. Clinical evaluation of a dedicated next generation sequencing panel for routine glioma diagnostics. Acta Neuropathol. Commun. 2018, 6, 126. [Google Scholar] [CrossRef]
- Nikiforova, M.N.; Wald, A.I.; Roy, S.; Durso, M.B.; Nikiforov, Y.E. Targeted next-generation sequenc ing panel (ThyroSeq) for detection of mutations in thyroid cancer. J. Clin. Endocrinol. Metab. 2013, 98, E1852–E1860. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI databas of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Ballester, L.Y.; Fuller, G.N.; Powell, S.Z.; Sulman, E.P.; Patel, K.P.; Luthra, R.; Routbort, M.J. Retrospective analysis of molecular and Immunohistochemical characterization of 381 primary brain tumors. J. Neuropathol. Exp. Neurol. 2017, 76, 179–188. [Google Scholar] [CrossRef]
- Zacher, A.; Kaulich, K.; Stepanow, S.; Wolter, M.; Köhrer, K.; Felsberg, J.; Malzkorn, B.; Reifenberger, G. Molecular Diagnostics of Gliomas Using Next Generation Sequencing of a Glioma-Tailored Gene Panel. Brain Pathol. 2017, 27, 146–159. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.A.; Zhang, H.; Roberts, P.S.; Jozwiak, S.; Wieslawa, G.; Lewin-Kowalik, J.; Kotulska, K.; Kwiatkowski, D.J. Pathogenesis of tuberous sclerosis-subependymal giant cell astrocytomas: Biallelic inactivation of TSC1 or TSC2 leads to mTOR activation. J. Neuropathol. Exp. Neurol. 2004, 63, 1236–1242. [Google Scholar] [CrossRef] [PubMed]
- Buczkowicz, P.; Hoeman, C.; Rakopoulos, P.; Pajovic, S.; Letourneau, L.; Dzamba, M.; Morrison, A.; Lewis, P.; Bouffet, E.; Bartels, U.; et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat. Genet. 2014, 46, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251–253. [Google Scholar] [PubMed]
- Virk, S.M.; Gibson, R.M.; Quinones-Mateu, M.E.; Barnholtz-Sloan, J.S. Identification of variants in primary and recurrent glioblastoma using a cancer-specific gene panel and whole exome sequencing. PLoS ONE 2015, 10, e0124178. [Google Scholar] [CrossRef]
- Crowley, E.; Di Nicolantonio, F.; Loupakis, F.; Bardelli, A. Liquid biopsy: Monitoring cancer-genetics in the blood. Nat. Rev. Clin. Oncol. 2013, 10, 472–484. [Google Scholar] [CrossRef]
- Skog, J.; Würdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Sena- Esteves, M.; Curry, W.T.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic bio- markers. Nat. Cell. Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Ulz, P.; Thallinger, G.G.; Auer, M.; Graf, R.; Kashofer, K.; Jahn, S.W.; Abete, L.; Pristauz, G.; Petru, E.; Geigl, J.B.; et al. Inferring expressed genes by whole-genome sequencing of plasma DNA. Nat. Genet. 2016, 48, 1273–1278. [Google Scholar] [CrossRef]
- El Messaoudi, S.; Rolet, F.; Mouliere, F.; Thierry, A.R. Circulating cell free DNA: Preanalytical considerations. Clin. Chim. Acta 2013, 424, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Klotzek, S.; Lewandowski, M.; Fleischhacker, M.; Jung, K. Changes in concentration of DNA in serum and plasma during storage of blood samples. Clin. Chem. 2003, 49, 1028–1029. [Google Scholar] [CrossRef] [PubMed]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [PubMed]
- Forshew, T.; Murtaza, M.; Parkinson, C.; Gale, D.; Tsui, D.W.Y.; Kaper, F.; Dawson, S.J.; Piskorz, A.M.; Jimenez-Linan, M.; Bentley, D.; et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci. Transl. Med. 2012, 4, 136ra68. [Google Scholar] [CrossRef]
- Merker, J.D.; Oxnard, G.R.; Compton, C.; Diehn, M.; Hurley, P.; Lazar, A.J.; Lindeman, N.; Lockwood, C.M.; Rai, A.J.; Schilsky, R.L.; et al. Circulating tumor DNA analysis in patients with cancer: American Society of Clinical Oncology and College of American Pathologists joint review. J. Clin. Oncol. 2014, 36, 1631–1641. [Google Scholar] [CrossRef] [PubMed]
- De Mattos-Arruda, L.; Mayor, R.; Ng, C.K.; Weigelt, B.; Martinez-Ricarte, F.; Torrejon, D.; Oliveira, M.; Arias, A.; Raventos, C.; Tang, J.; et al. Cerebrospinal fluid-derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nat. Commun. 2015, 6, 8839. [Google Scholar] [CrossRef]
- Wang, Y.; Springer, S.; Zhang, M.; McMahon, K.W.; Kinde, I.; Dobbyn, L.; Ptak, J.; Brem, H.; Chaichana, K.; Gallia, G.L.; et al. Detection of tumor-derived DNA in cerebrospinal fluid of patients with primary tumors of the brain and spinal cord. Proc. Natl. Acad. Sci. USA 2015, 112, 9704–9709. [Google Scholar] [CrossRef]
