Topical Therapy with Antisense Tumor Necrosis Factor Alpha Using Novel β-Glucan-Based Drug Delivery System Ameliorates Intestinal Inflammation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
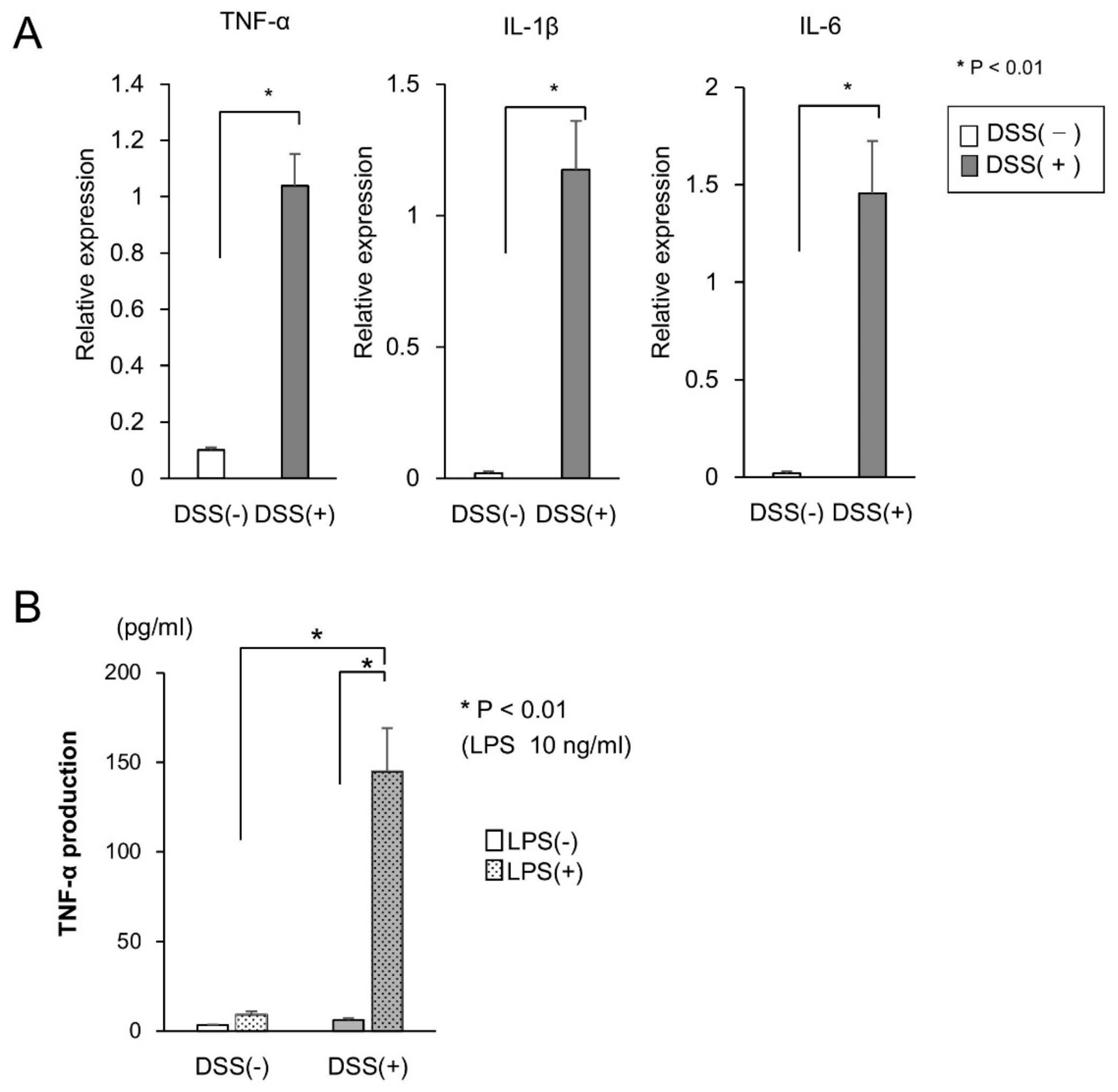
2.1. Cytokine Production in the Mucosa of DSS-Treated Mice
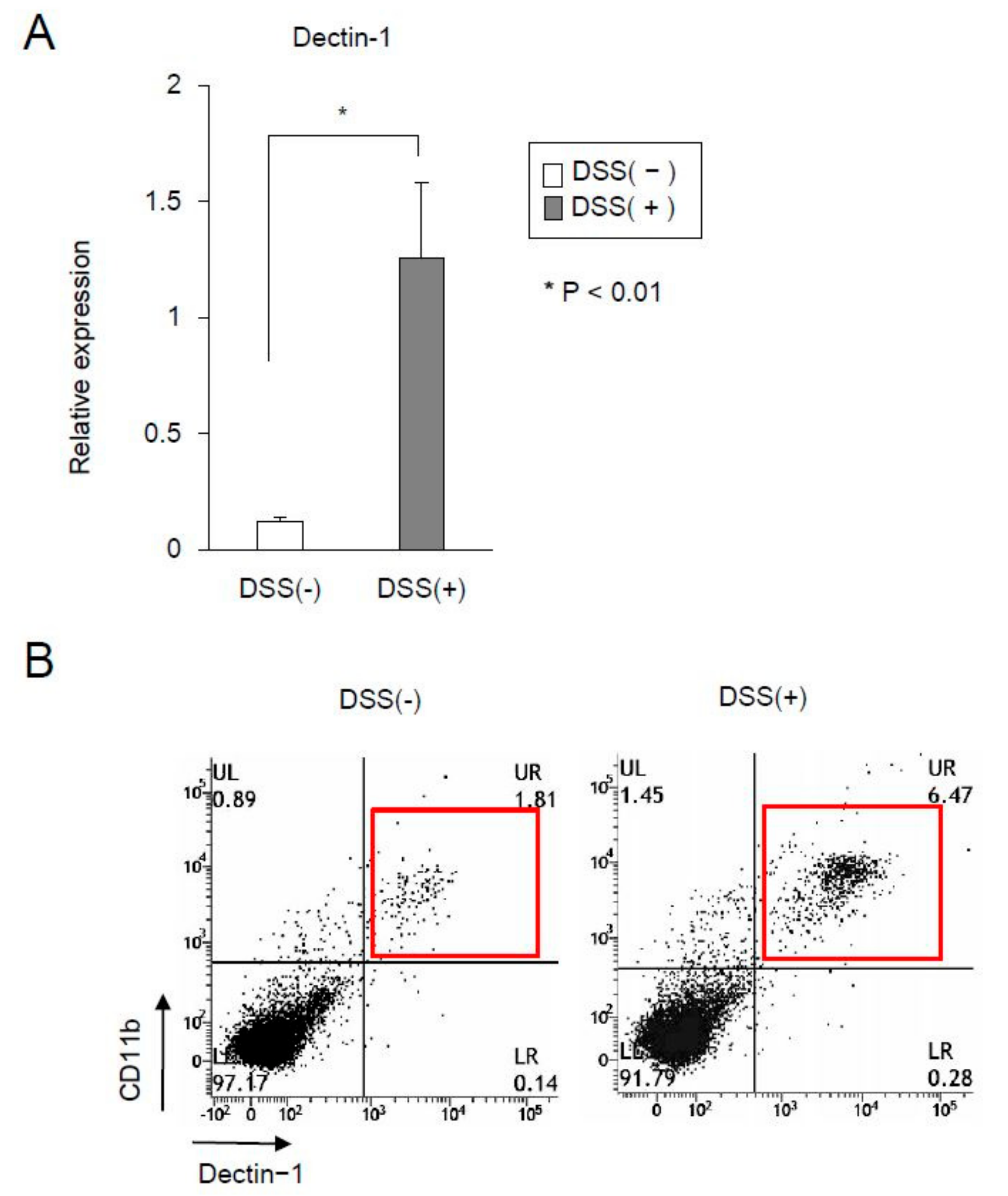
2.2. The Expression of Dectin-1 in CD11b+ Cells Significantly Increased in the Mucosa of DSS-Treated Mice
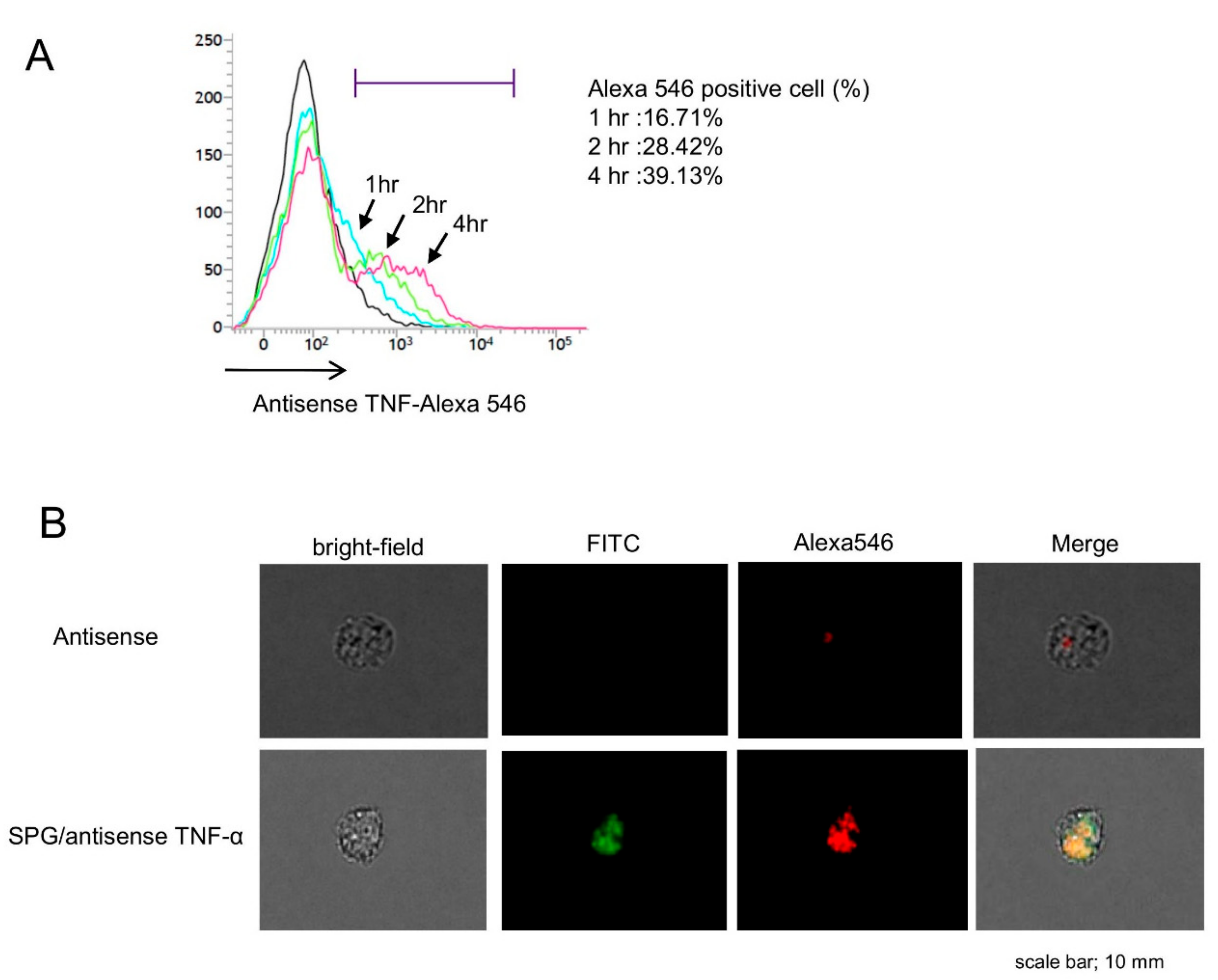
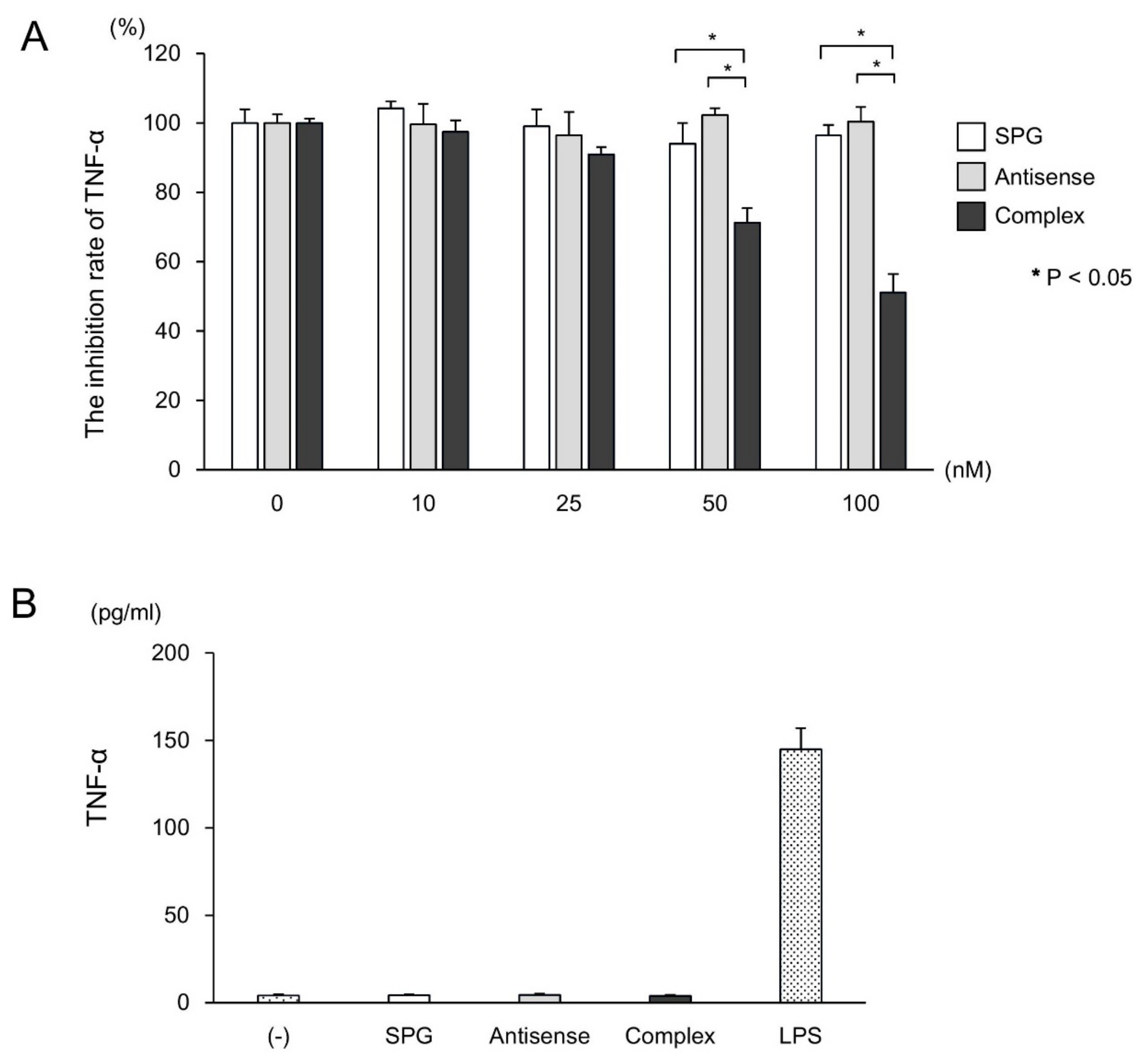
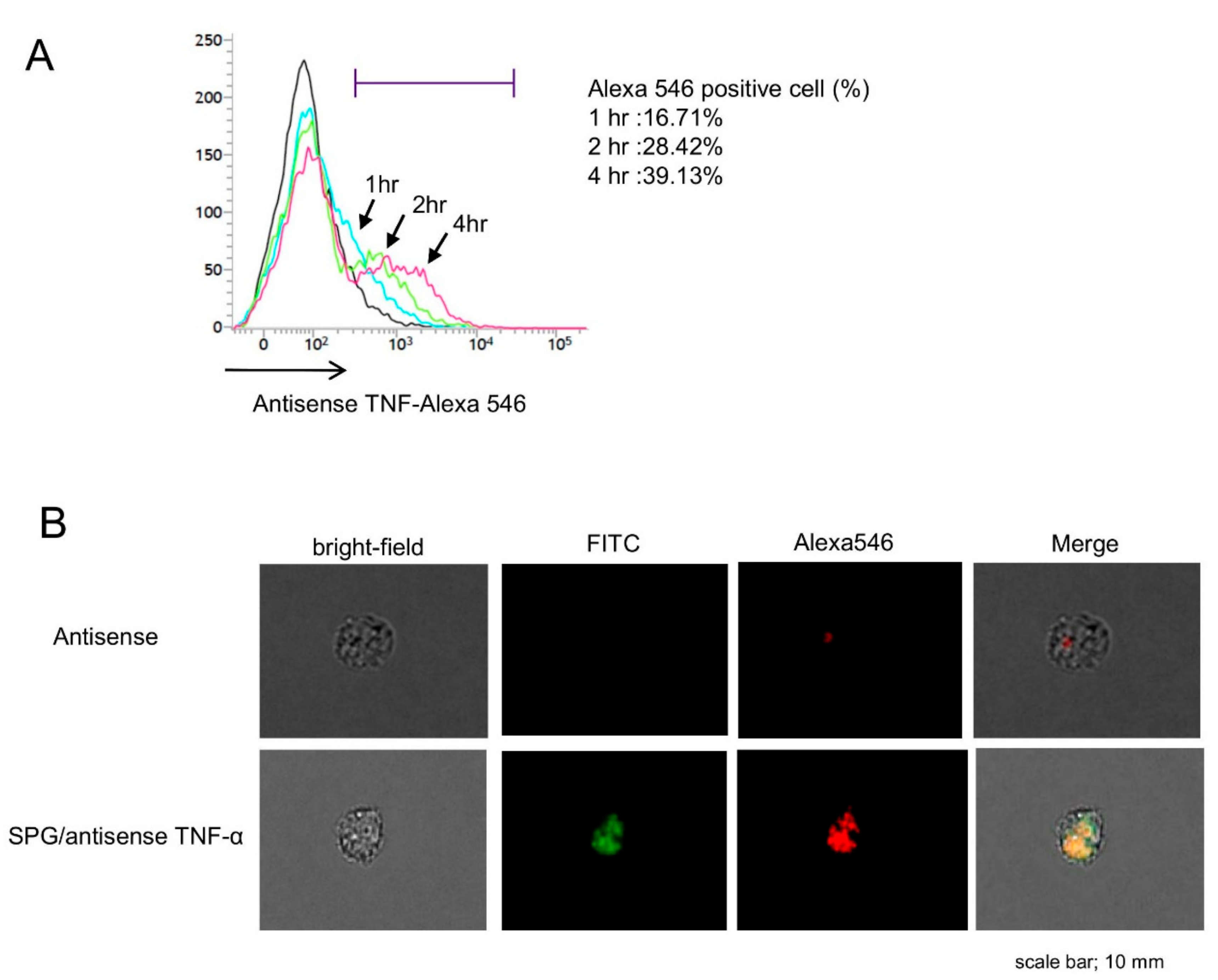
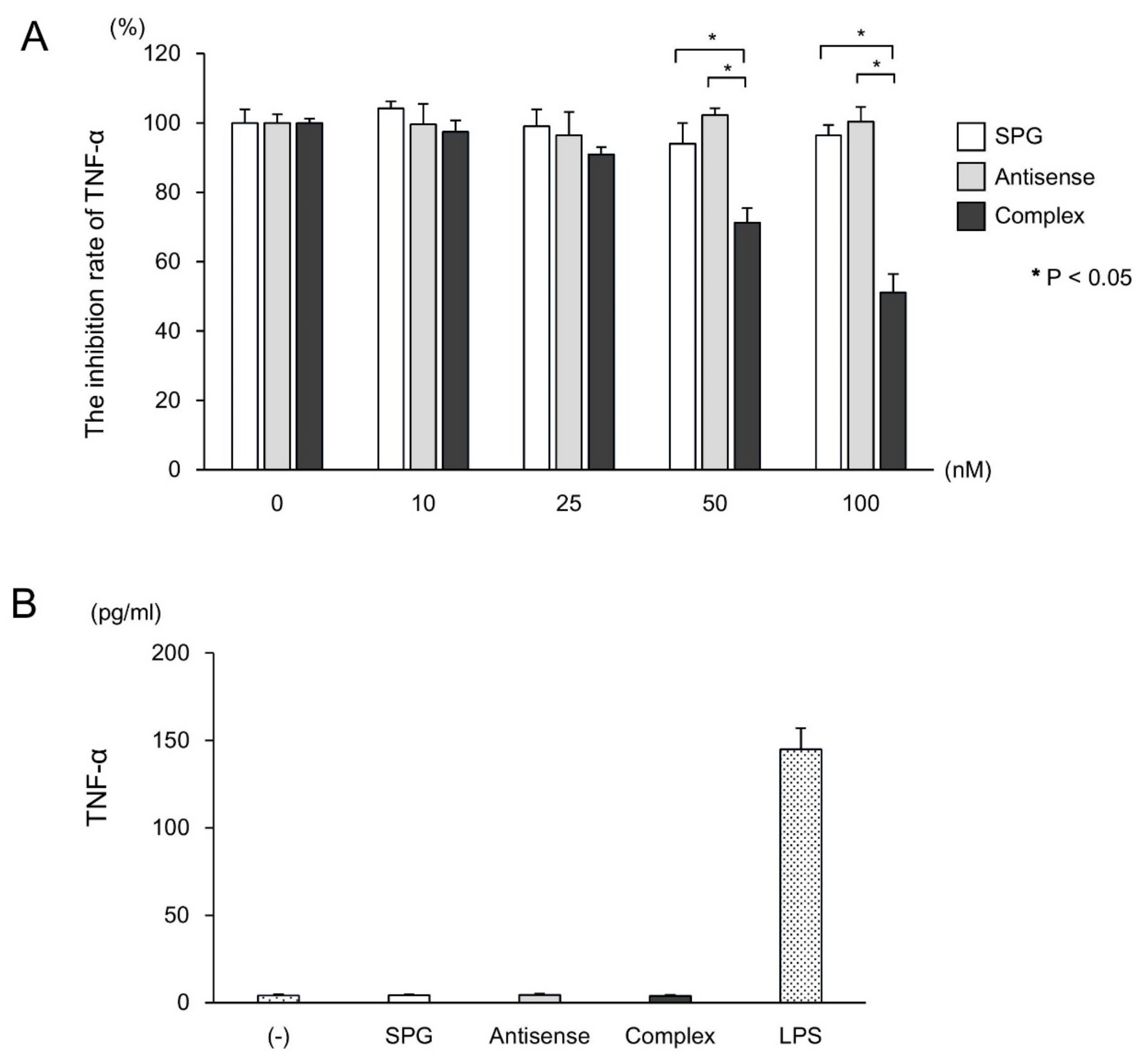
2.3. SPG-Antisense TNF-α Inhibited the Production of TNF-α in CD11b+ Cells
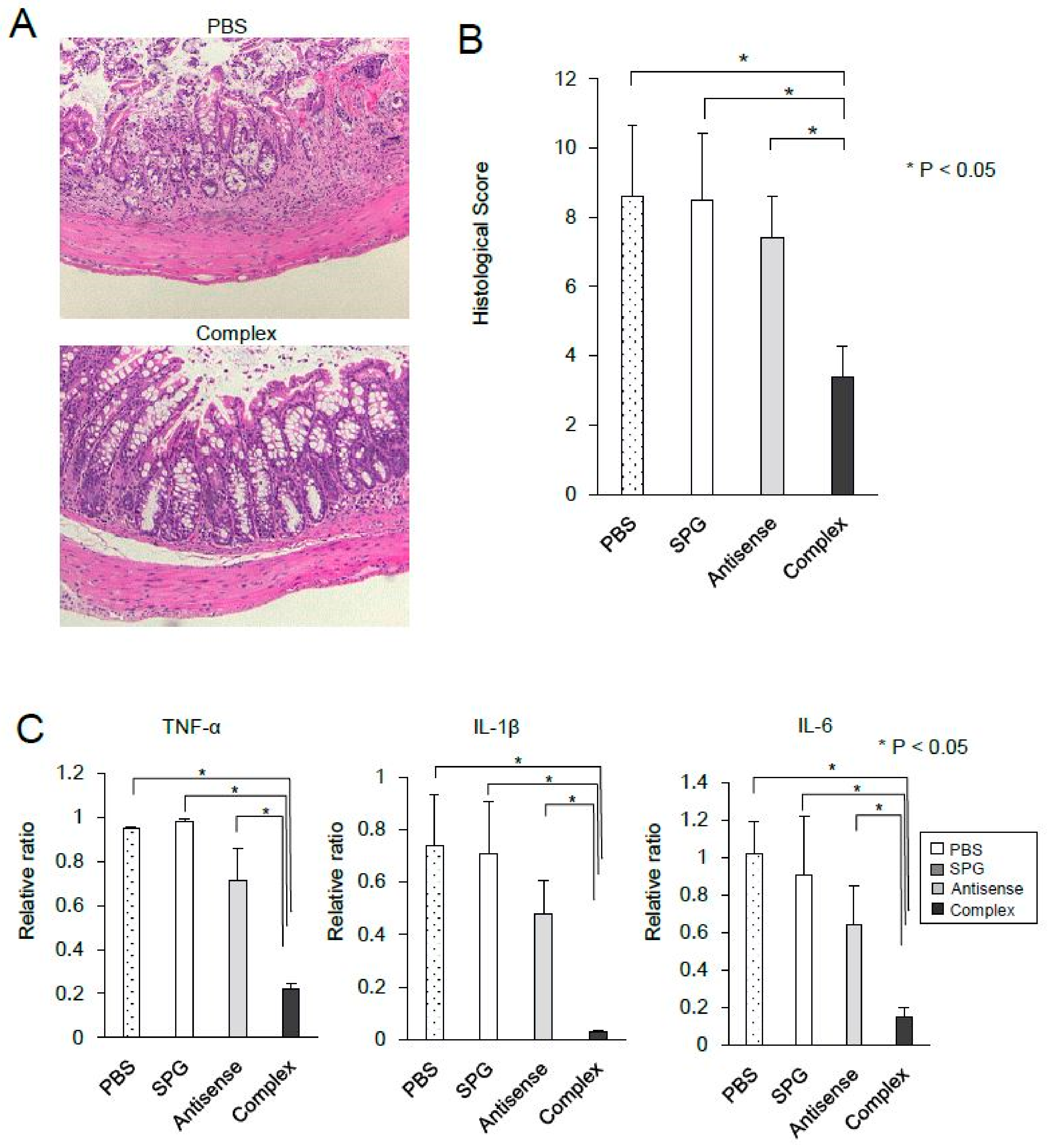
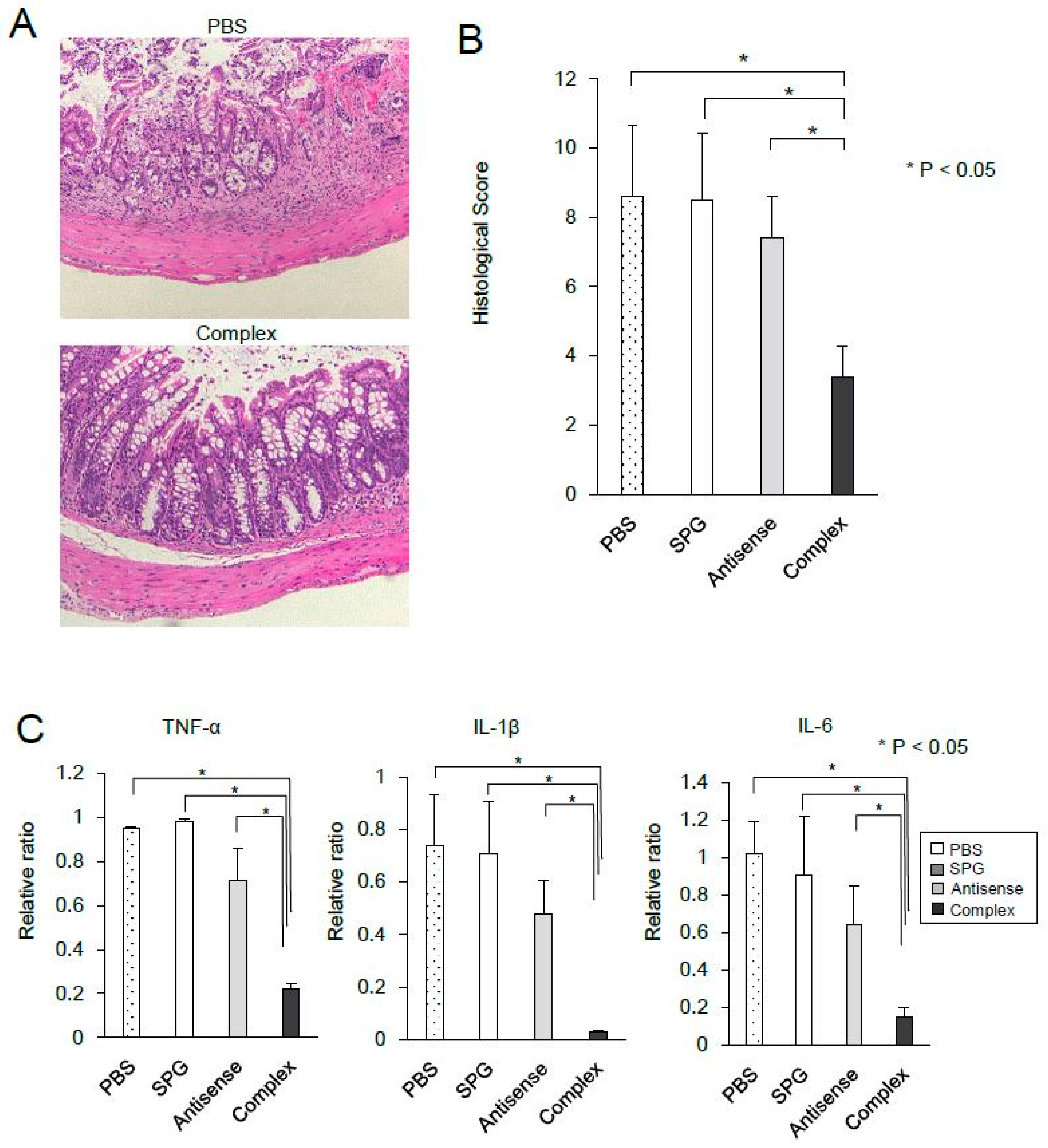
2.4. Administration of SPG-Antisense TNF-α Improved Colitis in DSS-Treated Mice
3. Discussion
4. Materials & Methods
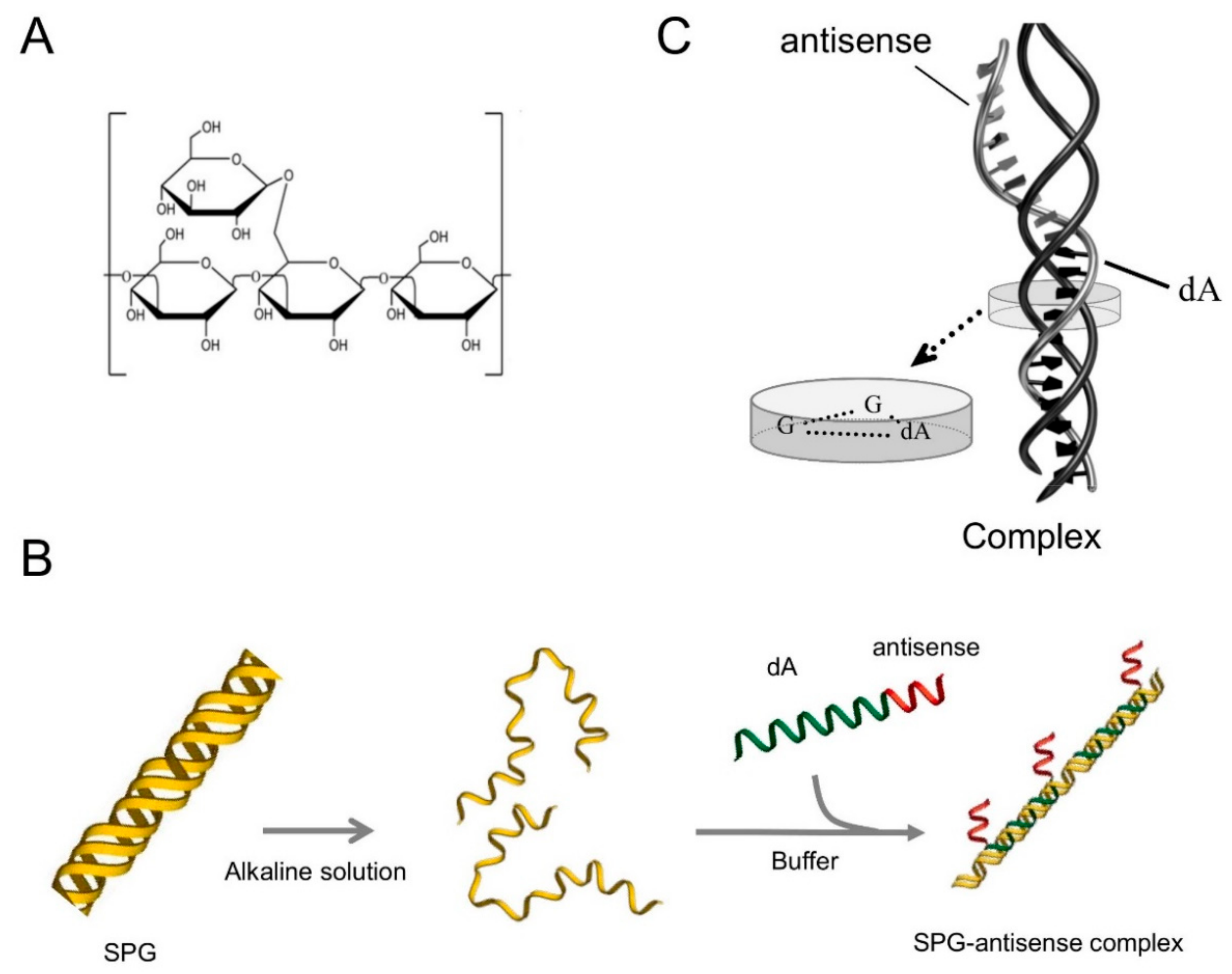
4.1. Preparation of SPG-Antisense TNF-α
4.2. Mice
4.3. DSS-Induced Colitis Model
4.4. Cell Culture and Cytokine Assay
4.5. Real-Time PCR
4.6. Flow Cytometric Analysis and Immunofluorescence
4.7. Treatment Using SPG-Antisense TNF-α
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Podolsky, D.K. Inflammatory bowel disease. N. Engl. J. Med. 2002, 347, 417–429. [Google Scholar]
- Xavier, R.J.; Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007, 448, 427–434. [Google Scholar]
- MacDonald, T.T.; Hutchings, P.; Choy, M.Y.; Murch, S.; Cooke, A. Tumour necrosis factor-alpha and interferon-gamma production measured at the single cell level in normal and inflamed human intestine. Clin. Exp. Immunol. 1990, 81, 301–305. [Google Scholar]
