Components from the Human c-myb Transcriptional Regulation System Reactivate Epigenetically Repressed Transgenes


Abstract
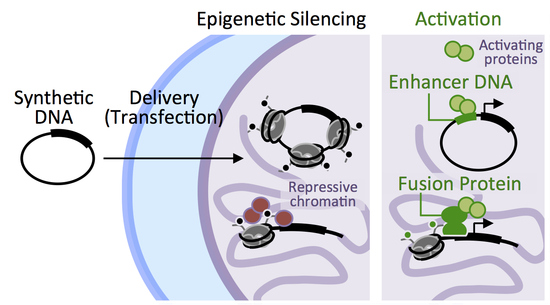
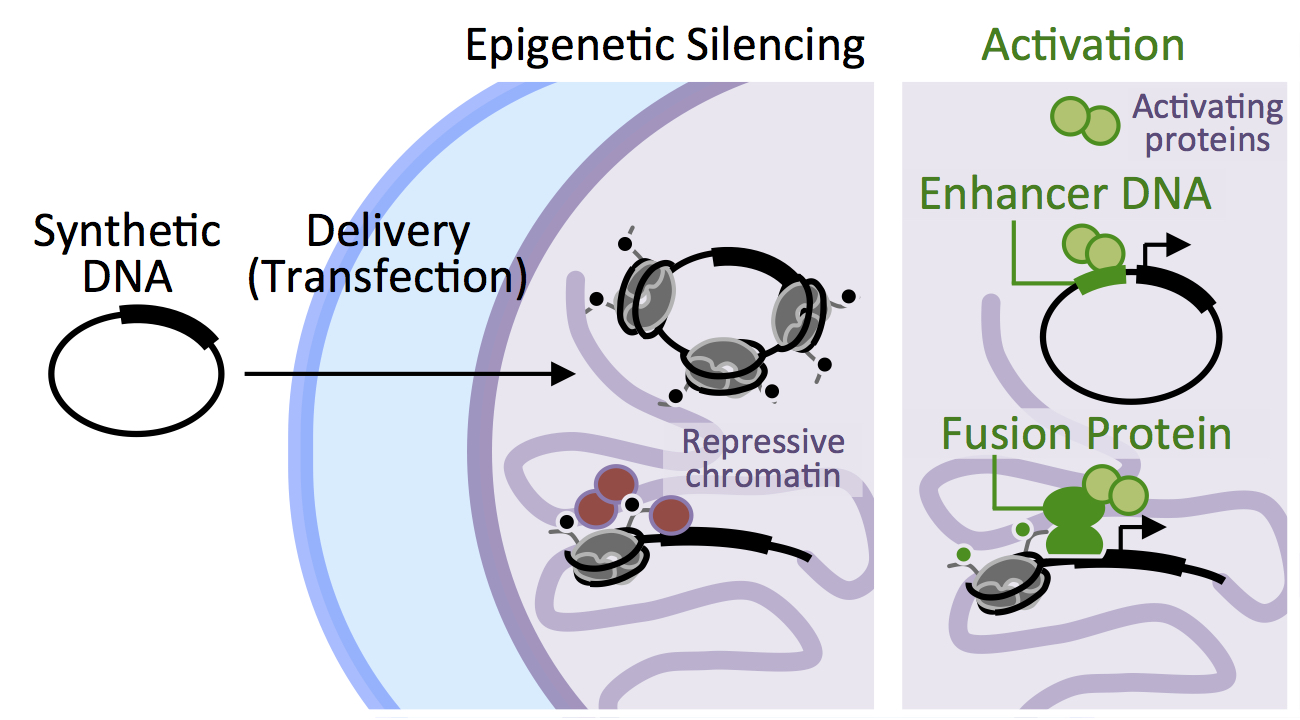{kind=link}
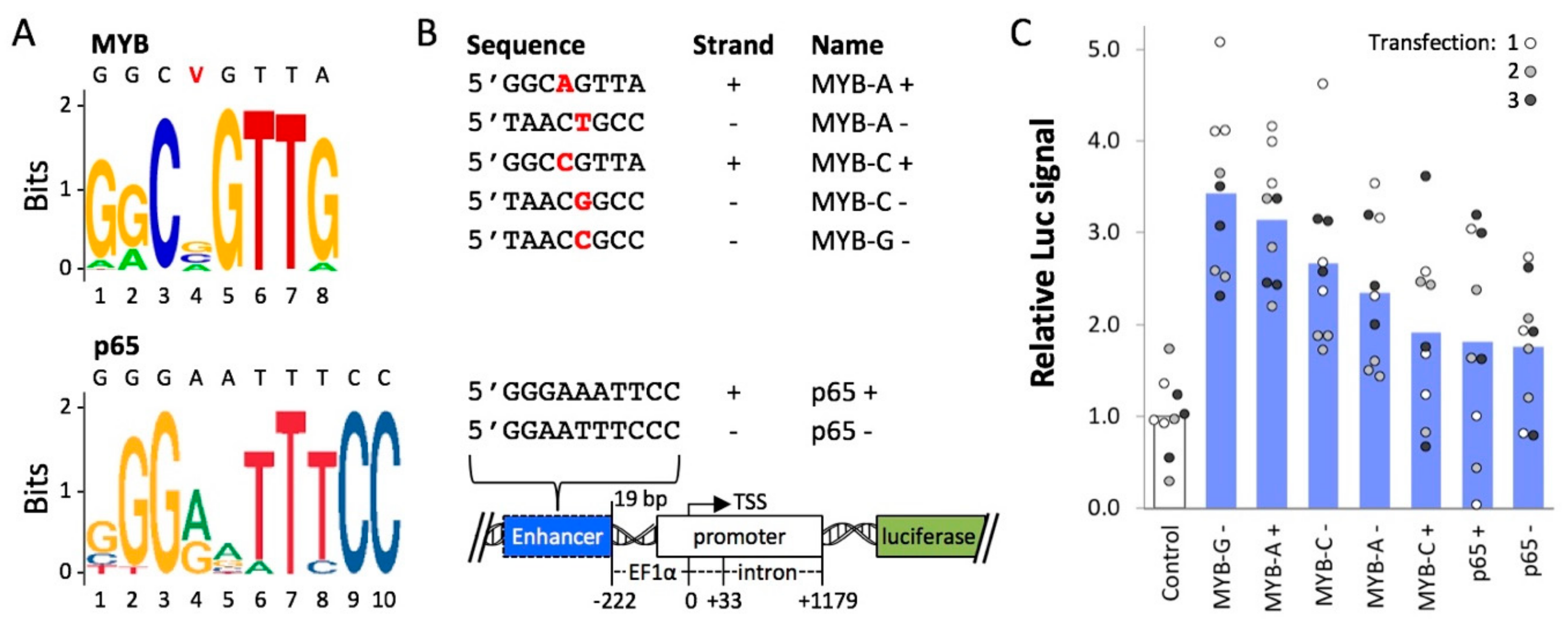{kind=link}
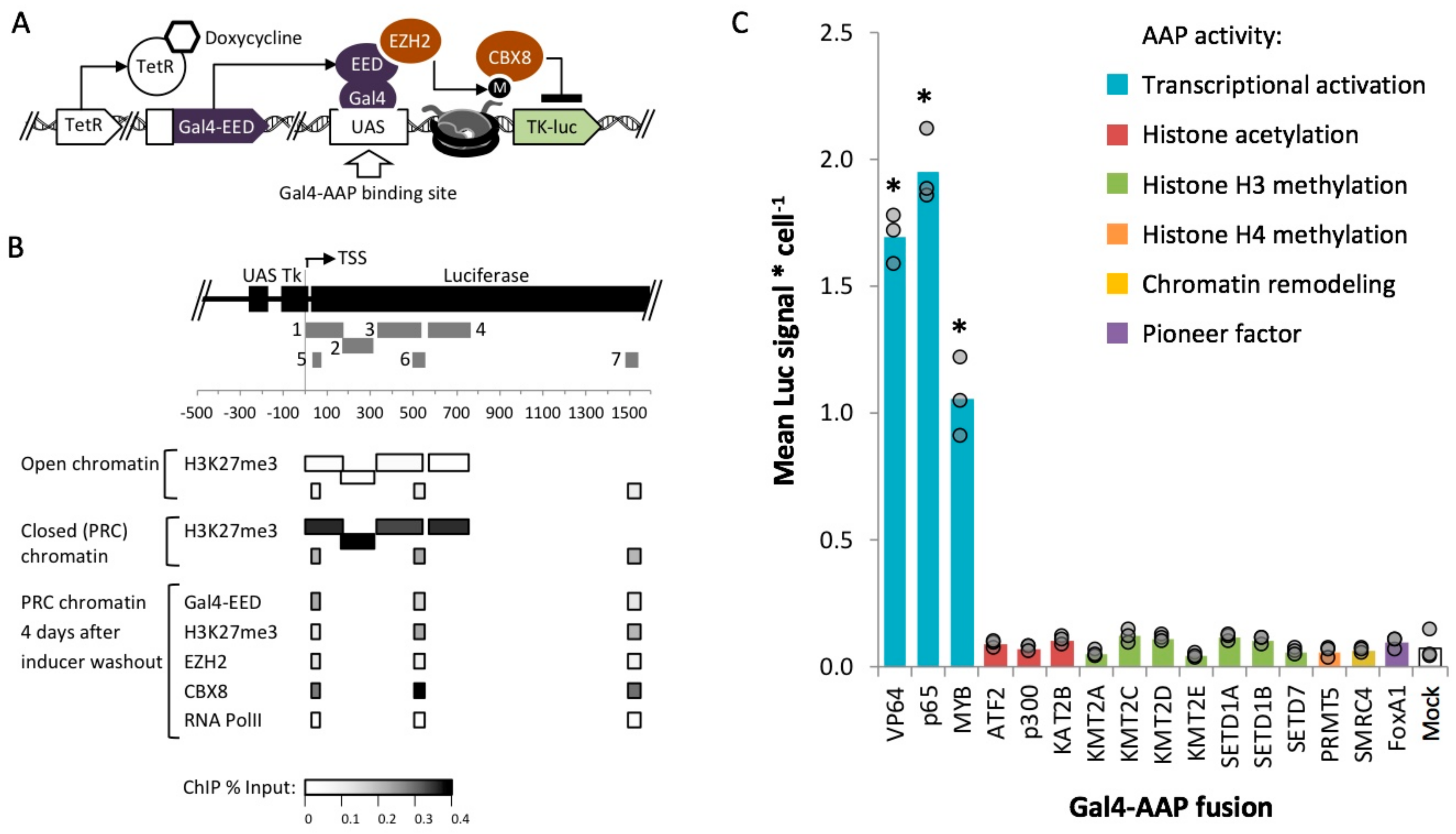{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Identification of Activation Associated Proteins
2.2. Cis-Regulatory Elements Recognized by Transcriptional Activators p65 and MYB Enhance Expression from an Extra-Chromosomal Transgene
2.3. Identification of Fusion Activators with Robust Activity within Polycomb Heterochromatin
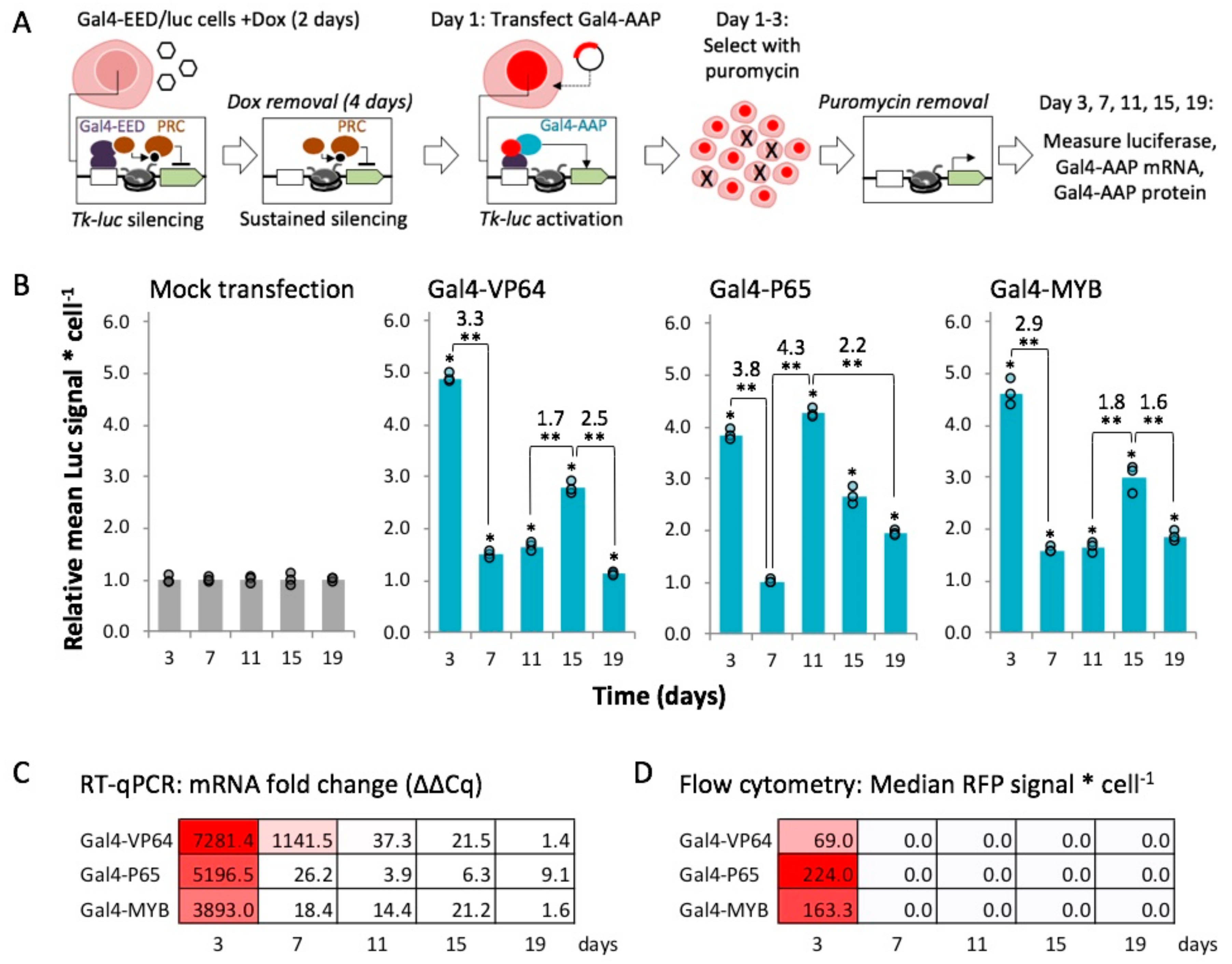
2.4. Gal4-MYB-Induced Activation at Tk-Luciferase Resists Complete re-Silencing over Time
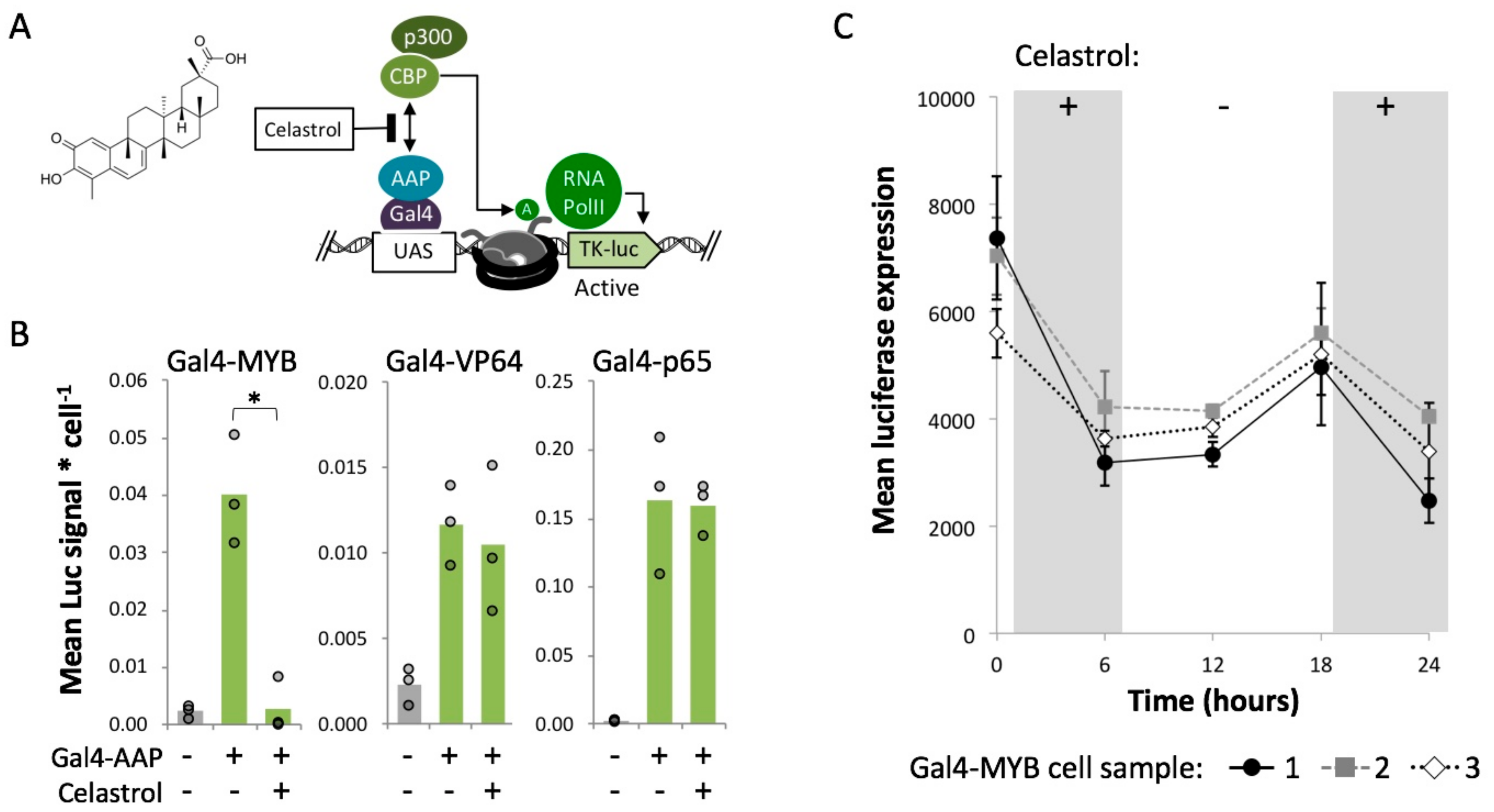
2.5. MYB-Mediated Activation within Closed Chromatin Requires Interaction with a Histone Acetyltransferase
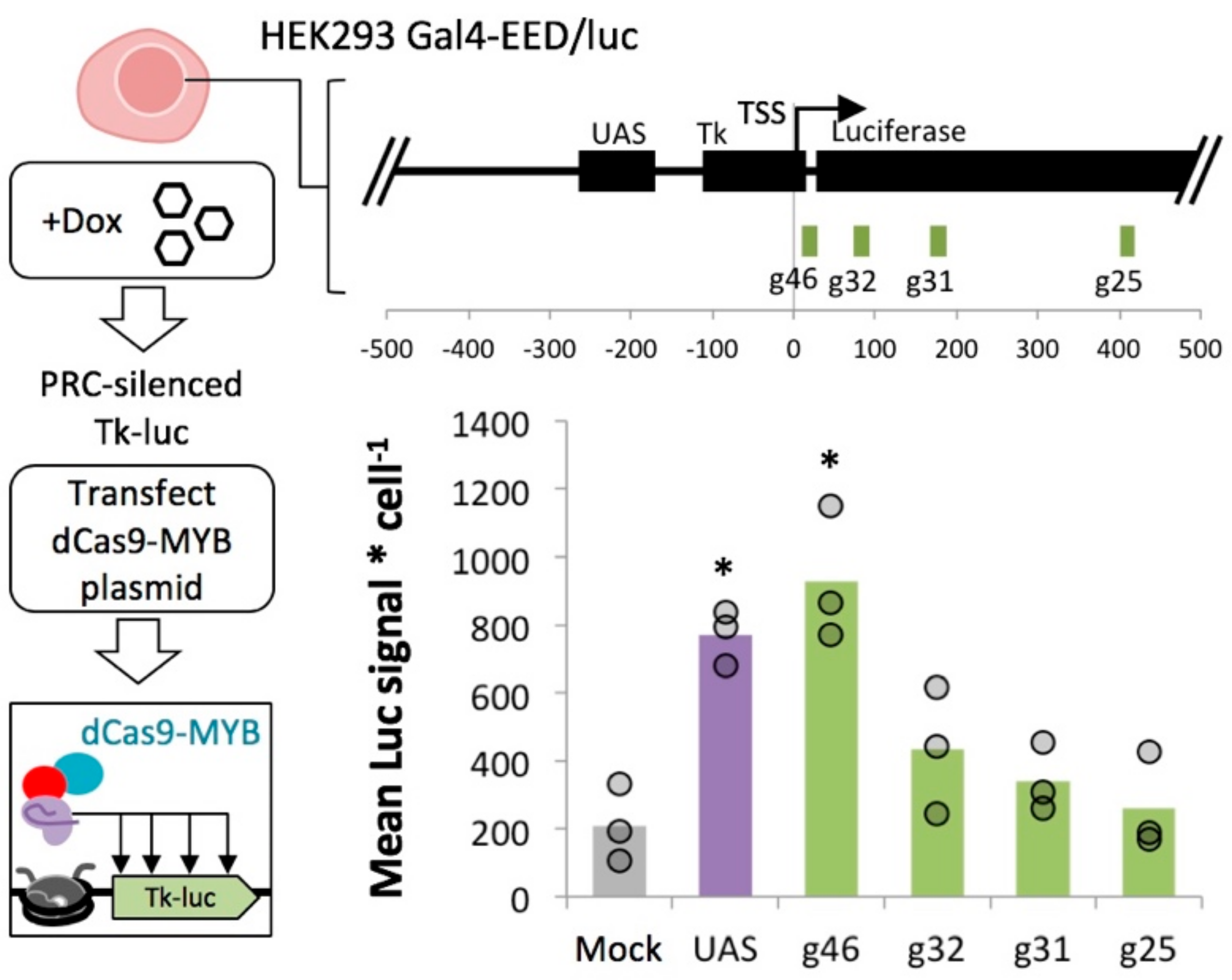
2.6. MYB-Mediated Activation in Polycomb Heterochromatin Relies upon Proximity to the Transcriptional Start Site
3. Discussion
4. Materials and Methods
4.1. Construction and Testing of Plasmids Containing MYB- and p65 Motifs
4.2. Construction of MV14 and Gal4-AAP Plasmids
4.3. Cell Culturing and Transfections
4.4. Luciferase Assays
4.5. RT-qPCR
4.6. Flow Cytometry
4.7. Construction of dCas9-MYB and Design of sgRNAs
4.8. Celastrol Treatments
4.9. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAP | activation associated protein |
| CMV | cytomegalovirus |
| CR | chromatin remodeler |
| Gal4 | Gal4 DNA binding domain |
| HAT | histone acetyltransferase |
| NLS | nuclear localization signal |
| ORF | open reading frame |
| PF | pioneer factor |
| PolII | RNA polymerase II |
| PRC | Polycomb repressive complex |
| TAD | transcriptional activation domain |
| UAS | upstream activation sequence |
References
- Brooks, A.R.; Harkins, R.N.; Wang, P.; Qian, H.S.; Liu, P.; Rubanyi, G.M. Transcriptional silencing is associated with extensive methylation of the CMV promoter following adenoviral gene delivery to muscle. J. Gene Med. 2004, 6, 395–404. [Google Scholar] [CrossRef]
- Suzuki, M.; Cerullo, V.; Bertin, T.K.; Cela, R.; Clarke, C.; Guenther, M.; Brunetti-Pierri, N.; Lee, B. MyD88-dependent silencing of transgene expression during the innate and adaptive immune response to helper-dependent adenovirus. Hum. Gene Ther. 2010, 21, 325–336. [Google Scholar] [CrossRef]
