Multiple Cell Signalling Pathways of Human Proinsulin C-Peptide in Vasculopathy Protection

 , ,
, ,  ,
, 
 ,
,  and
and
Abstract
1. Introduction
2. Functions and Features of Human Proinsulin C-Peptide
3. Cell Signalling Pathways of Human Proinsulin C-Peptide in Vasculopathy Protection
4. Development of Innovative and Effective Therapeutic Options in Diabetic Neuropathy
5. Development of Innovative and Effective Therapeutic Options in Diabetic Nephropathy
6. Conclusions
Funding
Conflicts of Interest
Abbreviations
| AGEs | Advanced glycation end-products |
| AMPKa | AMP-activated protein kinase a |
| Ang | Angiotensin |
| Ang II | Angiotensin II |
| CRP | C-reactive protein |
| DM | Diabetes mellitus |
| ER | Endoplasmic reticulum |
| Erk-1/-2 | Extracellular signal–regulated kinase -1/-2 |
| HGM | Hyperglycemic memory |
| HUVECs | Human umbilical vein endothelial cells |
| IL-10 | Interleukin-10 |
| IL-6 | Interleukin-6 |
| iNOS | Inducible nitric oxide synthase |
| L-NAME | NG-nitro-L-arginine methyl ester |
| MAPK | Mitogen-activated protein kinases |
| NCV | Nerve conduction velocity |
| NF-jβ | Translocation of nuclear factor jβ |
| NF-κβ | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NO | Nitric oxide |
| NOS | Nitric oxide synthase |
| ONOO− | Peroxynitrite |
| oxLDL | Oxidized low-density lipoprotein |
| PI | Proinsulin |
| PI:C | PI-to-serum C-peptide |
| PKC | Protein kinase C |
| ROS | Reactive oxygen species |
| Saos-2 | Human Osteoblast-like cells |
| STZ | Streptozotocin |
| T1DM | Type 1 diabetes mellitus |
| T2DM | Type 2 diabetes mellitus |
| TNFα | Tumor necrosis factor alpha |
| VEGF | Vascular endothelial growth factor |
References
- Vieira, R.; Souto, S.B.; Sanchez-Lopez, E.; Machado, A.L.; Severino, P.; Jose, S.; Santini, A.; Silva, A.M.; Fortuna, A.; Garcia, M.L.; et al. Sugar-Lowering Drugs for Type 2 Diabetes Mellitus and Metabolic Syndrome-Strategies for In Vivo Administration: Part-II. J. Clin. Med. 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Vieira, R.; Souto, S.B.; Sanchez-Lopez, E.; Machado, A.L.; Severino, P.; Jose, S.; Santini, A.; Fortuna, A.; Garcia, M.L.; Silva, A.M.; et al. Sugar-Lowering Drugs for Type 2 Diabetes Mellitus and Metabolic Syndrome-Review of Classical and New Compounds: Part-I. Pharmaceuticals (Basel) 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Souto, E.B.; Souto, S.B.; Campos, J.R.; Severino, P.; Pashirova, T.N.; Zakharova, L.Y.; Silva, A.M.; Durazzo, A.; Lucarini, M.; Izzo, A.A.; et al. Nanoparticle Delivery Systems in the Treatment of Diabetes Complications. Molecules 2019, 24. [Google Scholar] [CrossRef]
- Alaa El Din, R.; Ibrahim, H.M.; Ali, A.H. C-peptide Attenuates Progression of Atherosclerosis in Late Stages of Type 2 Diabetes in Male Albino Rats. Cardiol. Angiol. Int. J. 2016, 1–9. [Google Scholar] [CrossRef]
- Kitamura, T.; Kimura, K.; Jung, B.D.; Makondo, K.; Sakane, N.; Yoshida, T.; Saito, M. Proinsulin C-peptide activates cAMP response element-binding proteins through the p38 mitogen-activated protein kinase pathway in mouse lung capillary endothelial cells. Biochem. J. 2002, 366, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Wallerath, T.; Kunt, T.; Forst, T.; Closs, E.I.; Lehmann, R.; Flohr, T.; Gabriel, M.; Schäfer, D.; Göpfert, A.; Pfützner, A. Stimulation of endothelial nitric oxide synthase by proinsulin C-peptide. Nitric Oxide 2003, 9, 95–102. [Google Scholar] [CrossRef]
- Zhong, Z.; Davidescu, A.; Ehren, I.; Ekberg, K.; Jörnvall, H.; Wahren, J.; Chibalin, A. C-peptide stimulates ERK1/2 and JNK MAP kinases via activation of protein kinase C in human renal tubular cells. Diabetologia 2005, 48, 187–197. [Google Scholar] [CrossRef]
- Van Sloten, T.T.; Henry, R.M.; Dekker, J.M.; Nijpels, G.; Unger, T.; Schram, M.T.; Stehouwer, C.D. Endothelial dysfunction plays a key role in increasing cardiovascular risk in type 2 diabetes: The Hoorn study. Hypertension 2014. [Google Scholar] [CrossRef]
- Smith-Palmer, J.; Brändle, M.; Trevisan, R.; Federici, M.O.; Liabat, S.; Valentine, W. Assessment of the association between glycemic variability and diabetes-related complications in type 1 and type 2 diabetes. Diabetes Res. Clin. Pract. 2014, 105, 273–284. [Google Scholar] [CrossRef]
- Huang, E.S.; Laiteerapong, N.; Liu, J.Y.; John, P.M.; Moffet, H.H.; Karter, A.J. Rates of complications and mortality in older patients with diabetes mellitus: The diabetes and aging study. JAMA Intern. Med. 2014, 174, 251–258. [Google Scholar] [CrossRef]
- Durazzo, A.; Lucarini, M.; Novellino, E.; Souto, E.B.; Daliu, P.; Santini, A. Abelmoschus esculentus (L.): Bioactive Components’ Beneficial Properties-Focused on Antidiabetic Role-For Sustainable Health Applications. Molecules 2018, 24, 38. [Google Scholar] [CrossRef] [PubMed]
