Enteropathogenic Escherichia coli (EPEC) Recruitment of PAR Polarity Protein Atypical PKCζ to Pedestals and Cell–Cell Contacts Precedes Disruption of Tight Junctions in Intestinal Epithelial Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
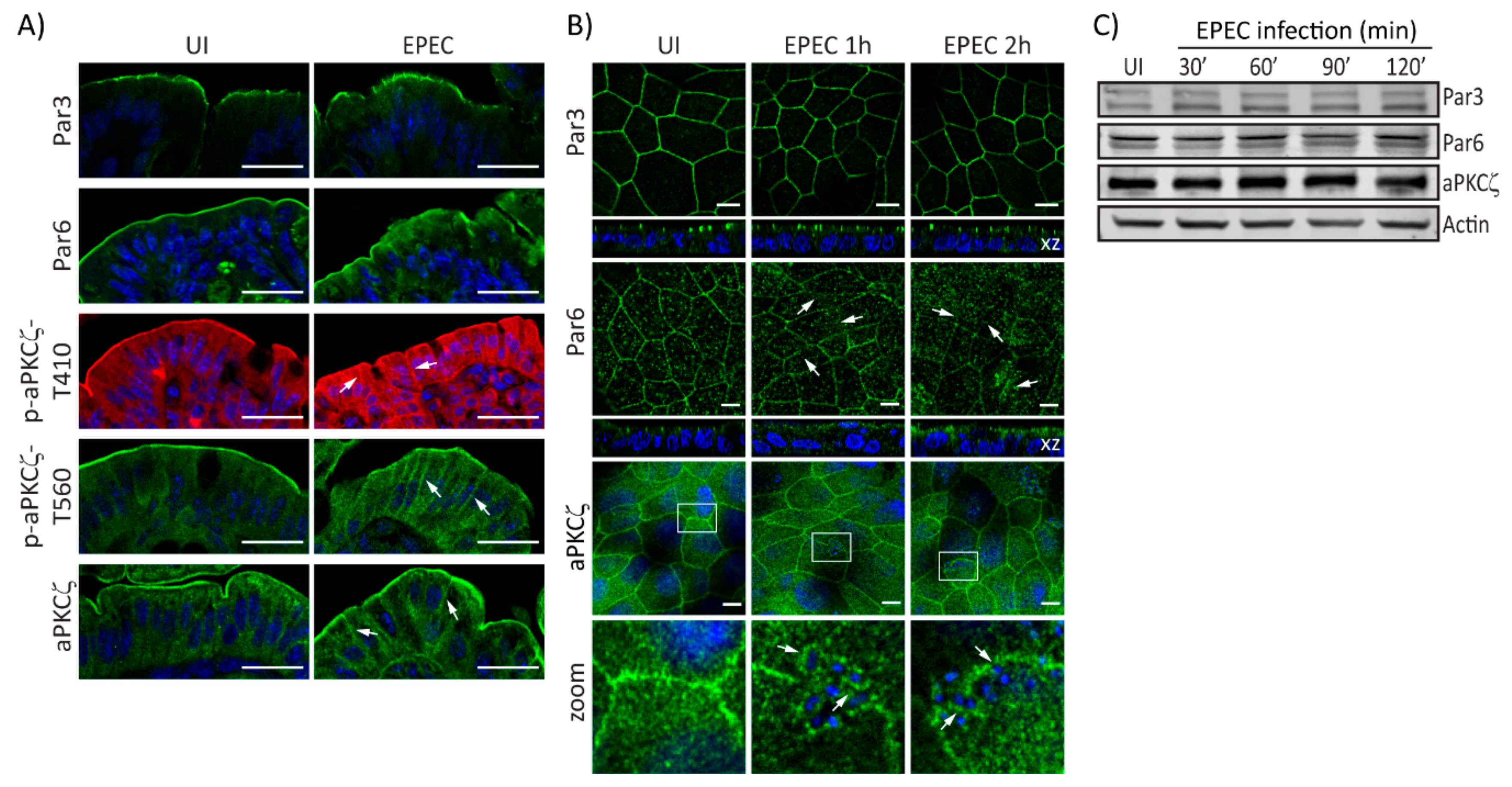
2.1. EPEC Disrupts PAR Polarity Complexes In Vivo and In Vitro
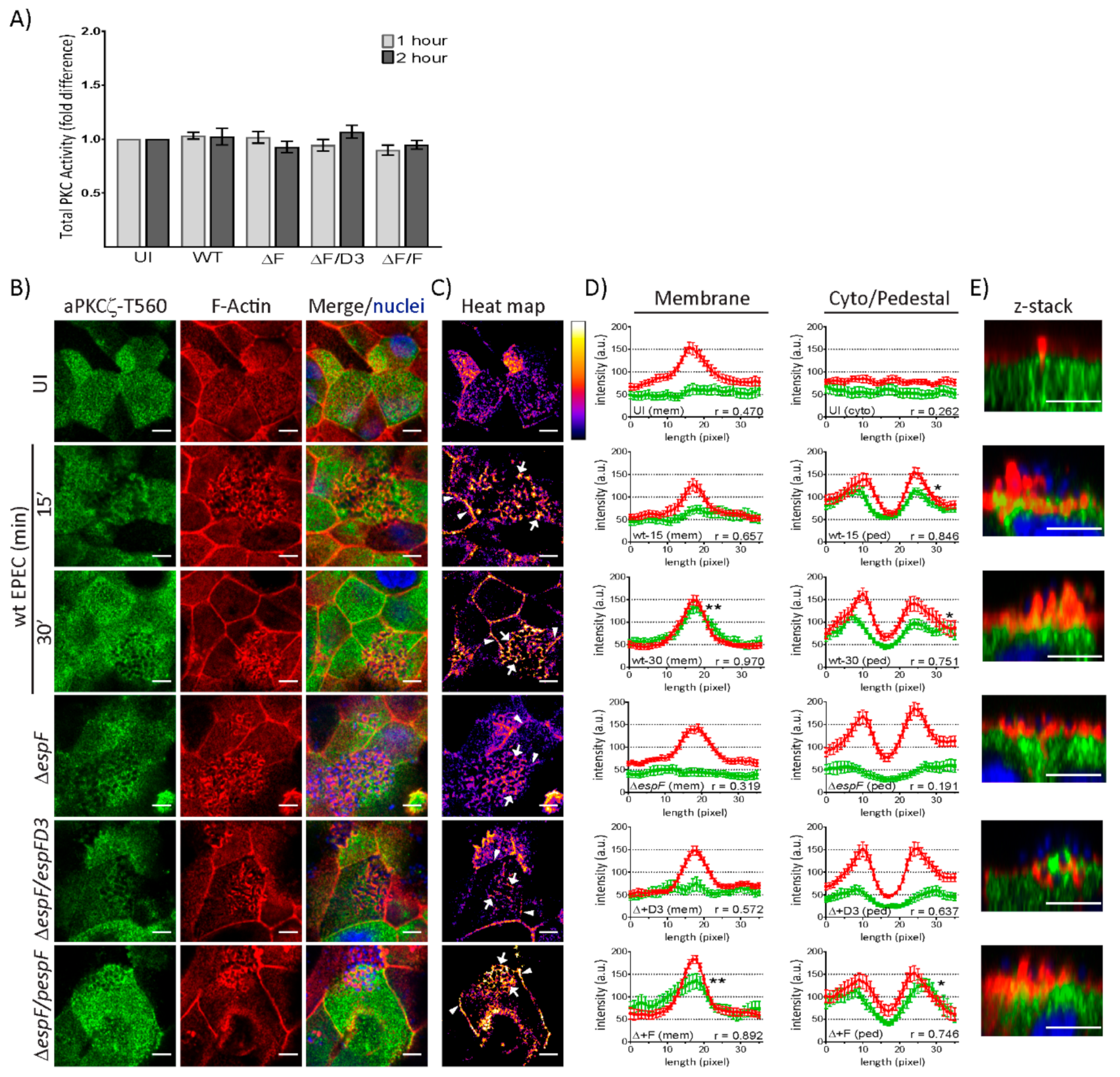
2.2. EspF, Via Its SNX9-Binding Domain, Impacts the Structural Organization of aPKCζ and F-actin at Pedestals
2.3. EspF and Its SNX9-Binding Domain Contribute to the Recruitment of Phosphorylated aPKCζ–T560 to Pedestals without Altering Kinase Activity
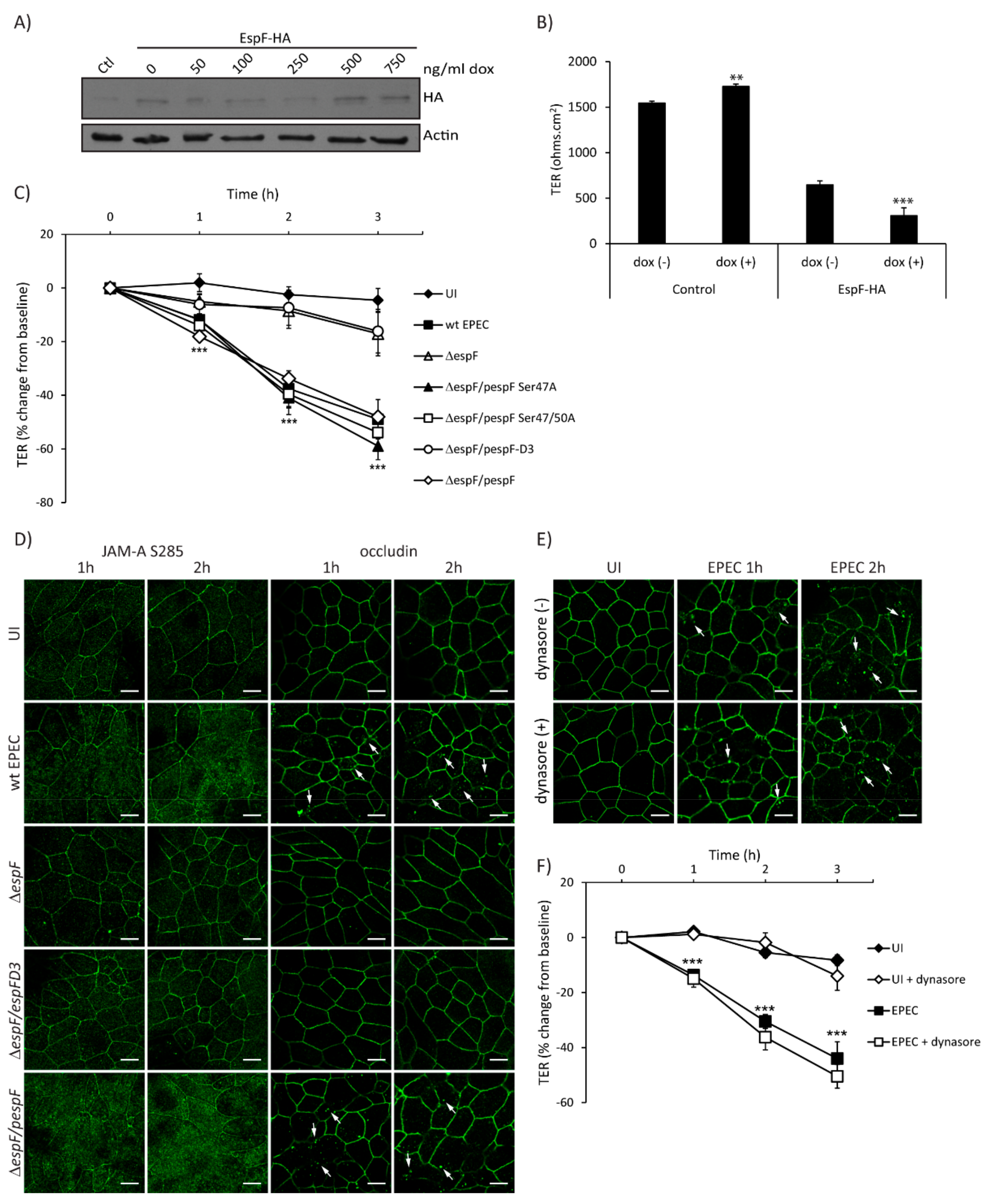
2.4. SNX9-Binding Domain of EspF Is Essential to Disrupt TJ Structure and Function in SKCO-15 Monolayers
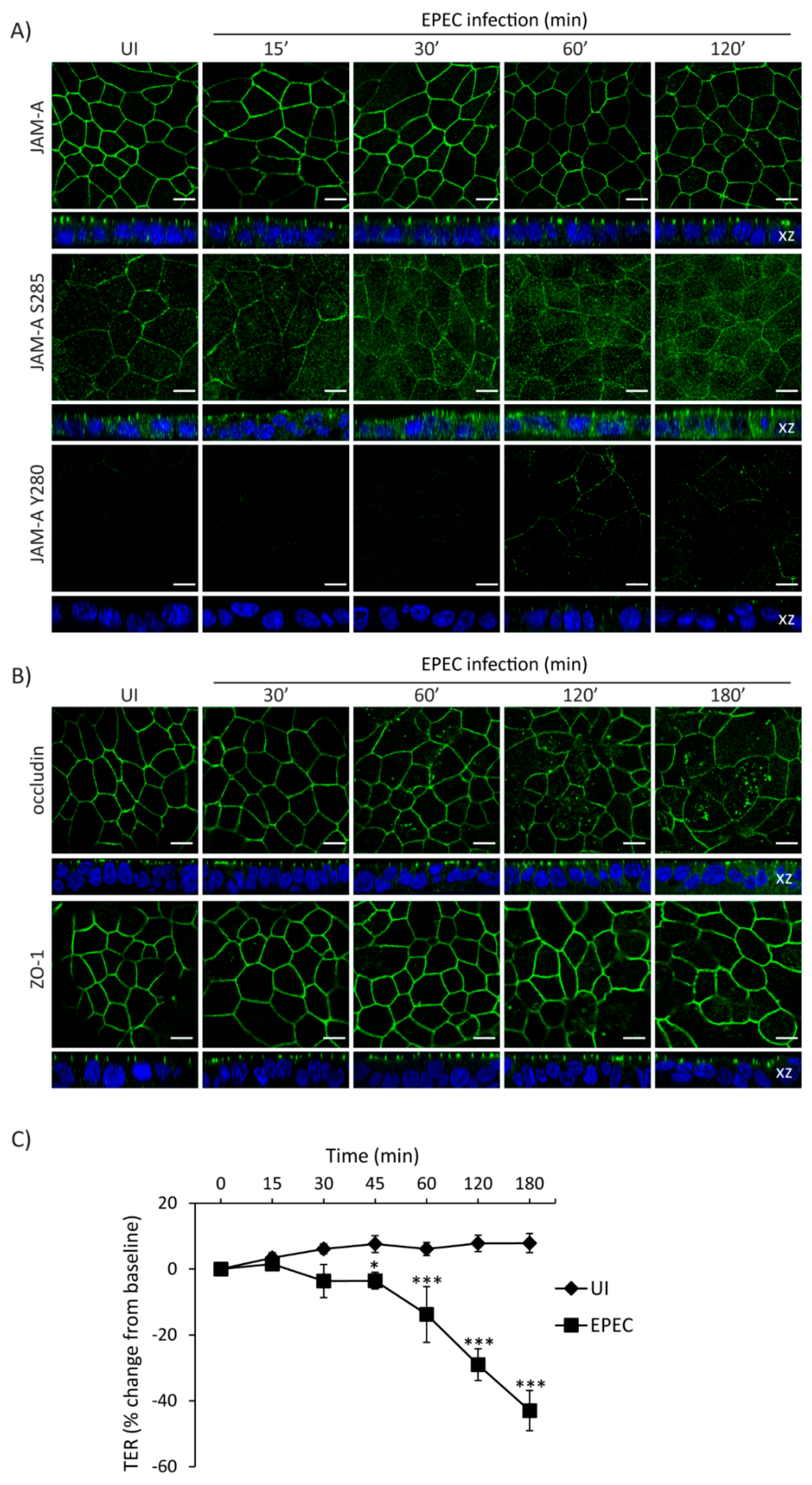
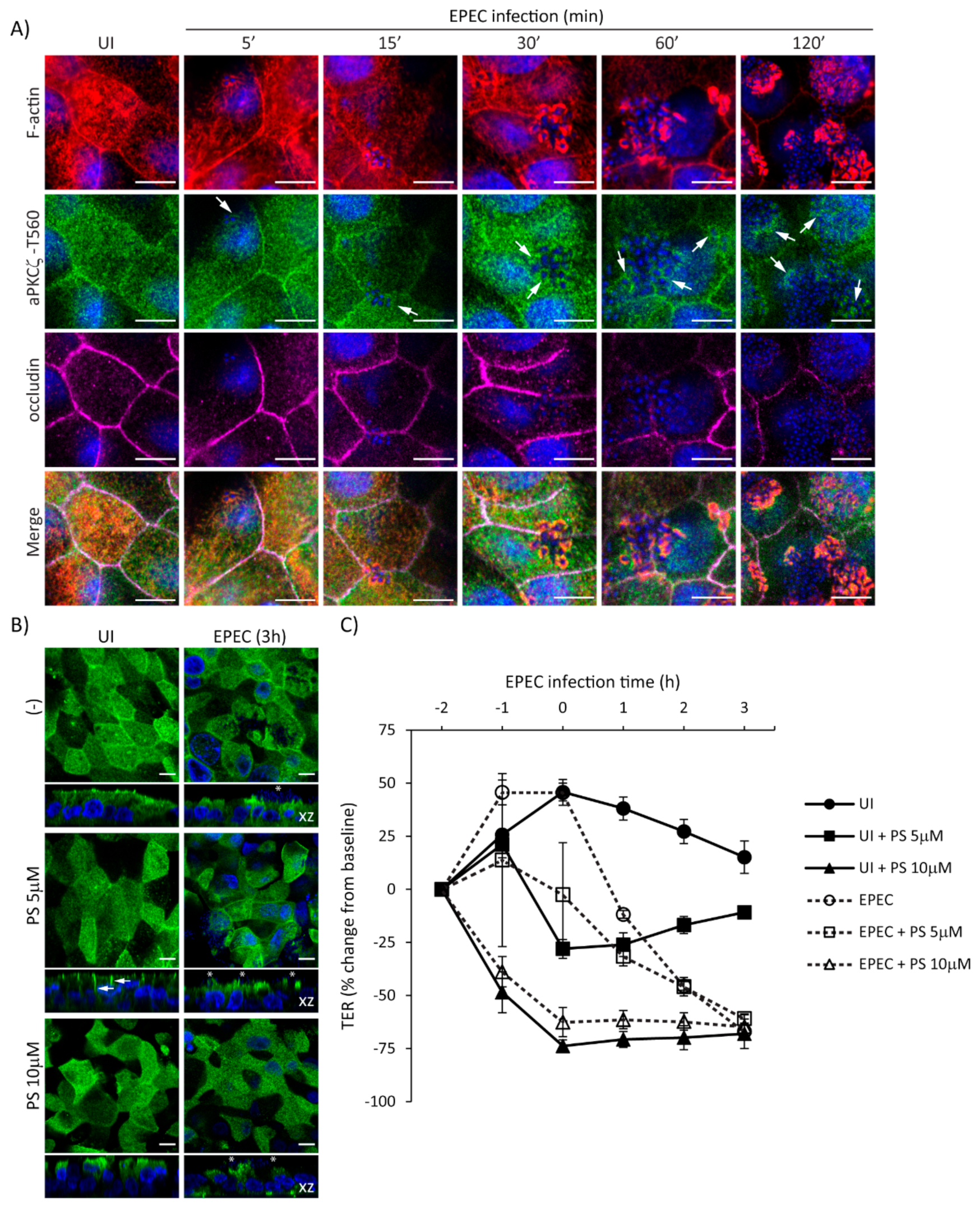
2.5. Temporal Disruption of TJ Proteins Mediated by EPEC Infection
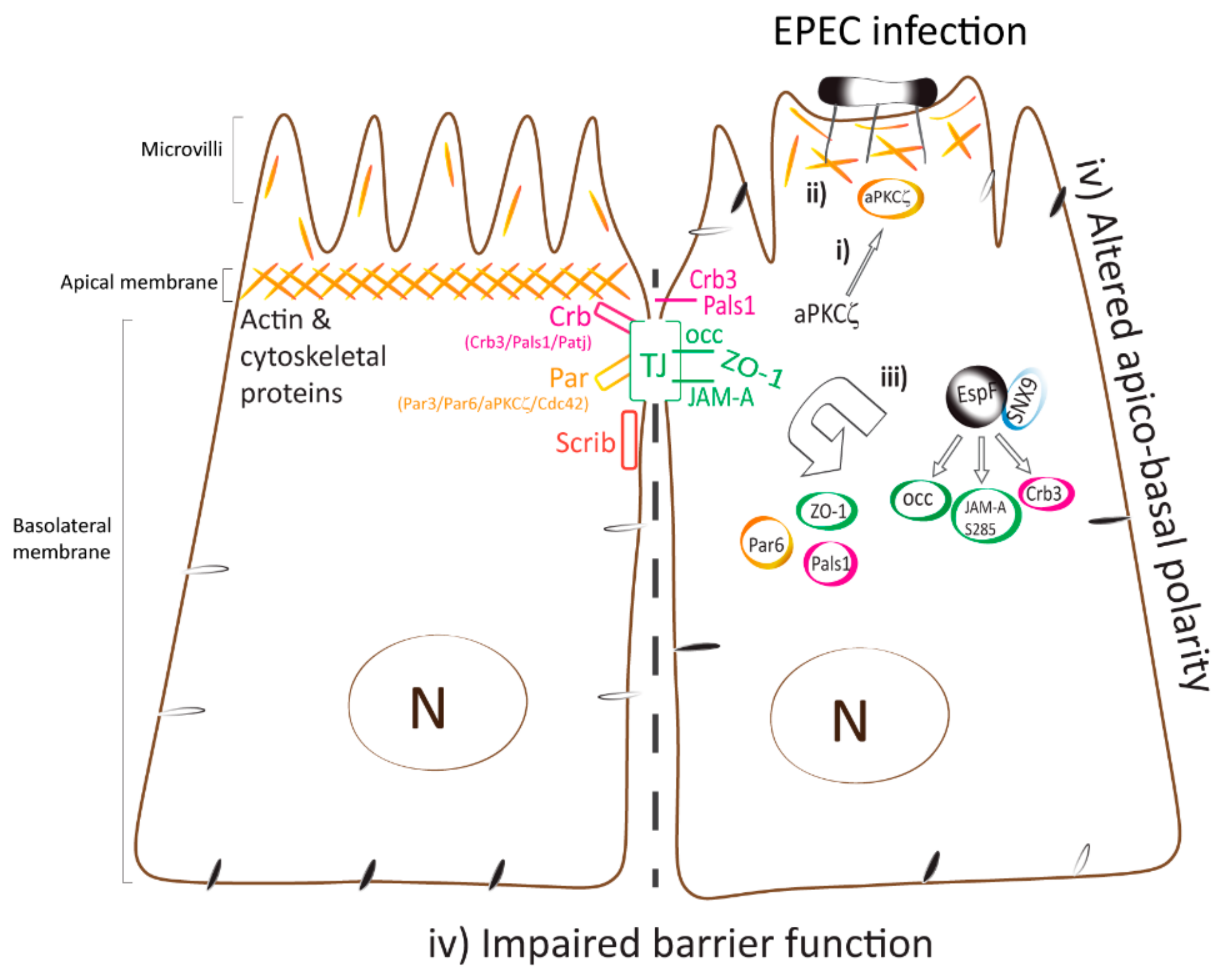
3. Discussion
4. Materials and Methods
4.1. Tissue Culture
4.2. Antibodies and Reagents
4.3. Bacterial Culture
4.4. Murine Infection
4.5. Immunofluorescence
4.6. Imaging
4.7. Western Blot Analysis
4.8. Generation of Tet-On System
4.9. Measurement of Transepithelial Electrical Resistance
4.10. PKC activity Assays
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| aPKCζ | Atypical protein kinase C zeta |
| EPEC | Enteropathogenic Escherichia coli |
| F-actin | Filamentous actin |
| JAM-A S285 | Junctional adhesion molecule-A phosphorylated at Serine 285 |
| JAM-A Y280 | Junctional adhesion molecule-A phosphorylated at Tyrosine 280 |
| p-aPKCζ-T410 | aPKCζ phosphorylated at Threonine 410 |
| p-aPKCζ-T560 | aPKCζ phosphorylated at Threonine 560 |
| PS | aPKCζ pseudosubstrate inhibitor |
| SNX9 | Sorting nexin 9 |
| TJ | Tight junctions |
| TER | Transepithelial electrical resistance |
| ZO-1 | Zonula Occludens -1 |
References
- Pearson, J.S.; Giogha, C.; Wong Fok Lung, T.; Hartland, E.L. The genetics of enteropathogenic escherichia coli virulence. Annu. Rev. Genet. 2016, 50, 493–513. [Google Scholar] [CrossRef]
- McNamara, B.P.; Koutsouris, A.; O’Connell, C.B.; Nougayrede, J.P.; Donnenberg, M.S.; Hecht, G. Translocated EspF protein from enteropathogenic escherichia coli disrupts host intestinal barrier function. J. Clin. Invest. 2001, 107, 621–629. [Google Scholar] [CrossRef]
- Muza-Moons, M.M.; Schneeberger, E.E.; Hecht, G.A. Enteropathogenic escherichia coli infection leads to appearance of aberrant tight junctions strands in the lateral membrane of intestinal epithelial cells. Cell. Microbiol. 2004, 6, 783–793. [Google Scholar] [CrossRef]
- Tsukita, S.; Tanaka, H.; Tamura, A. The claudins: From Tight junctions to biological systems. Trends Biochem. Sci. 2019, 44, 141–152. [Google Scholar] [CrossRef]
- Van Itallie, C.M.; Anderson, J.M. Architecture of tight junctions and principles of molecular composition. Semin. Cell Dev. Biol. 2014, 36, 157–165. [Google Scholar] [CrossRef]
- Wen, W.; Zhang, M. Protein complex assemblies in epithelial cell polarity and asymmetric cell division. J. Mol. Biol. 2018, 430, 3504–3520. [Google Scholar] [CrossRef]
- Rodriguez-Boulan, E.; Macara, I.G. Organization and execution of the epithelial polarity programme. Nat. Rev. Mol. Cell Biol. 2014, 15, 225–242. [Google Scholar] [CrossRef]