- Leung, D.C.; Lorincz, M.C. Silencing of endogenous retroviruses: When and why do histone marks predominate? Trends Biochem. Sci. 2012, 37, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Ross, P.J.; Kennedy, M.A.; Parks, R.J. Host cell detection of noncoding stuffer DNA contained in helper-dependent adenovirus vectors leads to epigenetic repression of transgene expression. J. Virol. 2009, 83, 8409–8417. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J. Silencing and variegation of gammaretrovirus and lentivirus vectors. Hum. Gene Ther. 2005, 16, 1241–1246. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Liu, F.; Xiong, Z.; Qi, R.; Luo, Z.; Gong, X.; Nie, Q.; Sun, Q.; Liu, Y.-F.; Qing, W.; et al. Heterochromatin protects retinal pigment epithelium cells from oxidative damage by silencing p53 target genes. Proc. Natl. Acad. Sci. USA 2018, 115, E3987–E3995. [Google Scholar] [CrossRef] [PubMed]
- Erhardt, S.; Lyko, F.; Ainscough, J.F.-X.; Surani, M.A.; Paro, R. Polycomb-group proteins are involved in silencing processes caused by a transgenic element from the murine imprinted H19/Igf2 region in Drosophila. Dev. Genes Evol. 2003, 213, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Dufourt, J.; Brasset, E.; Desset, S.; Pouchin, P.; Vaury, C. Polycomb group-dependent, heterochromatin protein 1-independent, chromatin structures silence retrotransposons in somatic tissues outside ovaries. DNA Res. 2011, 18, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Otte, A.P.; Kwaks, T.H.J. Gene repression by Polycomb group protein complexes: A distinct complex for every occasion? Curr. Opin. Genet. Dev. 2003, 13, 448–454. [Google Scholar] [CrossRef]
- Johansen, J.; Tornøe, J.; Møller, A.; Johansen, T.E. Increased in vitro and in vivo transgene expression levels mediated through cis-acting elements. J. Gene Med. 2003, 5, 1080–1089. [Google Scholar] [CrossRef]