- Daliu, P.; Santini, A.; Novellino, E. A decade of nutraceutical patents: Where are we now in 2018? Expert Opin. Ther. Pat. 2018, 28, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Daliu, P.; Santini, A.; Novellino, E. From pharmaceuticals to nutraceuticals: Bridging disease prevention and management. Expert Rev. Clin. Pharmacol. 2019, 12, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Campos, J.R.; Severino, P.; Ferreira, C.S.; Zielinska, A.; Santini, A.; Souto, S.B.; Souto, E.B. Linseed Essential Oil-Source of Lipids as Active Ingredients for Pharmaceuticals and Nutraceuticals. Curr. Med. Chem. 2019, 26, 4537–4558. [Google Scholar] [CrossRef]
- Abenavoli, L.; Izzo, A.A.; Milic, N.; Cicala, C.; Santini, A.; Capasso, R. Milk thistle (Silybum marianum): A concise overview on its chemistry, pharmacological, and nutraceutical uses in liver diseases. Phytother. Res. 2018, 32, 2202–2213. [Google Scholar] [CrossRef] [PubMed]
- Santini, A.; Novellino, E. Nutraceuticals-shedding light on the grey area between pharmaceuticals and food. Expert Rev. Clin. Pharmacol. 2018, 11, 545–547. [Google Scholar] [CrossRef]
- Santini, A.; Novellino, E. Nutraceuticals in hypercholesterolaemia: An overview. Br. J. Pharmacol. 2017, 174, 1450–1463. [Google Scholar] [CrossRef]
- Santini, A.; Tenore, G.C.; Novellino, E. Nutraceuticals: A paradigm of proactive medicine. Eur. J. Pharm. Sci. 2017, 96, 53–61. [Google Scholar] [CrossRef]
- Santini, A.; Cammarata, S.M.; Capone, G.; Ianaro, A.; Tenore, G.C.; Pani, L.; Novellino, E. Nutraceuticals: Opening the debate for a regulatory framework. Br. J. Clin. Pharmacol. 2018, 84, 659–672. [Google Scholar] [CrossRef]
- Durazzo, A.; Lucarini, M.; Souto, E.B.; Cicala, C.; Caiazzo, E.; Izzo, A.A.; Novellino, E.; Santini, A. Polyphenols: A concise overview on the chemistry, occurrence, and human health. Phytother. Res. 2019, 33, 2221–2243. [Google Scholar] [CrossRef]
- Santini, A.; Novellino, E. To Nutraceuticals and Back: Rethinking a Concept. Foods 2017, 6. [Google Scholar] [CrossRef]
- Santini, A.; Novellino, E. Nutraceuticals: Beyond the diet before the drugs. Curr. Bioact. Comp. 2014, 10, 1–12. [Google Scholar] [CrossRef]
- Durazzo, A.; D’Addezio, L.; Camilli, E.; Piccinelli, R.; Turrini, A.; Marletta, L.; Marconi, S.; Lucarini, M.; Lisciani, S.; Gabrielli, P.; et al. From Plant Compounds to Botanicals and Back: A Current Snapshot. Molecules 2018, 23. [Google Scholar] [CrossRef] [PubMed]
- Durazzo, A. Extractable and Non-extractable polyphenols: An overview. In Non-Extractable Polyphenols and Carotenoids: Importance in Human Nutrition and Health; Saura-Calixto, F., Pérez-Jiménez, J., Eds.; Royal Society of Chemistry: London, UK, 2018; pp. 1–37. [Google Scholar]
- Daliu, P.; Annunziata, G.; Tenore, G.C.; Santini, A. Abscisic acid identification in Okra, Abelmoschus esculentus L. (Moench): Perspective nutraceutical use for the treatment of diabetes. Nat. Prod. Res. 2020, 34, 3–9. [Google Scholar] [CrossRef]
- Durazzo, A.; Lucarini, M. Extractable and Non-Extractable Antioxidants. Molecules 2019, 24. [Google Scholar] [CrossRef] [PubMed]
- Salehi, B.; Ata, A.; Anil Kumar, V.N.; Sharopov, F.; Ramírez-Alarcón, K.; Ruiz-Ortega, A.; Abdulmajid Ayatollahi, S.; Tsouh Fokou, P.V.; Kobarfard, F.; Amiruddin Zakaria, Z.; et al. Antidiabetic potential of medicinal plants and their active components. Biomolecules 2019, 9. [Google Scholar] [CrossRef]
- Lucarini, M.; Durazzo, A.; Kiefer, J.; Santini, A.; Lombardi-Boccia, G.; Souto, E.B.; Romani, A.; Lampe, A.; Ferrari Nicoli, S.; Gabrielli, P.; et al. Grape Seeds: Chromatographic Profile of Fatty Acids and Phenolic Compounds and Qualitative Analysis by FTIR-ATR Spectroscopy. Foods 2020, 9, 10. [Google Scholar] [CrossRef]
- Yeung, A.W.K.; Tzvetkov, N.T.; Durazzo, A.; Lucarini, M.; Souto, E.B.; Santini, A.; Gan, R.Y.; Jozwik, A.; Grzybek, W.; Echeverría, J.; et al. Natural products in diabetes research: Quantitative literature analysis. Curr. Med. Chem. 2020. under review. [Google Scholar]
- Ding, H.; Triggle, C.R. Endothelial cell dysfunction and the vascular complications associated with type 2 diabetes: Assessing the health of the endothelium. Vasc. Health Risk Manag. 2005, 1, 55. [Google Scholar] [CrossRef]
- Bakker, W.; Eringa, E.C.; Sipkema, P.; van Hinsbergh, V.W. Endothelial dysfunction and diabetes: Roles of hyperglycemia, impaired insulin signaling and obesity. Cell Tissue Res. 2009, 335, 165. [Google Scholar] [CrossRef]