- Hurd, T.W.; Gao, L.; Roh, M.H.; Macara, I.G.; Margolis, B. Direct interaction of two polarity complexes implicated in epithelial tight junction assembly. Nat. Cell Biol. 2003, 5, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Roh, M.H.; Fan, S.; Liu, C.J.; Margolis, B. The Crumbs3-Pals1 complex participates in the establishment of polarity in mammalian epithelial cells. J. Cell. Sci. 2003, 116, 2895–2906. [Google Scholar] [CrossRef] [PubMed]
- Fogg, V.C.; Liu, C.J.; Margolis, B. Multiple regions of Crumbs3 are required for tight junction formation in MCF10A Cells. J. Cell. Sci. 2005, 118, 2859–2869. [Google Scholar] [CrossRef] [PubMed]
- Straight, S.W.; Shin, K.; Fogg, V.C.; Fan, S.; Liu, C.J.; Roh, M.; Margolis, B. Loss of PALS1 expression leads to tight junction and polarity defects. Mol. Biol. Cell 2004, 15, 1981–1990. [Google Scholar] [CrossRef] [PubMed]
- Tilston-Lunel, A.M.; Haley, K.E.; Schlecht, N.F.; Wang, Y.; Chatterton, A.L.; Moleirinho, S.; Watson, A.; Hundal, H.S.; Prystowsky, M.B.; Gunn-Moore, F.J.; et al. Crumbs 3b promotes tight junctions in an Ezrin-dependent manner in mammalian cells. J. Mol. Cell. Biol. 2016, 8, 439–455. [Google Scholar] [CrossRef] [PubMed]
- Nagai-Tamai, Y.; Mizuno, K.; Hirose, T.; Suzuki, A.; Ohno, S. Regulated protein-protein interaction between aPKC and PAR-3 plays an essential role in the polarization of epithelial cells. Genes Cells 2002, 7, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Yamanaka, T.; Hirose, T.; Manabe, N.; Mizuno, K.; Shimizu, M.; Akimoto, K.; Izumi, Y.; Ohnishi, T.; Ohno, S. Atypical protein kinase c is involved in the evolutionarily conserved par protein complex and plays a critical role in establishing epithelia-specific junctional structures. J. Cell Biol. 2001, 152, 1183–1196. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Macara, I.G. Par-3 controls tight junction assembly through the Rac exchange factor Tiam1. Nat. Cell Biol. 2005, 7, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, T.; Horikoshi, Y.; Suzuki, A.; Sugiyama, Y.; Kitamura, K.; Maniwa, R.; Nagai, Y.; Yamashita, A.; Hirose, T.; Ishikawa, H.; et al. PAR-6 regulates aPKC activity in a novel way and mediates cell-cell contact-induced formation of the epithelial junctional complex. Genes Cells 2001, 6, 721–731. [Google Scholar] [CrossRef]
- Jain, S.; Suzuki, T.; Seth, A.; Samak, G.; Rao, R. Protein kinase czeta phosphorylates occludin and promotes assembly of epithelial tight junctions. Biochem. J. 2011, 437, 289–299. [Google Scholar] [CrossRef]
- Qin, Y.; Capaldo, C.; Gumbiner, B.M.; Macara, I.G. The mammalian scribble polarity protein regulates epithelial cell adhesion and migration through E-cadherin. J. Cell Biol. 2005, 171, 1061–1071. [Google Scholar] [CrossRef]
- Stucke, V.M.; Timmerman, E.; Vandekerckhove, J.; Gevaert, K.; Hall, A. The MAGUK protein MPP7 binds to the polarity protein hDlg1 and facilitates epithelial tight junction formation. Mol. Biol. Cell 2007, 18, 1744–1755. [Google Scholar] [CrossRef]
- Ivanov, A.I.; Young, C.; Den Beste, K.; Capaldo, C.T.; Humbert, P.O.; Brennwald, P.; Parkos, C.A.; Nusrat, A. Tumor suppressor scribble regulates assembly of tight junctions in the intestinal epithelium. Am. J. Pathol. 2010, 176, 134–145. [Google Scholar] [CrossRef][Green Version]