- Cheng, J.K.; Alper, H.S. Transcriptomics-Guided Design of Synthetic Promoters for a Mammalian System. ACS Synth. Biol. 2016, 5, 1455–1465. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, D.; Patel, K.; Hall, M.; Elmer, J. Enhancement of transgene expression by nuclear transcription factor Y and CCCTC-binding factor. Biotechnol. Prog. 2018, 34, 1581–1588. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Guo, X.; Li, Y.-M.; Wang, X.-Y.; Yang, X.-J.; Wang, Y.-F.; Wang, T.-Y. Enhanced transgene expression using cis-acting elements combined with the EF1 promoter in a mammalian expression system. Eur. J. Pharm. Sci. 2018, 123, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.L.; Katsoupi, P.; Tseveleki, V.; Taoufik, E. Bioinformatically Informed Design of Synthetic Mammalian Promoters. Methods Mol. Biol. 2017, 1651, 93–112. [Google Scholar] [PubMed]
- Saxena, P.; Bojar, D.; Fussenegger, M. Design of Synthetic Promoters for Gene Circuits in Mammalian Cells. In Mammalian Synthetic Promoters; Humana Press: New York, NY, USA, 2017; pp. 263–273. [Google Scholar]
- Morita, S.; Noguchi, H.; Horii, T.; Nakabayashi, K.; Kimura, M.; Okamura, K.; Sakai, A.; Nakashima, H.; Hata, K.; Nakashima, K.; et al. Targeted DNA demethylation in vivo using dCas9-peptide repeat and scFv-TET1 catalytic domain fusions. Nat. Biotechnol. 2016, 34, 1060–1065. [Google Scholar] [CrossRef]
- Liu, P.; Chen, M.; Liu, Y.; Qi, L.S.; Ding, S. CRISPR-Based Chromatin Remodeling of the Endogenous Oct4 or Sox2 Locus Enables Reprogramming to Pluripotency. Cell Stem Cell 2018, 22, 252–261.e4. [Google Scholar] [CrossRef]
- Hilton, I.B.; D’Ippolito, A.M.; Vockley, C.M.; Thakore, P.I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef]
- Chavez, A.; Scheiman, J.; Vora, S.; Pruitt, B.W.; Tuttle, M.; Iyer, E.; Kiani, S.; Guzman, C.D.; Wiegand, D.J.; Ter-Ovanesyan, D.; et al. Highly-efficient Cas9-mediated transcriptional programming 2014. Nat. Methods 2015, 12, 326–328. [Google Scholar] [CrossRef]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef]
- Huang, Y.-H.; Su, J.; Lei, Y.; Brunetti, L.; Gundry, M.C.; Zhang, X.; Jeong, M.; Li, W.; Goodell, M.A. DNA epigenome editing using CRISPR-Cas SunTag-directed DNMT3A. Genome Biol. 2017, 18, 176. [Google Scholar] [CrossRef]
- Black, J.B.; Adler, A.F.; Wang, H.-G.; D’Ippolito, A.M.; Hutchinson, H.A.; Reddy, T.E.; Pitt, G.S.; Leong, K.W.; Gersbach, C.A. Targeted Epigenetic Remodeling of Endogenous Loci by CRISPR/Cas9-Based Transcriptional Activators Directly Converts Fibroblasts to Neuronal Cells. Cell Stem Cell 2016, 19, 406–414. [Google Scholar] [CrossRef]