- Janowska, J.; Chudek, J.; Olszanecka-Glinianowicz, M.; Semik-Grabarczyk, E.; Zahorska-Markiewicz, B. Interdependencies among selected pro-inflammatory markers of endothelial dysfunction, C-peptide, anti-inflammatory interleukin-10 and glucose metabolism disturbance in obese women. Int. J. Med. Sci. 2016, 13, 490. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, M.P.; Lee, Y.-J.; Jung, S.-H.; Kim, Y.H.; Hwang, J.Y.; Han, E.-T.; Park, W.S.; Hong, S.-H.; Kim, Y.-M.; Ha, K.-S. C-peptide protects against hyperglycemic memory and vascular endothelial cell apoptosis. J. Endocrinol. 2016, 231, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Escudero, C.A.; Herlitz, K.; Troncoso, F.; Guevara, K.; Acurio, J.; Aguayo, C.; Godoy, A.S.; González, M. Pro-angiogenic role of insulin: From physiology to pathology. Front. Physiol. 2017, 8, 204. [Google Scholar] [CrossRef] [PubMed]
- Dubó, S.; Gallegos, D.; Cabrera, L.; Sobrevia, L.; Zúñiga, L.; González, M. Cardiovascular action of insulin in health and disease: Endothelial l-arginine transport and cardiac voltage-dependent potassium channels. Front. Physiol. 2016, 7, 74. [Google Scholar] [CrossRef] [PubMed]
- Sobrevia, L.; Salsoso, R.; Fuenzalida, B.; Barros, E.; Toledo, L.; Silva, L.; Pizarro, C.; Subiabre, M.; Villalobos, R.; Araos, J. Insulin is a key modulator of fetoplacental endothelium metabolic disturbances in gestational diabetes mellitus. Front. Physiol. 2016, 7, 119. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Opland, D.M.; Way, K.J.; Ueki, K.; Bodyak, N.; Kang, P.M.; Izumo, S.; Kulkarni, R.N.; Wang, B.; Liao, R. Regulation of vascular endothelial growth factor expression and vascularization in the myocardium by insulin receptor and PI3K/Akt pathways in insulin resistance and ischemia. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 787–793. [Google Scholar] [CrossRef]
- Shibuya, M. Vascular endothelial growth factor and its receptor system: Physiological functions in angiogenesis and pathological roles in various diseases. J. Biochem. 2013, 153, 13–19. [Google Scholar] [CrossRef]
- Cumsille, P.; Coronel, A.; Conca, C.; Quiñinao, C.; Escudero, C. Proposal of a hybrid approach for tumor progression and tumor-induced angiogenesis. Theor. Biol. Med Model. 2015, 12, 13. [Google Scholar] [CrossRef][Green Version]
- Wahren, J.; Kallas, Å.; Sima, A.A.F. The Clinical Potential of C-Peptide Replacement in Type 1 Diabetes. J. Diabetes 2012, 61, 761–772. [Google Scholar] [CrossRef]
- Wahren, J.; Johansson, B.L.; Wallberg-Henriksson, H.; Linde, B.; Fernqvist-Forbes, E.; Zierath, J.R. C-peptide revisited—New physiological effects and therapeutic implications. J. Int. Med. 1996, 240, 115–124. [Google Scholar] [CrossRef]
- Lim, Y.C.; Bhatt, M.P.; Kwon, M.H.; Park, D.; Lee, S.; Choe, J.; Hwang, J.; Kim, Y.M.; Ha, K.S. Prevention of VEGF-mediated microvascular permeability by C-peptide in diabetic mice. Cardiovasc. Res. 2014, 101, 155–164. [Google Scholar] [CrossRef]
- Bhatt, M.P.; Lim, Y.C.; Hwang, J.; Na, S.; Kim, Y.M.; Ha, K.S. C-peptide prevents hyperglycemia-induced endothelial apoptosis through inhibition of reactive oxygen species-mediated transglutaminase 2 activation. Diabetes 2013, 62, 243–253. [Google Scholar] [CrossRef]
- Lim, Y.-C.; Bhatt, M.P.; Kwon, M.-H.; Park, D.; Na, S.; Kim, Y.-M.; Ha, K.-S. Proinsulin C-Peptide Prevents Impaired Wound Healing by Activating Angiogenesis in Diabetes. J. Investig. Dermatol. 2015, 135, 269–278. [Google Scholar] [CrossRef]
- Mughal, R.S.; Scragg, J.L.; Lister, P.; Warburton, P.; Riches, K.; O’Regan, D.J.; Ball, S.G.; Turner, N.A.; Porter, K.E. Cellular mechanisms by which proinsulin C-peptide prevents insulin-induced neointima formation in human saphenous vein. Diabetologia 2010, 53, 1761–1771. [Google Scholar] [CrossRef]
- Walcher, D.; Babiak, C.; Poletek, P.; Rosenkranz, S.; Bach, H.; Betz, S.; Durst, R.; Grüb, M.; Hombach, V.; Strong, J.; et al. C-Peptide Induces Vascular Smooth Muscle Cell Proliferation. Circ. Res. 2006, 99, 1181–1187. [Google Scholar] [CrossRef]
- Marx, N. C-peptide as a mediator of lesion development in early diabetes—A novel hypothesis. Trends Cardiovasc. Med. 2008, 18, 67–71. [Google Scholar] [CrossRef]
- Forst, T.; Hach, T.; Kunt, T.; Weber, M.M.; Pfützner, A. Molecular Effects of C-Peptide in Microvascular Blood Flow Regulation. Rev. Diabet. Stud. Rds. 2009, 6, 159–167. [Google Scholar] [CrossRef]
- Wahren, J. C-peptide: New findings and therapeutic implications in diabetes. Clinic. Psihol. Funct. Imaging 2004, 24, 180–189. [Google Scholar] [CrossRef]
- Pramanik, A.; Ekberg, K.; Zhong, Z.; Shafqat, J.; Henriksson, M.; Jansson, O.; Tibell, A.; Tally, M.; Wahren, J.; Jörnvall, H.; et al. C-Peptide Binding to Human Cell Membranes: Importance of Glu27. BBiochem. Biophys. Res. Commun. 2001, 284, 94–98. [Google Scholar] [CrossRef]
- Shafqat, J.; Juntti-Berggren, L.; Zhong, Z.; Ekberg, K.; Köhler, M.; Berggren, P.-O.; Johansson, J.; Wahren, J.; Jörnvall, H. Proinsulin C-Peptide and Its Analogues Induce Intracellular Ca2+ INCREASES in Human Renal Tubular Cells; Springer: Berlin, Germany, 2002; Volume 59, pp. 1185–1189. [Google Scholar]