- Murch, S.H.; Braegger, C.P.; Walker-Smith, J.A.; MacDonald, T.T. Location of tumour necrosis factor alpha by immunohistochemistry in chronic inflammatory bowel disease. Gut 1993, 34, 1705–1709. [Google Scholar]
- Powrie, F.; Leach, M.W.; Mauze, S.; Menon, S.; Caddle, L.B.; Coffman, R.L. Inhibition of Th1 responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity 1994, 1, 553–562. [Google Scholar]
- Targan, S.R.; Hanauer, S.B.; van Deventer, S.J.; Mayer, L.; Present, D.H.; Braakman, T.; DeWoody, K.L.; Schaible, T.F.; Rutgeerts, P.J. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn’s disease. Crohn’s Disease cA2 Study Group. N. Engl. J. Med. 1997, 337, 1029–1035. [Google Scholar]
- Danese, S.; Vuitton, L.; Peyrin-Biroulet, L. Biologic agents for IBD: Practical insights. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 537–545. [Google Scholar]
- Sandborn, W.J.; Hanauer, S.B. Antitumor necrosis factor therapy for inflammatory bowel disease: A review of agents, pharmacology, clinical results, and safety. Inflamm. Bowel Dis. 1999, 5, 119–133. [Google Scholar]
- Myers, K.J.; Murthy, S.; Flanigan, A.; Witchell, D.R.; Butler, M.; Murray, S.; Siwkowski, A.; Goodfellow, D.; Madsen, K.; Baker, B. Antisense oligonucleotide blockade of tumor necrosis factor-alpha in two murine models of colitis. J. Pharmacol. Exp. Ther. 2003, 304, 411–424. [Google Scholar]