- Gao, X.; Tsang, J.C.H.; Gaba, F.; Wu, D.; Lu, L.; Liu, P. Comparison of TALE designer transcription factors and the CRISPR/dCas9 in regulation of gene expression by targeting enhancers. Nucleic Acids Res. 2014, 42, e155. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yin, C.; Zhang, T.; Li, F.; Yang, W.; Kaminski, R.; Fagan, P.R.; Putatunda, R.; Young, W.-B.; Khalili, K.; et al. CRISPR/gRNA-directed synergistic activation mediator (SAM) induces specific, persistent and robust reactivation of the HIV-1 latent reservoirs. Sci. Rep. 2015, 5, 16277. [Google Scholar] [CrossRef] [PubMed]
- Daer, R.M.; Cutts, J.P.; Brafman, D.A.; Haynes, K.A. The Impact of Chromatin Dynamics on Cas9-Mediated Genome Editing in Human Cells. ACS Synth. Biol. 2017, 6, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Clapier, C.R.; Iwasa, J.; Cairns, B.R.; Peterson, C.L. Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat. Rev. Mol. Cell Biol. 2017, 18, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Zaret, K.S.; Carroll, J.S. Pioneer transcription factors: Establishing competence for gene expression. Genes Dev. 2011, 25, 2227–2241. [Google Scholar] [CrossRef] [PubMed]
- Magnani, L.; Eeckhoute, J.; Lupien, M. Pioneer factors: Directing transcriptional regulators within the chromatin environment. Trends Genet. 2011, 27, 465–474. [Google Scholar] [CrossRef]
- Kramer, B.P.; Viretta, A.U.; Daoud-El-Baba, M.; Aubel, D.; Weber, W.; Fussenegger, M. An engineered epigenetic transgene switch in mammalian cells. Nat. Biotechnol. 2004, 22, 867–870. [Google Scholar] [CrossRef]
- Kwaks, T.H.J.; Sewalt, R.G.A.B.; Van Blokland, R.; Siersma, T.J.; Kasiem, M.; Kelder, A.; Otte, A.P. Targeting of a histone acetyltransferase domain to a promoter enhances protein expression levels in mammalian cells. J. Biotechnol. 2005, 115, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Santillan, D.A.; Theisler, C.M.; Ryan, A.S.; Popovic, R.; Stuart, T.; Zhou, M.-M.; Alkan, S.; Zeleznik-Le, N.J. Bromodomain and histone acetyltransferase domain specificities control mixed lineage leukemia phenotype. Cancer Res. 2006, 66, 10032–10039. [Google Scholar] [CrossRef]
- Cano-Rodriguez, D.; Gjaltema, R.A.F.; Jilderda, L.J.; Jellema, P.; Dokter-Fokkens, J.; Ruiters, M.H.J.; Rots, M.G. Writing of H3K4Me3 overcomes epigenetic silencing in a sustained but context-dependent manner. Nat. Commun. 2016, 7, 12284. [Google Scholar] [CrossRef] [PubMed]