- Hills, C.E.; Brunskill, N.J. Cellular and physiological effects of C-peptide. Clin. Sci. 2009, 116, 565–574. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Naruse, K.; Hamada, Y.; Nakashima, E.; Kato, K.; Akiyama, N.; Kamiya, H.; Watarai, A.; Nakae, M.; Oiso, Y. Human proinsulin C-peptide prevents proliferation of rat aortic smooth muscle cells cultured in high-glucose conditions. Diabetologia 2005, 48, 2396–2401. [Google Scholar] [CrossRef]
- Marx, N.; Walcher, D. C-Peptide and atherogenesis: C-Peptide as a mediator of lesion development in patients with type 2 diabetes mellitus? Exp. Diabetes Res. 2008, 2008. [Google Scholar] [CrossRef]
- Johansson, B.-L.; Sundell, J.; Ekberg, K.; Jonsson, C.; Seppanen, M.; Raitakari, O.; Luotolahti, M.; Nuutila, P.; Wahren, J.; Knuuti, J. C-peptide improves adenosine-induced myocardial vasodilation in type 1 diabetes patients. Am. J. Physiol. Endocrinol. Metabol. 2004, 286, E14–E19. [Google Scholar] [CrossRef]
- Kitamura, T.; Kimura, K.; Makondo, K.; Furuya, D.; Suzuki, M.; Yoshida, T.; Saito, M. Proinsulin C-peptide increases nitric oxide production by enhancing mitogen-activated protein-kinase-dependent transcription of endothelial nitric oxide synthase in aortic endothelial cells of Wistar rats. Diabetologia 2003, 46, 1698–1705. [Google Scholar] [CrossRef]
- Luppi, P.; Cifarelli, V.; Tse, H.; Piganelli, J.; Trucco, M. Human C-peptide antagonises high glucose-induced endothelial dysfunction through the nuclear factor-κB pathway. Diabetologia 2008, 51, 1534. [Google Scholar] [CrossRef]
- Hoeben, A.; Landuyt, B.; Highley, M.S.; Wildiers, H.; Van Oosterom, A.T.; De Bruijn, E.A. Vascular endothelial growth factor and angiogenesis. Pharmacol. Rev. 2004, 56, 549–580. [Google Scholar] [CrossRef]
- Scheppke, L.; Aguilar, E.; Gariano, R.F.; Jacobson, R.; Hood, J.; Doukas, J.; Cao, J.; Noronha, G.; Yee, S.; Weis, S. Retinal vascular permeability suppression by topical application of a novel VEGFR2/Src kinase inhibitor in mice and rabbits. Pharmacol. Rev. 2008, 118, 2337–2346. [Google Scholar] [CrossRef]
- Caldwell, R.B.; Bartoli, M.; Behzadian, M.A.; El-Remessy, A.E.; Al-Shabrawey, M.; Platt, D.H.; Caldwell, R.W. Vascular endothelial growth factor and diabetic retinopathy: Pathophysiological mechanisms and treatment perspectives. Diabetes Metab. Res. Rev. 2003, 19, 442–455. [Google Scholar] [CrossRef]
- Al-Shabrawey, M.; Rojas, M.; Sanders, T.; Behzadian, A.; El-Remessy, A.; Bartoli, M.; Parpia, A.K.; Liou, G.; Caldwell, R.B. Role of NADPH oxidase in retinal vascular inflammation. Investig. Ophthalmol. Vis. Sci. 2008, 49, 3239–3244. [Google Scholar] [CrossRef]
- Li, J.; Wang, J.J.; Yu, Q.; Chen, K.; Mahadev, K.; Zhang, S.X. Inhibition of reactive oxygen species by Lovastatin downregulates vascular endothelial growth factor expression and ameliorates blood-retinal barrier breakdown in db/db mice: Role of NADPH oxidase 4. Diabetes 2010, 59, 1528–1538. [Google Scholar] [CrossRef]
- Abid, M.; Kachra, Z.; Spokes, K.C.; Aird, W.C. NADPH oxidase activity is required for endothelial cell proliferation and migration. FEBS Lett. 2000, 486, 252–256. [Google Scholar] [CrossRef]
- Monaghan-Benson, E.; Burridge, K. The regulation of vascular endothelial growth factor-induced microvascular permeability requires Rac and reactive oxygen species. J. Biol. Chem. 2009, 284, 25602–25611. [Google Scholar] [CrossRef]
- Spindler, V.; Schlegel, N.; Waschke, J. Role of GTPases in control of microvascular permeability. Cardiovasc. Res. 2010, 87, 243–253. [Google Scholar] [CrossRef]
- Tiruppathi, C.; Ahmmed, G.U.; Vogel, S.M.; Malik, A.B. Ca2+ signaling, TRP channels, and endothelial permeability. Microcirculation 2006, 13, 693–708. [Google Scholar] [CrossRef]
- Bhatt, M.P.; Lim, Y.-C.; Kim, Y.-M.; Ha, K.-S. C-peptide activates AMPKα and prevents ROS-mediated mitochondrial fission and endothelial apoptosis in diabetes. Diabetes 2013, 62, 3851–3862. [Google Scholar] [CrossRef]
- Ushio-Fukai, M.; Nakamura, Y. Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy. Cancer Lett. 2008, 266, 37–52. [Google Scholar] [CrossRef]
- Yang, B.; Cao, D.J.; Sainz, I.; Colman, R.W.; Guo, Y.L. Different roles of ERK and p38 MAP kinases during tube formation from endothelial cells cultured in 3-dimensional collagen matrices. J. Cell. Physiol. 2004, 200, 360–369. [Google Scholar] [CrossRef]
- El-Remessy, A.; Al-Shabrawey, M.; Platt, D.; Bartoli, M.; Behzadian, M.; Ghaly, N.; Tsai, N.; Motamed, K.; Caldwell, R. Peroxynitrite Mediates VEGF’s Angiogenic Signal and Function via a Nitration-Independent Mechanism in Endothelial Cells. FASEB J. 2007, 21, 2528–2539. [Google Scholar] [CrossRef]