- Zuo, L.; Huang, Z.; Dong, L.; Xu, L.; Zhu, Y.; Zeng, K.; Zhang, C.; Chen, J.; Zhang, J. Targeting delivery of anti-TNFalpha oligonucleotide into activated colonic macrophages protects against experimental colitis. Gut 2010, 59, 470–479. [Google Scholar]
- Huang, Y.; Guo, J.; Gui, S. Orally targeted galactosylated chitosan poly(lactic-co-glycolic acid) nanoparticles loaded with TNF-a siRNA provide a novel strategy for the experimental treatment of ulcerative colitis. Eur. J. Pharm. Sci. 2018, 125, 232–243. [Google Scholar]
- Sakurai, K.; Mizu, M.; Shinkai, S. Polysaccharide--polynucleotide complexes. 2. Complementary polynucleotide mimic behavior of the natural polysaccharide schizophyllan in the macromolecular complex with single-stranded RNA and DNA. Biomacromolecules 2001, 2, 641–650. [Google Scholar]
- Noda, K.; Takeuchi, S.; Yajima, A.; Akiya, K.; Kasamatsu, T.; Tomoda, Y.; Ozawa, M.; Sekiba, K.; Sugimori, H.; Hashimoto, S.; et al. Clinical effect of sizofiran combined with irradiation in cervical cancer patients: A randomized controlled study. Cooperative Study Group on SPG for Gynecological Cancer. Jpn. J. Clin. Oncol. 1992, 22, 17–25. [Google Scholar]
- Minari, J.; Mochizuki, S.; Matsuzaki, T.; Adachi, Y.; Ohno, N.; Sakurai, K. Enhanced cytokine secretion from primary macrophages due to Dectin-1 mediated uptake of CpG DNA/beta-1,3-glucan complex. Bioconjug. Chem. 2011, 22, 9–15. [Google Scholar]
- Adachi, Y.; Ishii, T.; Ikeda, Y.; Hoshino, A.; Tamura, H.; Aketagawa, J.; Tanaka, S.; Ohno, N. Characterization of beta-glucan recognition site on C-type lectin, dectin 1. Infect. Immun. 2004, 72, 4159–4171. [Google Scholar]