- McGinty, R.K.; Kim, J.; Chatterjee, C.; Roeder, R.G.; Muir, T.W. Chemically ubiquitylated histone H2B stimulates hDot1L-mediated intranucleosomal methylation. Nature 2008, 453, 812–816. [Google Scholar] [CrossRef] [PubMed]
- O’Geen, H.; Ren, C.; Nicolet, C.M.; Perez, A.A.; Halmai, J.; Le, V.M.; Mackay, J.P.; Farnham, P.J.; Segal, D.J. dCas9-based epigenome editing suggests acquisition of histone methylation is not sufficient for target gene repression. Nucleic Acids Res. 2017, 45, 9901–9916. [Google Scholar] [CrossRef] [PubMed]
- Aloia, L.; Di Stefano, B.; Di Croce, L. Polycomb complexes in stem cells and embryonic development. Development 2013, 140, 2525–2534. [Google Scholar] [CrossRef]
- Poynter, S.T.; Kadoch, C. Polycomb and trithorax opposition in development and disease. Wiley Interdiscip. Rev. Dev. Biol. 2016, 5, 659–688. [Google Scholar] [CrossRef]
- Schuettengruber, B.; Bourbon, H.-M.; Di Croce, L.; Cavalli, G. Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell 2017, 171, 34–57. [Google Scholar] [CrossRef]
- Simon, J.A.; Kingston, R.E. Occupying chromatin: Polycomb mechanisms for getting to genomic targets, stopping transcriptional traffic, and staying put. Mol. Cell 2013, 49, 808–824. [Google Scholar] [CrossRef]
- Beerli, R.R.; Segal, D.J.; Dreier, B.; Barbas, C.F. 3rd Toward controlling gene expression at will: Specific regulation of the erbB-2/HER-2 promoter by using polydactyl zinc finger proteins constructed from modular building blocks. Proc. Natl. Acad. Sci. USA 1998, 95, 14628–14633. [Google Scholar] [CrossRef]
- Liu, P.Q.; Rebar, E.J.; Zhang, L.; Liu, Q.; Jamieson, A.C.; Liang, Y.; Qi, H.; Li, P.X.; Chen, B.; Mendel, M.C.; et al. Regulation of an endogenous locus using a panel of designed zinc finger proteins targeted to accessible chromatin regions. Activation of vascular endothelial growth factor A. J. Biol. Chem. 2001, 276, 11323–11334. [Google Scholar] [CrossRef]
- Weston, K.; Michael Bishop, J. Transcriptional activation by the v-myb oncogene and its cellular progenitor, c-myb. Cell 1989, 58, 85–93. [Google Scholar] [CrossRef]