- Fukuhara, S.; Sako, K.; Noda, K.; Zhang, J.; Minami, M.; Mochizuki, N. Angiopoietin-1/Tie2 receptor signaling in vascular quiescence and angiogenesis. Histol. Histopathol. 2010, 25, 387–396. [Google Scholar]
- Thurston, G.; Rudge, J.S.; Ioffe, E.; Zhou, H.; Ross, L.; Croll, S.D.; Glazer, N.; Holash, J.; McDonald, D.M.; Yancopoulos, G.D. Angiopoietin-1 protects the adult vasculature against plasma leakage. Nat. Med. 2000, 6, 460. [Google Scholar] [CrossRef]
- Harfouche, R.; Malak, N.A.; Brandes, R.P.; Karsan, A.; Irani, K.; Hussain, S.N. Roles of reactive oxygen species in angiopoietin-1/tie-2 receptor signaling. FASEB J. 2005, 19, 1728–1730. [Google Scholar] [CrossRef]
- Cifarelli, V.; Geng, X.; Styche, A.; Lakomy, R.; Trucco, M.; Luppi, P. C-peptide reduces high-glucose-induced apoptosis of endothelial cells and decreases NAD (P) H-oxidase reactive oxygen species generation in human aortic endothelial cells. Diabetologia 2011, 54, 2702. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Chan, P.-S. Oxidative stress and diabetic retinopathy. J. Diabetes Res. 2007, 2007. [Google Scholar] [CrossRef]
- Kaplan, M.; Hamoud, S.; Tendler, Y.; Meilin, E.; Lazarovitch, A.; Nitecki, S.; Hayek, T. A significant correlation between C-reactive protein levels in blood monocytes derived macrophages versus content in carotid atherosclerotic lesions. J. Inflamm. 2014, 11, 7. [Google Scholar] [CrossRef]
- Hattori, Y.; Matsumura, M.; Kasai, K. Vascular smooth muscle cell activation by C-reactive protein. Cardiovasc. Res. 2003, 58, 186–195. [Google Scholar] [CrossRef]
- Yeo, E.-S.; Hwang, J.-Y.; Park, J.E.; Choi, Y.J.; Huh, K.B.; Kim, W.Y. Tumor necrosis factor (TNF-α) and C-reactive protein (CRP) are positively associated with the risk of chronic kidney disease in patients with type 2 diabetes. Yonsei Med. J. 2010, 51, 519–525. [Google Scholar] [CrossRef]
- Zhong, Z.; Kotova, O.; Davidescu, A.; Ehren, I.; Ekberg, K.; Jörnvall, H.; Wahren, J.; Chibalin, A. C-peptide stimulates Na+, K+-ATPase via activation of ERK1/2 MAP kinases in human renal tubular cells. Cell. Mol. Life Sci. 2004, 61, 2782–2790. [Google Scholar] [CrossRef]
- Joshua, I.G.; Zhang, Q.; Falcone, J.C.; Bratcher, A.P.; Rodriguez, W.E.; Tyagi, S.C. Mechanisms of endothelial dysfunction with development of type 1 diabetes mellitus: Role of insulin and C-peptide. J. Cell. Biochem. 2005, 96, 1149–1156. [Google Scholar] [CrossRef]
- Lee, J.-H.; Lee, W.; Kwon, O.H.; Kim, J.-H.; Kwon, O.W.; Kim, K.H.; Lim, J.-B. Cytokine profile of peripheral blood in type 2 diabetes mellitus patients with diabetic retinopathy. Ann. Clin. Lab. Sci. 2008, 38, 361–367. [Google Scholar]
- Didion, S.P.; Kinzenbaw, D.A.; Schrader, L.I.; Chu, Y.; Faraci, F.M. Endogenous Interleukin-10 Inhibits Angiotensin II–Induced Vascular Dysfunction. Hypertension 2009, 54, 619–624. [Google Scholar] [CrossRef]
- Hong, E.-G.; Ko, H.J.; Cho, Y.-R.; Kim, H.-J.; Ma, Z.; Yu, T.Y.; Friedline, R.H.; Kurt-Jones, E.; Finberg, R.; Fischer, M.A.; et al. Interleukin-10 Prevents Diet-Induced Insulin Resistance by Attenuating Macrophage and Cytokine Response in Skeletal Muscle. Diabetes 2009, 58, 2525–2535. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A.; Ihnat, M.A.; Thorpe, J.E. The “metabolic memory”: Is more than just tight glucose control necessary to prevent diabetic complications? J. Clin. Endocrinol. Metab. 2009, 94, 410–415. [Google Scholar] [CrossRef]
- Cooper, M.E.; El-Osta, A. Epigenetics: Mechanisms and implications for diabetic complications. Circ. Res. 2010, 107, 1403–1413. [Google Scholar] [CrossRef] [PubMed]
- Pirola, L.; Balcerczyk, A.; Okabe, J.; El-Osta, A. Epigenetic phenomena linked to diabetic complications. Nat. Rev. Endocrinol. 2010, 6, 665. [Google Scholar] [CrossRef]
- Fadini, G.P.; Albiero, M.; Menegazzo, L.; Boscaro, E.; Pagnin, E.; Iori, E.; Cosma, C.; Lapolla, A.; Pengo, V.; Stendardo, M. The redox enzyme p66Shc contributes to diabetes and ischemia-induced delay in cutaneous wound healing. Diabetes 2010, 59, 2306–2314. [Google Scholar] [CrossRef]
- Kim, C.-S.; Jung, S.-B.; Naqvi, A.; Hoffman, T.A.; DeRicco, J.; Yamamori, T.; Cole, M.P.; Jeon, B.-H.; Irani, K. p53 impairs endothelium-dependent vasomotor function through transcriptional upregulation of p66shc. Circ. Res. 2008, 103, 1441–1450. [Google Scholar] [CrossRef]
- Paneni, F.; Mocharla, P.; Akhmedov, A.; Costantino, S.; Osto, E.; Volpe, M.; Lüscher, T.F.; Cosentino, F. Gene Silencing of the Mitochondrial Adaptor p66Shc Suppresses Vascular Hyperglycemic Memory in DiabetesNovelty and Significance. Circ. Res. 2012, 111, 278–289. [Google Scholar] [CrossRef]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813. [Google Scholar] [CrossRef]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef]
- Paneni, F.; Volpe, M.; Lüscher, T.F.; Cosentino, F. SIRT1, p66Shc, and Set7/9 in vascular hyperglycemic memory: Bringing all the strands together. Diabetes 2013, 62, 1800–1807. [Google Scholar] [CrossRef]