- Miller, A.M.; Shah, R.H.; Pentsova, E.I.; Pourmaleki, M.; Briggs, S.; Distefano, N.; Zheng, Y.; Skakodub, A.; Mehta, S.A.; Campos, C.; et al. Tracking tumour evolution in glioma through liquid biopsies of cerebrospinal fluid. Nature 2019, 565, 654–658. [Google Scholar] [CrossRef]
- Chen, W.W.; Balaj, L.; Liau, L.M.; Samuels, M.L.; Kotsopoulos, S.K.; Maguire, C.A.; Loguidice, L.; Soto, H.; Garrett, M.; Zhu, L.D.; et al. BEAMing and Droplet Digital PCR Analysis of Mutant IDH1 mRNA in Glioma Patient Serum and Cerebrospinal Fluid Extracellular Vesicles. Mol. Ther. Nucleic Acids 2013, 2, e109. [Google Scholar] [CrossRef]
- Liu, B.L.; Cheng, J.X.; Zhang, W.; Zhang, X.; Wang, R.; Lin, H.; Huo, J.L.; Cheng, H. Quantitative detection of multiple gene promoter hypermethylation in tumor tissue, serum, and cerebrospinal fluid predicts prognosis of malignant gliomas. Neuro Oncol. 2010, 12, 540–548. [Google Scholar] [CrossRef]
- Drusco, A.; Bottoni, A.; Lagana, A.; Acunzo, M.; Fassan, M.; Cascione, L.; Antenucci, A.; Kumchala, P.; Vicentini, C.; Gardiman, M.P.; et al. A differentially expressed set of microRNAs in cerebro-spinal fluid (CSF) can diagnose CNS malignancies. Oncotarget 2015, 6, 20829–20839. [Google Scholar] [CrossRef] [PubMed]
- Kopkova, A.; Sana, J.; Fadrus, P.; Slaby, O. Cerebrospinal fluid microRNAs as diagnostic biomarkers in brain tumors. Clin. Chem. Lab. Med. 2018, 56, 869–879. [Google Scholar] [CrossRef] [PubMed]
- Murtaza, M.; Dawson, S.J.; Tsui, D.W.Y.; Gale, D.; Forshew, T.; Piskorz, A.M.; Parkinson, C.; Chin, S.F.; Kingsbury, Z.; Wong, A.S.; et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013, 497, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Akers, J.C.; Hua, W.; Li, H.; Ramakrishnan, V.; Yang, Z.; Quan, K.; Zhu, W.; Li, J.; Figueroa, J.; Hirshman, B.R.; et al. A cerebrospinal fluid microRNA signature as biomarker for glioblastoma. Oncotarget 2017, 8, 68769–68779. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Huang, J.; Liu, X.; Cheng, Q.; Luo, C.; Liu, Z. CTLA-4 correlates with immune and clinical characteristics of glioma. Cancer Cell. Int. 2020, 20, 7. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Kurzrock, R. The Marriage Between Genomics and Immunotherapy: Mismatch Meets Its Match. Oncologist 2019, 24, 1–3. [Google Scholar] [CrossRef]
- Deng, X.; Lin, D.; Chen, B.; Zhang, X.; Xu, X.; Yang, Z.; Shen, X.; Yang, L.; Lu, X.; Sheng, H.; et al. Development and Validation of an IDH1-Associated Immune Prognostic Signature for Diffuse Lower-Grade Glioma. Front. Oncol. 2019, 9, 1310. [Google Scholar] [CrossRef]
- Hao, Z.; Guo, D. EGFR mutation: Novel prognostic factor associated with immune infiltration in lower-grade glioma; an exploratory study. BMC Cancer 2019, 19, 1184. [Google Scholar] [CrossRef]
- Chen, R.Q.; Liu, F.; Qiu, X.Y.; Chen, X.Q. The Prognostic and Therapeutic Value of PD-L1 in Glioma. Front. Pharmacol. 2019, 9, 1503. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arcella, A.; Limanaqi, F.; Ferese, R.; Biagioni, F.; Oliva, M.A.; Storto, M.; Fanelli, M.; Gambardella, S.; Fornai, F. Dissecting Molecular Features of Gliomas: Genetic Loci and Validated Biomarkers. Int. J. Mol. Sci. 2020, 21, 685. https://doi.org/10.3390/ijms21020685
Arcella A, Limanaqi F, Ferese R, Biagioni F, Oliva MA, Storto M, Fanelli M, Gambardella S, Fornai F. Dissecting Molecular Features of Gliomas: Genetic Loci and Validated Biomarkers. International Journal of Molecular Sciences. 2020; 21(2):685. https://doi.org/10.3390/ijms21020685
Chicago/Turabian StyleArcella, Antonietta, Fiona Limanaqi, Rosangela Ferese, Francesca Biagioni, Maria Antonietta Oliva, Marianna Storto, Mirco Fanelli, Stefano Gambardella, and Francesco Fornai. 2020. "Dissecting Molecular Features of Gliomas: Genetic Loci and Validated Biomarkers" International Journal of Molecular Sciences 21, no. 2: 685. https://doi.org/10.3390/ijms21020685
APA StyleArcella, A., Limanaqi, F., Ferese, R., Biagioni, F., Oliva, M. A., Storto, M., Fanelli, M., Gambardella, S., & Fornai, F. (2020). Dissecting Molecular Features of Gliomas: Genetic Loci and Validated Biomarkers. International Journal of Molecular Sciences, 21(2), 685. https://doi.org/10.3390/ijms21020685