- Elsum, I.A.; Martin, C.; Humbert, P.O. Scribble regulates an EMT polarity pathway through modulation of MAPK-ERK signaling to mediate junction formation. J. Cell. Sci. 2013, 126, 3990–3999. [Google Scholar] [CrossRef] [PubMed]
- Shifflett, D.E.; Clayburgh, D.R.; Koutsouris, A.; Turner, J.R.; Hecht, G.A. Enteropathogenic E. Coli disrupts tight junction barrier function and structure in Vivo. Lab. Invest. 2005, 85, 1308–1324. [Google Scholar] [CrossRef] [PubMed]
- Guttman, J.A.; Li, Y.; Wickham, M.E.; Deng, W.; Vogl, A.W.; Finlay, B.B. Attaching and effacing pathogen-induced tight junction disruption in Vivo. Cell. Microbiol. 2006, 8, 634–645. [Google Scholar] [CrossRef] [PubMed]
- Dean, P.; Kenny, B. Intestinal barrier dysfunction by Enteropathogenic Escherichia Coli is mediated by two effector molecules and a bacterial surface protein. Mol. Microbiol. 2004, 54, 665–675. [Google Scholar] [CrossRef]
- Singh, A.P.; Aijaz, S. Generation of a MDCK cell line with constitutive expression of the Enteropathogenic E. Coli effector protein map as an in Vitro model of pathogenesis. Bioengineered 2015, 6, 335–341. [Google Scholar] [CrossRef]
- Thanabalasuriar, A.; Koutsouris, A.; Weflen, A.; Mimee, M.; Hecht, G.; Gruenheid, S. The bacterial virulence factor NleA is required for the disruption of intestinal tight junctions by Enteropathogenic Escherichia Coli. Cell. Microbiol. 2010, 12, 31–41. [Google Scholar] [CrossRef]
- Tomson, F.L.; Viswanathan, V.K.; Kanack, K.J.; Kanteti, R.P.; Straub, K.V.; Menet, M.; Kaper, J.B.; Hecht, G. Enteropathogenic Escherichia Coli EspG disrupts microtubules and in conjunction with Orf3 enhances perturbation of the tight junction barrier. Mol. Microbiol. 2005, 56, 447–464. [Google Scholar] [CrossRef]
- Matsuzawa, T.; Kuwae, A.; Abe, A. Enteropathogenic Escherichia Coli Type III effectors EspG and EspG2 alter epithelial paracellular permeability. Infect. Immun. 2005, 73, 6283–6289. [Google Scholar] [CrossRef]
- Glotfelty, L.G.; Zahs, A.; Hodges, K.; Shan, K.; Alto, N.M.; Hecht, G.A. Enteropathogenic E. Coli effectors EspG1/G2 disrupt microtubules, contribute to tight junction perturbation and inhibit restoration. Cell. Microbiol. 2014, 16, 1767–1783. [Google Scholar] [CrossRef]
- Thanabalasuriar, A.; Kim, J.; Gruenheid, S. The inhibition of COPII trafficking is important for intestinal epithelial tight junction disruption during Enteropathogenic Escherichia Coli and Citrobacter Rodentium infection. Microbes Infect. 2013, 15, 738–744. [Google Scholar] [CrossRef]
- Muza-Moons, M.M.; Koutsouris, A.; Hecht, G. Disruption of cell polarity by Enteropathogenic Escherichia Coli enables basolateral membrane proteins to migrate apically and to potentiate physiological consequences. Infect. Immun. 2003, 71, 7069–7078. [Google Scholar] [CrossRef] [PubMed]
- Tapia, R.; Kralicek, S.E.; Hecht, G.A. EPEC Effector EspF Promotes Crumbs3 Endocytosis and Disrupts Epithelial Cell Polarity. Cell. Microbiol. 2017, 19, e12757. [Google Scholar] [CrossRef] [PubMed]
- Alto, N.M.; Weflen, A.W.; Rardin, M.J.; Yarar, D.; Lazar, C.S.; Tonikian, R.; Koller, A.; Taylor, S.S.; Boone, C.; Sidhu, S.S.; et al. The type III effector EspF coordinates membrane trafficking by the spatiotemporal activation of two eukaryotic signaling pathways. J. Cell Biol. 2007, 178, 1265–1278. [Google Scholar] [CrossRef] [PubMed]