- Herre, J.; Marshall, A.S.; Caron, E.; Edwards, A.D.; Williams, D.L.; Schweighoffer, E.; Tybulewicz, V.; Reis e Sousa, C.; Gordon, S.; Brown, G.D. Dectin-1 uses novel mechanisms for yeast phagocytosis in macrophages. Blood 2004, 104, 4038–4045. [Google Scholar]
- Ariizumi, K.; Shen, G.L.; Shikano, S.; Xu, S.; Ritter, R., 3rd; Kumamoto, T.; Edelbaum, D.; Morita, A.; Bergstresser, P.R.; Takashima, A. Identification of a novel, dendritic cell-associated molecule, dectin-1, by subtractive cDNA cloning. J. Biol. Chem. 2000, 275, 20157–20167. [Google Scholar]
- Holtmann, M.H.; Schutz, M.; Galle, P.R.; Neurath, M.F. Functional relevance of soluble TNF-alpha, transmembrane TNF-alpha and TNF-signal transduction in gastrointestinal diseases with special reference to inflammatory bowel diseases. Z. Gastroenterol. 2002, 40, 587–600. [Google Scholar]
- Breese, E.J.; Michie, C.A.; Nicholls, S.W.; Murch, S.H.; Williams, C.B.; Domizio, P.; Walker-Smith, J.A.; MacDonald, T.T. Tumor necrosis factor alpha-producing cells in the intestinal mucosa of children with inflammatory bowel disease. Gastroenterology 1994, 106, 1455–1466. [Google Scholar]
- Atreya, R.; Zimmer, M.; Bartsch, B.; Waldner, M.J.; Atreya, I.; Neumann, H.; Hildner, K.; Hoffman, A.; Kiesslich, R.; Rink, A.D.; et al. Antibodies against tumor necrosis factor (TNF) induce T-cell apoptosis in patients with inflammatory bowel diseases via TNF receptor 2 and intestinal CD14(+) macrophages. Gastroenterology 2011, 141, 2026–2038. [Google Scholar]
- Regueiro, M.; Siemanowski, B.; Kip, K.E.; Plevy, S. Infliximab dose intensification in Crohn’s disease. Inflamm. Bowel Dis. 2007, 13, 1093–1099. [Google Scholar]