- Vo, N.; Goodman, R.H. CREB-binding Protein and p300 in Transcriptional Regulation. J. Biol. Chem. 2001, 276, 13505–13508. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Rank, G.; Tan, Y.T.; Li, H.; Moritz, R.L.; Simpson, R.J.; Cerruti, L.; Curtis, D.J.; Patel, D.J.; Allis, C.D.; et al. PRMT5-mediated methylation of histone H4R3 recruits DNMT3A, coupling histone and DNA methylation in gene silencing. Nat. Struct. Mol. Biol. 2009, 16, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Antonysamy, S.; Bonday, Z.; Campbell, R.M.; Doyle, B.; Druzina, Z.; Gheyi, T.; Han, B.; Jungheim, L.N.; Qian, Y.; Rauch, C.; et al. Crystal structure of the human PRMT5:MEP50 complex. Proc. Natl. Acad. Sci. USA 2012, 109, 17960–17965. [Google Scholar] [CrossRef] [PubMed]
- Alver, B.H.; Kim, K.H.; Lu, P.; Wang, X.; Manchester, H.E.; Wang, W.; Haswell, J.R.; Park, P.J.; Roberts, C.W.M. The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nat. Commun. 2017, 8, 14648. [Google Scholar] [CrossRef]
- Clark, K.L.; Halay, E.D.; Lai, E.; Burley, S.K. Co-crystal structure of the HNF-3/fork head DNA-recognition motif resembles histone H5. Nature 1993, 364, 412–420. [Google Scholar] [CrossRef]
- Kadoch, C.; Williams, R.T.; Calarco, J.P.; Miller, E.L.; Weber, C.M.; Braun, S.M.G.; Pulice, J.L.; Chory, E.J.; Crabtree, G.R. Dynamics of BAF-Polycomb complex opposition on heterochromatin in normal and oncogenic states. Nat. Genet. 2017, 49, 213–222. [Google Scholar] [CrossRef]
- Kia, S.K.; Gorski, M.M.; Giannakopoulos, S.; Verrijzer, C.P. SWI/SNF mediates polycomb eviction and epigenetic reprogramming of the INK4b-ARF-INK4a locus. Mol. Cell. Biol. 2008, 28, 3457–3464. [Google Scholar] [CrossRef]
- Lau, P.N.I.; Cheung, P. Histone code pathway involving H3 S28 phosphorylation and K27 acetylation activates transcription and antagonizes polycomb silencing. Proc. Natl. Acad. Sci. USA 2011, 108, 2801–2806. [Google Scholar] [CrossRef]
- Gehani, S.S.; Agrawal-Singh, S.; Dietrich, N.; Christophersen, N.S.; Helin, K.; Hansen, K. Polycomb group protein displacement and gene activation through MSK-dependent H3K27me3S28 phosphorylation. Mol. Cell 2010, 39, 886–900. [Google Scholar] [CrossRef]
- Josefowicz, S.Z.; Shimada, M.; Armache, A.; Li, C.H.; Miller, R.M.; Lin, S.; Yang, A.; Dill, B.D.; Molina, H.; Park, H.-S.; et al. Chromatin Kinases Act on Transcription Factors and Histone Tails in Regulation of Inducible Transcription. Mol. Cell 2016, 64, 347–361. [Google Scholar] [CrossRef]