- Peralta-Ramirez, J.; Hernandez, J.M.; Manning-Cela, R.; Luna-Munoz, J.; Garcia-Tovar, C.; Nougayrede, J.P.; Oswald, E.; Navarro-Garcia, F. EspF interacts with nucleation-promoting factors to recruit junctional proteins into pedestals for pedestal maturation and disruption of paracellular permeability. Infect. Immun. 2008, 76, 3854–3868. [Google Scholar] [CrossRef] [PubMed]
- Marches, O.; Batchelor, M.; Shaw, R.K.; Patel, A.; Cummings, N.; Nagai, T.; Sasakawa, C.; Carlsson, S.R.; Lundmark, R.; Cougoule, C.; et al. EspF of Enteropathogenic Escherichia Coli binds sorting nexin 9. J. Bacteriol. 2006, 188, 3110–3115. [Google Scholar] [CrossRef]
- Viswanathan, V.K.; Lukic, S.; Koutsouris, A.; Miao, R.; Muza, M.M.; Hecht, G. Cytokeratin 18 interacts with the Enteropathogenic Escherichia Coli Secreted Protein F (EspF) and is redistributed after infection. Cell. Microbiol. 2004, 6, 987–997. [Google Scholar] [CrossRef]
- Nougayrede, J.P.; Foster, G.H.; Donnenberg, M.S. Enteropathogenic Escherichia Coli effector EspF Interacts with host protein Abcf2. Cell. Microbiol. 2007, 9, 680–693. [Google Scholar] [CrossRef]
- Kassa, E.G.; Zlotkin-Rivkin, E.; Friedman, G.; Ramachandran, R.P.; Melamed-Book, N.; Weiss, A.M.; Belenky, M.; Reichmann, D.; Breuer, W.; Pal, R.R.; et al. Enteropathogenic Escherichia Coli remodels host endosomes to promote endocytic turnover and breakdown of surface polarity. PLoS Pathog. 2019, 15, e1007851. [Google Scholar] [CrossRef]
- Weflen, A.W.; Alto, N.M.; Viswanathan, V.K.; Hecht, G.E. Coli secreted protein f promotes EPEC invasion of intestinal epithelial cells via an SNX9-Dependent mechanism. Cell. Microbiol. 2010, 12, 919–929. [Google Scholar] [CrossRef]
- Garber, J.J.; Mallick, E.M.; Scanlon, K.M.; Turner, J.R.; Donnenberg, M.S.; Leong, J.M.; Snapper, S.B. Attaching-and-Effacing pathogens exploit junction regulatory activities of N-WASP and SNX9 to disrupt the intestinal barrier. Cell. Mol. Gastroenterol. Hepatol. 2017, 5, 273–288. [Google Scholar] [CrossRef]
- Tomson, F.L.; Koutsouris, A.; Viswanathan, V.K.; Turner, J.R.; Savkovic, S.D.; Hecht, G. Differing roles of protein kinase C-Zeta in disruption of tight junction barrier by Enteropathogenic and Enterohemorrhagic Escherichia Coli. Gastroenterology 2004, 127, 859–869. [Google Scholar] [CrossRef] [PubMed]
- Savkovic, S.D.; Koutsouris, A.; Hecht, G. PKC zeta participates in activation of inflammatory response induced by Enteropathogenic E. Coli. Am. J. Physiol. Cell. Physiol. 2003, 285, C512–C521. [Google Scholar] [CrossRef] [PubMed]
- Keranen, L.M.; Dutil, E.M.; Newton, A.C. Protein Kinase C is regulated in Vivo by Three functionally distinct phosphorylations. Curr. Biol. 1995, 5, 1394–1403. [Google Scholar] [CrossRef]
- Tsutakawa, S.E.; Medzihradszky, K.F.; Flint, A.J.; Burlingame, A.L.; Koshland, D.E., Jr. Determination of in Vivo phosphorylation sites in Protein Kinase C. J. Biol. Chem. 1995, 270, 26807–26812. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Weight, C.M.; Luissint, A.C.; Hilgarth, R.S.; Brazil, J.C.; Ettel, M.; Nusrat, A.; Parkos, C.A. Role of JAM-A tyrosine phosphorylation in epithelial barrier dysfunction during intestinal inflammation. Mol. Biol. Cell 2019, 30, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Iden, S.; Misselwitz, S.; Peddibhotla, S.S.; Tuncay, H.; Rehder, D.; Gerke, V.; Robenek, H.; Suzuki, A.; Ebnet, K. aPKC Phosphorylates JAM-A at Ser285 to promote cell contact maturation and tight junction formation. J. Cell Biol. 2012, 196, 623–639. [Google Scholar] [CrossRef]