- Gisbert, J.P.; Panes, J. Loss of response and requirement of infliximab dose intensification in Crohn’s disease: A review. Am. J. Gastroenterol. 2009, 104, 760–767. [Google Scholar]
- Rostholder, E.; Ahmed, A.; Cheifetz, A.S.; Moss, A.C. Outcomes after escalation of infliximab therapy in ambulatory patients with moderately active ulcerative colitis. Aliment. Pharmacol. Ther. 2012, 35, 562–567. [Google Scholar]
- Olesen, C.M.; Coskun, M.; Peyrin-Biroulet, L.; Nielsen, O.H. Mechanisms behind efficacy of tumor necrosis factor inhibitors in inflammatory bowel diseases. Pharmacol. Ther. 2016, 159, 110–119. [Google Scholar]
- Karmiris, K.; Paintaud, G.; Noman, M.; Magdelaine-Beuzelin, C.; Ferrante, M.; Degenne, D.; Claes, K.; Coopman, T.; Van Schuerbeek, N.; Van Assche, G.; et al. Influence of trough serum levels and immunogenicity on long-term outcome of adalimumab therapy in Crohn’s disease. Gastroenterology 2009, 137, 1628–1640. [Google Scholar]
- Waters, H.; Vanderpoel, J.; McKenzie, S.; Lunacsek, O.; Franklin, M.; Lennert, B.; Goff, J.; Augustyn, D.H. Stability of infliximab dosing in inflammatory bowel disease: Results from a multicenter US chart review. J. Med. Econ. 2011, 14, 397–402. [Google Scholar]
- Figdor, C.G.; van Kooyk, Y.; Adema, G.J. C-type lectin receptors on dendritic cells and Langerhans cells. Nat. Rev. Immunol. 2002, 2, 77–84. [Google Scholar]
- Brown, G.D.; Taylor, P.R.; Reid, D.M.; Willment, J.A.; Williams, D.L.; Martinez-Pomares, L.; Wong, S.Y.; Gordon, S. Dectin-1 is a major beta-glucan receptor on macrophages. J. Exp. Med. 2002, 196, 407–412. [Google Scholar]