- Bagi, Z.; Feher, A.; Cassuto, J. Peroxynitrite disrupts endothelial caveolae leading to eNOS uncoupling and diminished flow-mediated dilation in coronary arterioles of diabetic patients (1082.3). FASEB J. 2014, 28, 1381–1393. [Google Scholar]
- El-Remessy, A.B.; Abou-Mohamed, G.; Caldwell, R.W.; Caldwell, R.B. High glucose-induced tyrosine nitration in endothelial cells: Role of eNOS uncoupling and aldose reductase activation. Investig. Ophthalmol. Vis. Sci. 2003, 44, 3135–3143. [Google Scholar] [CrossRef]
- Navarro-Antolín, J.; López-Muñoz, M.J.; Klatt, P.; Soria, J.; Michel, T.; Lamas, S. Formation of peroxynitrite in vascular endothelial cells exposed to cyclosporine A. FASEB J. 2001, 15, 1291–1293. [Google Scholar] [CrossRef]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef]
- Ceriello, A.; Esposito, K.; Ihnat, M.; Thorpe, J.; Giugliano, D. Long-term glycemic control influences the long-lasting effect of hyperglycemia on endothelial function in type 1 diabetes. J. Clin. Endocrinol. Metab. 2009, 94, 2751–2756. [Google Scholar] [CrossRef]
- Ihnat, M.; Thorpe, J.; Kamat, C.; Szabo, C.; Green, D.; Warnke, L.; Lacza, Z.; Cselenyak, A.; Ross, K.; Shakir, S. Reactive oxygen species mediate a cellular ‘memory’of high glucose stress signalling. Diabetologia 2007, 50, 1523–1531. [Google Scholar] [CrossRef]
- Bhatt, M.P.; Lim, Y.-C.; Ha, K.-S. C-peptide replacement therapy as an emerging strategy for preventing diabetic vasculopathy. Cardiovasc. Res. 2014, 104, 234–244. [Google Scholar] [CrossRef]
- Massi-Benedetti, M.; Orsini-Federici, M. Treatment of type 2 diabetes with combined therapy: What are the pros and cons? Diabetes Care 2008, 31, S131–S135. [Google Scholar] [CrossRef]
- Beckman, J.A.; Goldfine, A.B.; Gordon, M.B.; Garrett, L.A.; Keaney, J.F., Jr.; Creager, M.A. Oral antioxidant therapy improves endothelial function in Type 1 but not Type 2 diabetes mellitus. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H2392–H2398. [Google Scholar] [CrossRef]
- Atkinson, M.A.; Bluestone, J.A.; Eisenbarth, G.S.; Hebrok, M.; Herold, K.C.; Accili, D.; Pietropaolo, M.; Arvan, P.R.; Von Herrath, M.; Markel, D.S.; et al. How Does Type 1 Diabetes Develop? Notion Homicide Or Β-Cell Suicide Revisit. 2011, 60, 1370–1379. [Google Scholar] [CrossRef]
- Eizirik, D.L.; Miani, M.; Cardozo, A.K. Signalling danger: Endoplasmic reticulum stress and the unfolded protein response in pancreatic islet inflammation. Diabetologia 2013, 56, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Steiner, D.F. On the Discovery of Precursor Processing. In Proprotein Convertases; Mbikay, M., Seidah, N.G., Eds.; Humana Press: Totowa, NJ, USA, 2011; pp. 3–11. [Google Scholar] [CrossRef]
- Tersey, S.A.; Nishiki, Y.; Templin, A.T.; Cabrera, S.M.; Stull, N.D.; Colvin, S.C.; Evans-Molina, C.; Rickus, J.L.; Maier, B.; Mirmira, R.G. Islet β-Cell Endoplasmic Reticulum Stress Precedes the Onset of Type 1 Diabetes in the Nonobese Diabetic Mouse Model. Diabetes 2012, 61, 818–827. [Google Scholar] [CrossRef] [PubMed]
- Sims, E.K.; Chaudhry, Z.; Watkins, R.; Syed, F.; Blum, J.; Ouyang, F.; Perkins, S.M.; Mirmira, R.G.; Sosenko, J.; DiMeglio, L.A.; et al. Elevations in the Fasting Serum Proinsulin–to–C-Peptide Ratio Precede the Onset of Type 1 Diabetes. Diabetes Care 2016, 39, 1519–1526. [Google Scholar] [CrossRef] [PubMed]
- Montalcini, T.; Gallotti, P.; Coppola, A.; Zambianchi, V.; Fodaro, M.; Galliera, E.; Marazzi, M.G.; Romeo, S.; Giannini, S.; Corsi Romanelli, M.M.; et al. Association between low C-peptide and low lumbar bone mineral density in postmenopausal women without diabetes. Osteoporos. Int. 2015, 26, 1639–1646. [Google Scholar] [CrossRef]
- Russo, C.; Lazzaro, V.; Gazzaruso, C.; Maurotti, S.; Ferro, Y.; Pingitore, P.; Fumo, F.; Coppola, A.; Gallotti, P.; Zambianchi, V.; et al. Proinsulin C-peptide modulates the expression of ERK1/2, type I collagen and RANKL in human osteoblast-like cells (Saos-2). Mol. Cell. Endocrinol. 2017, 442, 134–141. [Google Scholar] [CrossRef]
- Sima, A.A.F.; Zhang, W.; Sugimoto, K.; Henry, D.; Li, Z.; Wahren, J.; Grunberger, G. C-peptide prevents and improves chronic Type I diabetic polyneuropathy in the BB/Wor rat. Diabetologia 2001, 44, 889–897. [Google Scholar] [CrossRef]
- Ido, Y.; Vindigni, A.; Chang, K.; Stramm, L.; Chance, R.; Heath, W.F.; DiMarchi, R.D.; Di Cera, E.; Williamson, J.R. Prevention of Vascular and Neural Dysfunction in Diabetic Rats by C-Peptide. Science 1997, 277, 563–566. [Google Scholar] [CrossRef]
- Zhang, W.; Yorek, M.; Pierson, C.R.; Murakawa, Y.; Breidenbach, A.; Sima, A.A.F. Human C-peptide Dose Dependently Prevents Early Neuropathy in the BB/Wor-rat. Mol. Cell. Endocrinol. 2001, 2, 187–193. [Google Scholar] [CrossRef]