- Savkovic, S.D.; Koutsouris, A.; Hecht, G. Attachment of a noninvasive enteric pathogen, Enteropathogenic Escherichia Coli, to cultured human intestinal epithelial monolayers induces transmigration of neutrophils. Infect. Immun. 1996, 64, 4480–4487. [Google Scholar] [CrossRef]
- Savkovic, S.D.; Koutsouris, A.; Hecht, G. Activation of NF-kappaB in intestinal epithelial cells by Enteropathogenic Escherichia Coli. Am. J. Physiol. 1997, 273, C1160–C1167. [Google Scholar] [CrossRef]
- Czerucka, D.; Dahan, S.; Mograbi, B.; Rossi, B.; Rampal, P. Implication of mitogen-activated protein kinases in T84 cell responses to Enteropathogenic Escherichia Coli infection. Infect. Immun. 2001, 69, 1298–1305. [Google Scholar] [CrossRef]
- Young, J.C.; Clements, A.; Lang, A.E.; Garnett, J.A.; Munera, D.; Arbeloa, A.; Pearson, J.; Hartland, E.L.; Matthews, S.J.; Mousnier, A.; et al. The Escherichia Coli Effector EspJ Blocks Src Kinase activity Via Amidation and ADP Ribosylation. Nat. Commun. 2014, 5, 5887. [Google Scholar] [CrossRef]
- Pollard, D.J.; Berger, C.N.; So, E.C.; Yu, L.; Hadavizadeh, K.; Jennings, P.; Tate, E.W.; Choudhary, J.S.; Frankel, G. Broad-spectrum regulation of nonreceptor tyrosine kinases by the bacterial ADP-Ribosyltransferase EspJ. MBio 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Phillips, N.; Hayward, R.D.; Koronakis, V. Phosphorylation of the Enteropathogenic E. Coli receptor by the Src-Family Kinase C-Fyn triggers actin pedestal formation. Nat. Cell Biol. 2004, 6, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Hayward, R.D.; Hume, P.J.; Humphreys, D.; Phillips, N.; Smith, K.; Koronakis, V. Clustering transfers the translocated escherichia coli receptor into lipid rafts to stimulate reversible activation of C-Fyn. Cell. Microbiol. 2009, 11, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Sutton, S.E.; Wallenfang, A.J.; Orchard, R.C.; Wu, X.; Feng, Y.; Chai, J.; Alto, N.M. Structural insights into Host GTPase isoform selection by a Family of bacterial GEF mimics. Nat. Struct. Mol. Biol. 2009, 16, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Berger, C.N.; Crepin, V.F.; Jepson, M.A.; Arbeloa, A.; Frankel, G. The Mechanisms used by Enteropathogenic Escherichia Coli to control filopodia dynamics. Cell. Microbiol. 2009, 11, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Ugalde-Silva, P.; Navarro-Garcia, F. Coordinated transient interaction of ZO-1 and Afadin is required for pedestal maturation induced by EspF from Enteropathogenic Escherichia Coli. Microbiologyopen 2019, 8, e931. [Google Scholar] [CrossRef] [PubMed]
- Hurd, T.W.; Fan, S.; Liu, C.J.; Kweon, H.K.; Hakansson, K.; Margolis, B. Phosphorylation-dependent binding of 14-3-3 to the polarity protein Par3 regulates cell polarity in Mammalian Epithelia. Curr. Biol. 2003, 13, 2082–2090. [Google Scholar] [CrossRef]
- Devriese, S.; Van den Bossche, L.; Van Welden, S.; Holvoet, T.; Pinheiro, I.; Hindryckx, P.; De Vos, M.; Laukens, D. T84 monolayers are superior to Caco-2 as a model system of Colonocytes. Histochem. Cell Biol. 2017, 148, 85–93. [Google Scholar]