- Taylor, P.R.; Tsoni, S.V.; Willment, J.A.; Dennehy, K.M.; Rosas, M.; Findon, H.; Haynes, K.; Steele, C.; Botto, M.; Gordon, S.; et al. Dectin-1 is required for beta-glucan recognition and control of fungal infection. Nat. Immunol. 2007, 8, 31–38. [Google Scholar]
- Saijo, S.; Fujikado, N.; Furuta, T.; Chung, S.H.; Kotaki, H.; Seki, K.; Sudo, K.; Akira, S.; Adachi, Y.; Ohno, N.; et al. Dectin-1 is required for host defense against Pneumocystis carinii but not against Candida albicans. Nat. Immunol. 2007, 8, 39–46. [Google Scholar]
- De Vries, H.S.; Plantinga, T.S.; van Krieken, J.H.; Stienstra, R.; van Bodegraven, A.A.; Festen, E.A.; Weersma, R.K.; Crusius, J.B.; Linskens, R.K.; Joosten, L.A.; et al. Genetic association analysis of the functional c.714T>G polymorphism and mucosal expression of dectin-1 in inflammatory bowel disease. PLoS ONE 2009, 4, e7818. [Google Scholar]
- Goodridge, H.S.; Wolf, A.J.; Underhill, D.M. Beta-glucan recognition by the innate immune system. Immunol. Rev. 2009, 230, 38–50. [Google Scholar]
- Heinsbroek, S.E.; Oei, A.; Roelofs, J.J.; Dhawan, S.; te Velde, A.; Gordon, S.; de Jonge, W.J. Genetic deletion of dectin-1 does not affect the course of murine experimental colitis. BMC Gastroenterol. 2012, 12, 33. [Google Scholar]
- Monteleone, G.; Neurath, M.F.; Ardizzone, S.; Di Sabatino, A.; Fantini, M.C.; Castiglione, F.; Scribano, M.L.; Armuzzi, A.; Caprioli, F.; Sturniolo, G.C.; et al. Mongersen, an oral SMAD7 antisense oligonucleotide, and Crohn’s disease. N. Engl. J. Med. 2015, 372, 1104–1113. [Google Scholar]