- Cotter, M.A.; Ekberg, K.; Wahren, J.; Cameron, N.E. Effects of Proinsulin C-Peptide in Experimental Diabetic Neuropathy. Vasc. Actions Modul. Nitric Oxide Synthase Inhib. 2003, 52, 1812–1817. [Google Scholar] [CrossRef]
- Ekberg, K.; Brismar, T.; Johansson, B.-L.; Jonsson, B.; Lindström, P.; Wahren, J. Amelioration of Sensory Nerve Dysfunction by C-Peptide in Patients With Type 1 Diabetes. Diabetes 2003, 52, 536–541. [Google Scholar] [CrossRef]
- Johansson, B.L.; Borg, K.; Fernqvist-Forbes, E.; Kernell, A.; Odergren, T.; Wahren, J. Beneficial effects of C-peptide on incipient nephropathy and neuropathy in patients with Type 1 diabetes mellitus. Diabet. Med. 2000, 17, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Johansson, B.L.; Borg, K.; Fernqvist-Forbes, E.; Odergren, T.; Remahl, S.; Wahren, J. C-peptide improves autonomic nerve function in IDDM patients. Diabetologia 1996, 39, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, S.; Kimura, K.; Kitamura, T.; Cañas, X.; Yoshida, T.; Saito, M. Proinsulin C peptide obviates sympathetically mediated suppression of splenic lymphocyte activity in rats. Diabetologia 2000, 43, 1512–1517. [Google Scholar] [CrossRef][Green Version]
- Stevens, M.J.; Dananberg, J.; Feldman, E.L.; Lattimer, S.A.; Kamijo, M.; Thomas, T.P.; Shindo, H.; Sima, A.A.; Greene, D.A. The linked roles of nitric oxide, aldose reductase and, (Na+,K+)-ATPase in the slowing of nerve conduction in the streptozotocin diabetic rat. J. Clin. Investig. 1994, 94, 853–859. [Google Scholar] [CrossRef]
- Thomas, P. Diabetic neuropathy: Mechanisms and future treatment options. J. Neurol. Neurosurg. Psychiatry 1999, 67, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, W.; Sima, A.A.F. C-peptide enhances insulin-mediated cell growth and protection against high glucose–induced apoptosis in SH-SY5Y cells. Diabetes/Metab. Res. Rev. 2003, 19, 375–385. [Google Scholar] [CrossRef]
- Afkarian, M.; Sachs, M.C.; Kestenbaum, B.; Hirsch, I.B.; Tuttle, K.R.; Himmelfarb, J.; De Boer, I.H. Kidney disease and increased mortality risk in type 2 diabetes. J. Am. Soc. Nephrol. 2013. ASN.2012070718. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou-Marketou, N.; Chrousos, G.P.; Kanaka-Gantenbein, C. Diabetic nephropathy in type 1 diabetes: A review of early natural history, pathogenesis, and diagnosis. J. Am. Soc. Nephrol. 2017, 33. [Google Scholar] [CrossRef]
- Luppi, P.; Drain, P. C-peptide antioxidant adaptive pathways in β cells and diabetes. J. Intern. Med. 2017, 281, 7–24. [Google Scholar] [CrossRef]
- Huggett, R.J.; Scott, E.M.; Gilbey, S.G.; Stoker, J.B.; Mackintosh, A.F.; Mary, D.A. Impact of type 2 diabetes mellitus on sympathetic neural mechanisms in hypertension. Circulation 2003, 108, 3097–3101. [Google Scholar] [CrossRef]
- Masuo, K.; Mikami, H.; Ogihara, T.; Tuck, M.L. Sympathetic nerve hyperactivity precedes hyperinsulinemia and blood pressure elevation in a young, nonobese Japanese population. Am. J. Hypertens. 1997, 10, 77–83. [Google Scholar] [CrossRef]
- Grassi, G.; Dell’Oro, R.; Quarti-Trevano, F.; Scopelliti, F.; Seravalle, G.; Paleari, F.; Gamba, P.; Mancia, G. Neuroadrenergic and reflex abnormalities in patients with metabolic syndrome. Diabetologia 2005, 48, 1359–1365. [Google Scholar] [CrossRef] [PubMed]
- Grassi, G.; Dell’Oro, R.; Facchini, A.; Trevano, F.Q.; Bolla, G.B.; Mancia, G. Effect of central and peripheral body fat distribution on sympathetic and baroreflex function in obese normotensives. J. Hypertens. 2004, 22, 2363–2369. [Google Scholar] [CrossRef]
- Scherrer, U.; Sartori, C. Insulin as a vascular and sympathoexcitatory hormone: Implications for blood pressure regulation, insulin sensitivity, and cardiovascular morbidity. Circulation 1997, 96, 4104–4113. [Google Scholar] [CrossRef]
- Bardgett, M.E.; McCarthy, J.J.; Stocker, S.D. Glutamatergic receptor activation in the rostral ventrolateral medulla mediates the sympathoexcitatory response to hyperinsulinemia. Hypertension 2010, 55, 284–290. [Google Scholar] [CrossRef]
- Mahfoud, F.; Schlaich, M.; Kindermann, I.; Ukena, C.; Cremers, B.; Brandt, M.C.; Hoppe, U.C.; Vonend, O.; Rump, L.C.; Sobotka, P.A. Effect of renal sympathetic denervation on glucose metabolism in patients with resistant hypertensionClinical perspective: A Pilot study. Circulation 2011, 123, 1940–1946. [Google Scholar] [CrossRef]
- Elbassuoni, E.A.; Aziz, N.M.; El-Tahawy, N.F. Protective effect of C-peptide on experimentally induced diabetic nephropathy and the possible link between C-peptide and Nitric Oxide. Appl. Physiol. Nutr. Metabol. 2018, 43, 617–624. [Google Scholar] [CrossRef]
- DiBona, G.F. Physiology in perspective: The Wisdom of the Body. Neural control of the kidney. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R633–R641. [Google Scholar] [CrossRef]