- Le Bivic, A.; Real, F.X.; Rodriguez-Boulan, E. Vectorial targeting of apical and basolateral plasma membrane proteins in a human adenocarcinoma epithelial cell line. Proc. Natl. Acad. Sci. USA 1989, 86, 9313–9317. [Google Scholar] [CrossRef]
- Yoo, B.K.; Yanda, M.K.; No, Y.R.; Yun, C.C. Human intestinal epithelial cell line SK-CO15 is a new model system to study Na(+)/H(+) exchanger 3. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G180–G188. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, X.; Guo, Y.; Xu, L.; Pei, D. Sorting nexin 33 induces mammalian cell micronucleated phenotype and actin polymerization by interacting with Wiskott-Aldrich syndrome protein. J. Biol. Chem. 2009, 284, 21659–21669. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kim, Y.; Lee, S.; Park, J.J.; Park, Z.Y.; Sun, W.; Kim, H.; Chang, S. SNX18 shares a redundant role with SNX9 and modulates endocytic trafficking at the plasma membrane. J. Cell. Sci. 2010, 123, 1742–1750. [Google Scholar] [CrossRef] [PubMed]
- Haberg, K.; Lundmark, R.; Carlsson, S.R. SNX18 is an SNX9 paralog that acts as a membrane Tubulator in AP-1-positive endosomal trafficking. J. Cell. Sci. 2008, 121, 1495–1505. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Yan, K.; Wan, C. Clever cooperation: Interactions between EspF and host proteins. Front. Microbiol. 2018, 9, 2831. [Google Scholar] [CrossRef]
- Badour, K.; McGavin, M.K.; Zhang, J.; Freeman, S.; Vieira, C.; Filipp, D.; Julius, M.; Mills, G.B.; Siminovitch, K.A. Interaction of the Wiskott-Aldrich syndrome protein with sorting Nexin 9 is required for CD28 endocytosis and cosignaling in T cells. Proc. Natl. Acad. Sci. USA 2007, 104, 1593–1598. [Google Scholar] [CrossRef]
- Merrifield, C.J.; Qualmann, B.; Kessels, M.M.; Almers, W. Neural Wiskott Aldrich Syndrome Protein (N-WASP) and the Arp2/3 complex are recruited to sites of clathrin-mediated endocytosis in cultured fibroblasts. Eur. J. Cell Biol. 2004, 83, 13–18. [Google Scholar] [CrossRef]
- Benesch, S.; Polo, S.; Lai, F.P.; Anderson, K.I.; Stradal, T.E.; Wehland, J.; Rottner, K. N-WASP deficiency impairs EGF internalization and Actin assembly at clathrin-coated pits. J. Cell. Sci. 2005, 118, 3103–3115. [Google Scholar] [CrossRef]
- Yarar, D.; Waterman-Storer, C.M.; Schmid, S.L. SNX9 couples actin assembly to phosphoinositide signals and is required for membrane remodeling during endocytosis. Dev. Cell. 2007, 13, 43–56. [Google Scholar] [CrossRef]
- Lundmark, R.; Carlsson, S.R. SNX9—A prelude to vesicle release. J. Cell. Sci. 2009, 122, 5–11. [Google Scholar] [CrossRef]
- Andreeva, A.Y.; Krause, E.; Muller, E.C.; Blasig, I.E.; Utepbergenov, D.I. Protein kinase C regulates the phosphorylation and cellular localization of occludin. J. Biol. Chem. 2001, 276, 38480–38486. [Google Scholar] [CrossRef]
- Aono, S.; Hirai, Y. Phosphorylation of claudin-4 is required for tight junction formation in a human keratinocyte cell line. Exp. Cell Res. 2008, 314, 3326–3339. [Google Scholar] [CrossRef] [PubMed]
- Sotillos, S.; Diaz-Meco, M.T.; Caminero, E.; Moscat, J.; Campuzano, S. DaPKC-dependent phosphorylation of crumbs is required for epithelial cell polarity in drosophila. J. Cell Biol. 2004, 166, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Hurov, J.B.; Watkins, J.L.; Piwnica-Worms, H. Atypical PKC phosphorylates PAR-1 kinases to regulate localization and activity. Curr. Biol. 2004, 14, 736–741. [Google Scholar] [CrossRef] [PubMed]