- Vainer, B.; Nielsen, O.H. Changed colonic profile of P-selectin, platelet-endothelial cell adhesion molecule-1 (PECAM-1), intercellular adhesion molecule-1 (ICAM-1), ICAM-2, and ICAM-3 in inflammatory bowel disease. Clin. Exp. Immunol. 2000, 121, 242–247. [Google Scholar]
- Rivera-Nieves, J.; Gorfu, G.; Ley, K. Leukocyte adhesion molecules in animal models of inflammatory bowel disease. Inflamm. Bowel Dis. 2008, 14, 1715–1735. [Google Scholar]
- Gewirtz, A.T.; Sitaraman, S. Alicaforsen. Isis Pharmaceuticals. Curr. Opin. Investig. Drugs 2001, 2, 1401–1406. [Google Scholar]
- van Deventer, S.J.; Tami, J.A.; Wedel, M.K. A randomised, controlled, double blind, escalating dose study of alicaforsen enema in active ulcerative colitis. Gut 2004, 53, 1646–1651. [Google Scholar]
- Sanchez-Munoz, F.; Fonseca-Camarillo, G.; Villeda-Ramirez, M.A.; Miranda-Perez, E.; Mendivil, E.J.; Barreto-Zuniga, R.; Uribe, M.; Bojalil, R.; Dominguez-Lopez, A.; Yamamoto-Furusho, J.K. Transcript levels of Toll-Like Receptors 5, 8 and 9 correlate with inflammatory activity in Ulcerative Colitis. BMC Gastroenterol. 2011, 11, 138. [Google Scholar]
- Takedatsu, H.; Mitsuyama, K.; Mochizuki, S.; Kobayashi, T.; Sakurai, K.; Takeda, H.; Fujiyama, Y.; Koyama, Y.; Nishihira, J.; Sata, M. A new therapeutic approach using a schizophyllan-based drug delivery system for inflammatory bowel disease. Mol. Ther. 2012, 20, 1234–1241. [Google Scholar]