- Tie, F.; Banerjee, R.; Stratton, C.A.; Prasad-Sinha, J.; Stepanik, V.; Zlobin, A.; Diaz, M.O.; Scacheri, P.C.; Harte, P.J. CBP-mediated acetylation of histone H3 lysine 27 antagonizes Drosophila Polycomb silencing. Development 2009, 136, 3131–3141. [Google Scholar] [CrossRef] [PubMed]
- Englert, N.A.; Luo, G.; Goldstein, J.A.; Surapureddi, S. Epigenetic Modification of Histone 3 Lysine 27. J. Biol. Chem. 2014, 290, 2264–2278. [Google Scholar] [CrossRef] [PubMed]
- Christensen, M.D.; Nitiyanandan, R.; Meraji, S.; Daer, R.; Godeshala, S.; Goklany, S.; Haynes, K.; Rege, K. An inhibitor screen identifies histone-modifying enzymes as mediators of polymer-mediated transgene expression from plasmid DNA. J. Control. Release 2018, 286, 210–223. [Google Scholar] [CrossRef] [PubMed]
- Gracey Maniar, L.E.; Maniar, J.M.; Chen, Z.-Y.; Lu, J.; Fire, A.Z.; Kay, M.A. Minicircle DNA vectors achieve sustained expression reflected by active chromatin and transcriptional level. Mol. Ther. 2013, 21, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Riu, E.; Chen, Z.-Y.; Xu, H.; He, C.-Y.; Kay, M.A. Histone modifications are associated with the persistence or silencing of vector-mediated transgene expression in vivo. Mol. Ther. 2007, 15, 1348–1355. [Google Scholar] [CrossRef]
- Mathelier, A.; Fornes, O.; Arenillas, D.J.; Chen, C.-Y.; Denay, G.; Lee, J.; Shi, W.; Shyr, C.; Tan, G.; Worsley-Hunt, R.; et al. JASPAR 2016: A major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2016, 44, D110–D115. [Google Scholar] [CrossRef]
- Zobel, A.; Kalkbrenner, F.; Vorbrueggen, G.; Moelling, K. Transactivation of the human c-myc gene by c-Myb. Biochem. Biophys. Res. Commun. 1992, 186, 715–722. [Google Scholar] [CrossRef][Green Version]
- Kawasaki, H.; Schiltz, L.; Chiu, R.; Itakura, K.; Taira, K.; Nakatani, Y.; Yokoyama, K.K. ATF-2 has intrinsic histone acetyltransferase activity which is modulated by phosphorylation. Nature 2000, 405, 195–200. [Google Scholar] [CrossRef]
- Yang, L.; Wang, H.; Luo, X.; Mao, P.; Tian, W.; Shi, Y.; Huang, G.; Zhang, J.; Ma, D. Virion protein 16 induces demethylation of DNA integrated within chromatin in a novel mammalian cell model. Acta Biochim. Biophys. Sin. 2011, 44, 154–161. [Google Scholar] [CrossRef][Green Version]