- Plant, P.J.; Fawcett, J.P.; Lin, D.C.; Holdorf, A.D.; Binns, K.; Kulkarni, S.; Pawson, T. A Polarity complex of mPar-6 and atypical PKC binds, phosphorylates and regulates mammalian Lgl. Nat. Cell Biol. 2003, 5, 301–308. [Google Scholar] [CrossRef]
- Tobias, I.S.; Newton, A.C. Protein scaffolds control localized protein kinase Czeta activity. J. Biol. Chem. 2016, 291, 13809–13822. [Google Scholar] [CrossRef]
- Chaki, S.P.; Barhoumi, R.; Rivera, G.M. Actin remodeling by Nck regulates endothelial lumen formation. Mol. Biol. Cell 2015, 26, 3047–3060. [Google Scholar] [CrossRef]
- Gruenheid, S.; DeVinney, R.; Bladt, F.; Goosney, D.; Gelkop, S.; Gish, G.D.; Pawson, T.; Finlay, B.B. Enteropathogenic E. Coli Tir binds Nck to initiate actin pedestal formation in host cells. Nat. Cell Biol. 2001, 3, 856–859. [Google Scholar] [CrossRef]
- Nieto-Pelegrin, E.; Kenny, B.; Martinez-Quiles, N. Nck adaptors, besides promoting N-WASP mediated actin-nucleation activity at pedestals, influence the cellular levels of Enteropathogenic Escherichia Coli Tir effector. Cell Adhes Migr. 2014, 8, 404–417. [Google Scholar] [CrossRef][Green Version]
- Worby, C.A.; Simonson-Leff, N.; Clemens, J.C.; Huddler, D., Jr.; Muda, M.; Dixon, J.E. Drosophila Ack targets its substrate, the sorting nexin DSH3PX1, to a protein complex involved in axonal guidance. J. Biol. Chem. 2002, 277, 9422–9428. [Google Scholar] [CrossRef]
- Worby, C.A.; Simonson-Leff, N.; Clemens, J.C.; Kruger, R.P.; Muda, M.; Dixon, J.E. The sorting nexin, DSH3PX1, connects the axonal guidance receptor, dscam, to the actin cytoskeleton. J. Biol. Chem. 2001, 276, 41782–41789. [Google Scholar] [CrossRef]
- Georgiou, M.; Marinari, E.; Burden, J.; Baum, B. Cdc42, Par6, and aPKC regulate Arp2/3-mediated endocytosis to control local adherens junction stability. Curr. Biol. 2008, 18, 1631–1638. [Google Scholar] [CrossRef] [PubMed]
- Wald, F.A.; Oriolo, A.S.; Mashukova, A.; Fregien, N.L.; Langshaw, A.H.; Salas, P.J. Atypical protein kinase C (Iota) activates Ezrin in the apical domain of intestinal epithelial cells. J. Cell. Sci. 2008, 121, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Rouven Bruckner, B.; Pietuch, A.; Nehls, S.; Rother, J.; Janshoff, A. Ezrin is a major regulator of membrane tension in epithelial cells. Sci. Rep. 2015, 5, 14700. [Google Scholar] [CrossRef] [PubMed]
- Crawley, S.W.; Mooseker, M.S.; Tyska, M.J. Shaping the intestinal brush border. J. Cell Biol. 2014, 207, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Tsai, F.C.; Bertin, A.; Bousquet, H.; Manzi, J.; Senju, Y.; Tsai, M.C.; Picas, L.; Miserey-Lenkei, S.; Lappalainen, P.; Lemichez, E.; et al. Ezrin enrichment on curved membranes requires a specific conformation or interaction with a curvature-sensitive partner. Elife 2018, 7, e37262. [Google Scholar] [CrossRef] [PubMed]
- Simonovic, I.; Arpin, M.; Koutsouris, A.; Falk-Krzesinski, H.J.; Hecht, G. Enteropathogenic Escherichia Coli activates ezrin, which participates in disruption of tight junction barrier function. Infect. Immun. 2001, 69, 5679–5688. [Google Scholar] [CrossRef]
- Law, H.T.; Chua, M.; Moon, K.M.; Foster, L.J.; Guttman, J.A. Mass spectrometry-based proteomics identification of Enteropathogenic Escherichia Coli pedestal constituents. J. Proteome Res. 2015, 14, 2520–2527. [Google Scholar] [CrossRef]