- Vollenweider, P.; Tappy, L.; Randin, D.; Schneiter, P.; Jéquier, E.; Nicod, P.; Scherrer, U. Differential effects of hyperinsulinemia and carbohydrate metabolism on sympathetic nerve activity and muscle blood flow in humans. J. Clin. Investig. 1993, 92, 147–154. [Google Scholar] [CrossRef]
- Mancia, G.; Bousquet, P.; Elghozi, J.L.; Esler, M.; Grassi, G.; Julius, S.; Reid, J.; Van Zwieten, P.A. The sympathetic nervous system and the metabolic syndrome. J. Hypertens. 2007, 25, 909–920. [Google Scholar] [CrossRef]
- Menditto, E.; Guerriero, F.; Orlando, V.; Crola, C.; Di Somma, C.; Illario, M.; Morisky, D.E.; Colao, A. Self-Assessment of Adherence to Medication: A Case Study in Campania Region Community-Dwelling Population. J. Aging Res. 2015, 2015, 682503. [Google Scholar] [CrossRef] [PubMed]
- Iolascon, G.; Gimigliano, F.; Moretti, A.; Riccio, I.; Di Gennaro, M.; Illario, M.; Monetti, V.M.; Orlando, V.; Menditto, E. Rates and reasons for lack of persistence with anti-osteoporotic drugs: Analysis of the Campania region database. Clin. Cases Miner. Bone Metab. 2016, 13, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Putignano, D.; Bruzzese, D.; Orlando, V.; Fiorentino, D.; Tettamanti, A.; Menditto, E. Differences in drug use between men and women: An Italian cross sectional study. BMC Womens Health 2017, 17, 73. [Google Scholar] [CrossRef]
- Menditto, E.; Cahir, C.; Aza-Pascual-Salcedo, M.; Bruzzese, D.; Poblador-Plou, B.; Malo, S.; Costa, E.; Gonzalez-Rubio, F.; Gimeno-Miguel, A.; Orlando, V.; et al. Adherence to chronic medication in older populations: Application of a common protocol among three European cohorts. Patient Prefer Adherence 2018, 12, 1975–1987. [Google Scholar] [CrossRef]
- Scala, D.; Menditto, E.; Armellino, M.F.; Manguso, F.; Monetti, V.M.; Orlando, V.; Antonino, A.; Makoul, G.; De Palma, M. Italian translation and cultural adaptation of the communication assessment tool in an outpatient surgical clinic. BMC Health Serv. Res. 2016, 16, 163. [Google Scholar] [CrossRef]


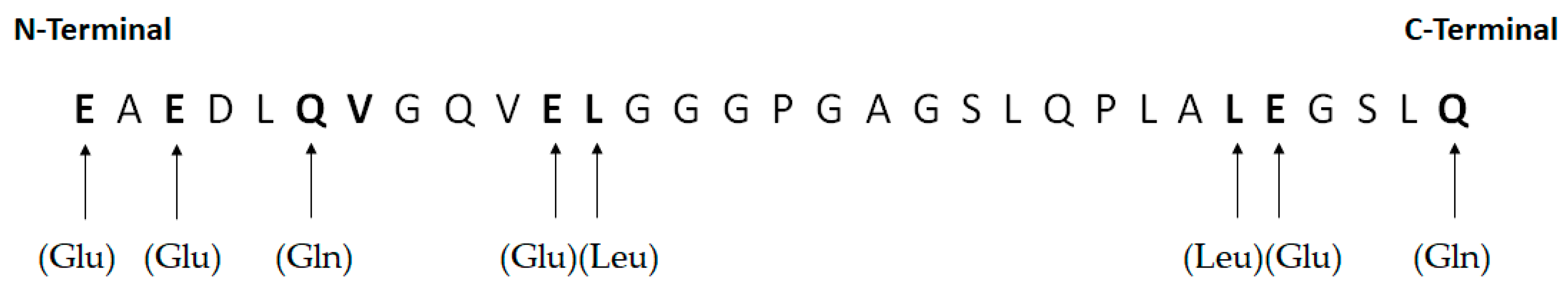{kind=link}
| Cytoprotective, Antiapoptotic and Anti-Inflammatory Effects of C-Peptide | ↑ 5rc-kinase ↑ Pl-3 kinase ↑Erk-1/-2 | ↑ VSMC proliferation | ↑ vascular damage | |
| ↑Pl-3 | ↑ Chemotaxis of leukocytes | |||
| ↓ ROS ↓ NF-kβ | ↓ proliferation and migration of VSMC | ↓ Vascular damage | ||
| ↓ IL-8 ↓ MCP-1 | ↓ leukocyte adhesion and migration into the vascular wall | |||
| ↓ P-selectin ↓ ICAM-1 ↓ VCAM-1 | ↓ Leukocyte endothelium interaction | |||
| ↑ Bcl-2 ↓ Caspase-3 | ↓ Apoptosis | |||
| Effects of C-Peptide on the Renal Function | NF-kβ PPARΥ | Transcriptional effects | ↑ IGF-1 ↓ TNFα ↓ TGFβ | Improved renal structure ↓ Apoptosis ↓ Mesangial expansion |
| ↓ Na+ ATPase ↓ K+ ATPase | ↓ Na+ excretion ↑ Energy status | Improved renal structure Improved glomerular function ↓ Apoptosis ↓ Mesangial expansion ↓ Glomerular hyperfiltration ↓ Albumin excretion ↑ RBC deformability | ||
| ↓ eNOS | ↑ Endothelial function | Improved glomerular function ↓ Glomerular hyperfiltration ↓ Albumin excretion ↑ RBC deformability | ||
| Effects of C-Peptide on Diabetic Neuropathy | c-fos c-jun | ↑ Neurotropic factors | ↑ NGF ↑ IGF-1 ↑ NT3 | Improved nerve structures |
| ↑ eNOS | ↑ Na+ ATPase ↑ K+ ATPase ↑ Endothelial function ↑ Nerve blood flow | Improved nerve function | ||
| ↑ Na+ ATPase ↑ K+ ATPase | ↓ Na+ retention ↑ Energy status | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Souto, S.B.; Campos, J.R.; Fangueiro, J.F.; Silva, A.M.; Cicero, N.; Lucarini, M.; Durazzo, A.; Santini, A.; Souto, E.B. Multiple Cell Signalling Pathways of Human Proinsulin C-Peptide in Vasculopathy Protection. Int. J. Mol. Sci. 2020, 21, 645. https://doi.org/10.3390/ijms21020645
Souto SB, Campos JR, Fangueiro JF, Silva AM, Cicero N, Lucarini M, Durazzo A, Santini A, Souto EB. Multiple Cell Signalling Pathways of Human Proinsulin C-Peptide in Vasculopathy Protection. International Journal of Molecular Sciences. 2020; 21(2):645. https://doi.org/10.3390/ijms21020645
Chicago/Turabian StyleSouto, Selma B., Joana R. Campos, Joana F. Fangueiro, Amélia M. Silva, Nicola Cicero, Massimo Lucarini, Alessandra Durazzo, Antonello Santini, and Eliana B. Souto. 2020. "Multiple Cell Signalling Pathways of Human Proinsulin C-Peptide in Vasculopathy Protection" International Journal of Molecular Sciences 21, no. 2: 645. https://doi.org/10.3390/ijms21020645
APA StyleSouto, S. B., Campos, J. R., Fangueiro, J. F., Silva, A. M., Cicero, N., Lucarini, M., Durazzo, A., Santini, A., & Souto, E. B. (2020). Multiple Cell Signalling Pathways of Human Proinsulin C-Peptide in Vasculopathy Protection. International Journal of Molecular Sciences, 21(2), 645. https://doi.org/10.3390/ijms21020645