- Becker, C.; Fantini, M.C.; Wirtz, S.; Nikolaev, A.; Kiesslich, R.; Lehr, H.A.; Galle, P.R.; Neurath, M.F. In vivo imaging of colitis and colon cancer development in mice using high resolution chromoendoscopy. Gut 2005, 54, 950–954. [Google Scholar]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sakisaka, H.; Takedatsu, H.; Mitsuyama, K.; Mochizuki, S.; Sakurai, K.; Sakisaka, S.; Hirai, F. Topical Therapy with Antisense Tumor Necrosis Factor Alpha Using Novel β-Glucan-Based Drug Delivery System Ameliorates Intestinal Inflammation. Int. J. Mol. Sci. 2020, 21, 683. https://doi.org/10.3390/ijms21020683
Sakisaka H, Takedatsu H, Mitsuyama K, Mochizuki S, Sakurai K, Sakisaka S, Hirai F. Topical Therapy with Antisense Tumor Necrosis Factor Alpha Using Novel β-Glucan-Based Drug Delivery System Ameliorates Intestinal Inflammation. International Journal of Molecular Sciences. 2020; 21(2):683. https://doi.org/10.3390/ijms21020683
Chicago/Turabian StyleSakisaka, Hideto, Hidetoshi Takedatsu, Keiichi Mitsuyama, Shinichi Mochizuki, Kazuo Sakurai, Shotaro Sakisaka, and Fumihito Hirai. 2020. "Topical Therapy with Antisense Tumor Necrosis Factor Alpha Using Novel β-Glucan-Based Drug Delivery System Ameliorates Intestinal Inflammation" International Journal of Molecular Sciences 21, no. 2: 683. https://doi.org/10.3390/ijms21020683
APA StyleSakisaka, H., Takedatsu, H., Mitsuyama, K., Mochizuki, S., Sakurai, K., Sakisaka, S., & Hirai, F. (2020). Topical Therapy with Antisense Tumor Necrosis Factor Alpha Using Novel β-Glucan-Based Drug Delivery System Ameliorates Intestinal Inflammation. International Journal of Molecular Sciences, 21(2), 683. https://doi.org/10.3390/ijms21020683