- Campellone, K.G.; Giese, A.; Tipper, D.J.; Leong, J.M. A tyrosine-phosphorylated 12-amino-acid sequence of Enteropathogenic Escherichia Coli Tir Binds the host adaptor protein Nck and is required for Nck localization to actin pedestals. Mol. Microbiol. 2002, 43, 1227–1241. [Google Scholar] [CrossRef]
- Kalman, D.; Weiner, O.D.; Goosney, D.L.; Sedat, J.W.; Finlay, B.B.; Abo, A.; Bishop, J.M. Enteropathogenic E. Coli acts through WASP and Arp2/3 complex to form actin pedestals. Nat. Cell Biol. 1999, 1, 389–391. [Google Scholar] [CrossRef]
- Lommel, S.; Benesch, S.; Rottner, K.; Franz, T.; Wehland, J.; Kuhn, R. Actin pedestal formation by Enteropathogenic Escherichia Coli and intracellular motility of Shigella Flexneri are abolished in N-WASP-defective cells. EMBO Rep. 2001, 2, 850–857. [Google Scholar] [CrossRef]
- Kralicek, S.E.; Nguyen, M.; Rhee, K.J.; Tapia, R.; Hecht, G. EPEC NleH1 is significantly more effective in reversing colitis and reducing mortality than NleH2 via differential effects on host signaling pathways. Lab. Invest. 2018, 98, 477–488. [Google Scholar] [CrossRef] [PubMed]
- McNamara, B.P.; Donnenberg, M.S. A novel proline-rich protein, EspF, is secreted from Enteropathogenic Escherichia Coli via the Type III export pathway. FEMS Microbiol. Lett. 1998, 166, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Kenny, B.; Jepson, M. Targeting of an Enteropathogenic Escherichia Coli (EPEC) effector protein to host mitochondria. Cell. Microbiol. 2000, 2, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Rhee, K.J.; Cheng, H.; Harris, A.; Morin, C.; Kaper, J.B.; Hecht, G. Determination of spatial and temporal colonization of Enteropathogenic E. Coli and Enterohemorrhagic E. Coli in mice using bioluminescent in Vivo imaging. Gut Microbes 2011, 2, 34–41. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tapia, R.; Kralicek, S.E.; Hecht, G.A. Enteropathogenic Escherichia coli (EPEC) Recruitment of PAR Polarity Protein Atypical PKCζ to Pedestals and Cell–Cell Contacts Precedes Disruption of Tight Junctions in Intestinal Epithelial Cells. Int. J. Mol. Sci. 2020, 21, 527. https://doi.org/10.3390/ijms21020527
Tapia R, Kralicek SE, Hecht GA. Enteropathogenic Escherichia coli (EPEC) Recruitment of PAR Polarity Protein Atypical PKCζ to Pedestals and Cell–Cell Contacts Precedes Disruption of Tight Junctions in Intestinal Epithelial Cells. International Journal of Molecular Sciences. 2020; 21(2):527. https://doi.org/10.3390/ijms21020527
Chicago/Turabian StyleTapia, Rocio, Sarah E. Kralicek, and Gail A. Hecht. 2020. "Enteropathogenic Escherichia coli (EPEC) Recruitment of PAR Polarity Protein Atypical PKCζ to Pedestals and Cell–Cell Contacts Precedes Disruption of Tight Junctions in Intestinal Epithelial Cells" International Journal of Molecular Sciences 21, no. 2: 527. https://doi.org/10.3390/ijms21020527
APA StyleTapia, R., Kralicek, S. E., & Hecht, G. A. (2020). Enteropathogenic Escherichia coli (EPEC) Recruitment of PAR Polarity Protein Atypical PKCζ to Pedestals and Cell–Cell Contacts Precedes Disruption of Tight Junctions in Intestinal Epithelial Cells. International Journal of Molecular Sciences, 21(2), 527. https://doi.org/10.3390/ijms21020527