- Milne, T.A.; Briggs, S.D.; Brock, H.W.; Martin, M.E.; Gibbs, D.; Allis, C.D.; Hess, J.L. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol. Cell 2002, 10, 1107–1117. [Google Scholar] [CrossRef]
- Nishioka, K.; Chuikov, S.; Sarma, K.; Erdjument-Bromage, H.; Allis, C.D.; Tempst, P.; Reinberg, D. Set9, a novel histone H3 methyltransferase that facilitates transcription by precluding histone tail modifications required for heterochromatin formation. Genes Dev. 2002, 16, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Serandour, A.A.; Avner, S.; Percevault, F.; Demay, F.; Bizot, M.; Lucchetti-Miganeh, C.; Barloy-Hubler, F.; Brown, M.; Lupien, M.; Metivier, R.; et al. Epigenetic switch involved in activation of pioneer factor FOXA1-dependent enhancers. Genome Res. 2011, 21, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Côté, J.; Xue, Y.; Zhou, S.; Khavari, P.A.; Biggar, S.R.; Muchardt, C.; Kalpana, G.V.; Goff, S.P.; Yaniv, M.; et al. Purification and biochemical heterogeneity of the mammalian SWI-SNF complex. EMBO J. 1996, 15, 5370–5382. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.H.; Bracken, A.P.; Pasini, D.; Dietrich, N.; Gehani, S.S.; Monrad, A.; Rappsilber, J.; Lerdrup, M.; Helin, K. A model for transmission of the H3K27me3 epigenetic mark. Nat. Cell Biol. 2008, 10, 1291–1300. [Google Scholar] [CrossRef] [PubMed]
- Pirrotta, V. Introduction to Polycomb Group Mechanisms. In Polycomb Group Proteins; Academic Press: Cambridge, MA, USA, 2017; pp. 1–3. [Google Scholar]
- Sandberg, M.L.; Sutton, S.E.; Pletcher, M.T.; Wiltshire, T.; Tarantino, L.M.; Hogenesch, J.B.; Cooke, M.P. c-Myb and p300 regulate hematopoietic stem cell proliferation and differentiation. Dev. Cell 2005, 8, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Pattabiraman, D.R.; Sun, J.; Dowhan, D.H.; Ishii, S.; Gonda, T.J. Mutations in multiple domains of c-Myb disrupt interaction with CBP/p300 and abrogate myeloid transforming ability. Mol. Cancer Res. 2009, 7, 1477–1486. [Google Scholar] [CrossRef]
- Raisner, R.; Kharbanda, S.; Jin, L.; Jeng, E.; Chan, E.; Merchant, M.; Haverty, P.M.; Bainer, R.; Cheung, T.; Arnott, D.; et al. Enhancer Activity Requires CBP/P300 Bromodomain-Dependent Histone H3K27 Acetylation. Cell Rep. 2018, 24, 1722–1729. [Google Scholar] [CrossRef]
- Ogryzko, V.V.; Louis Schiltz, R.; Russanova, V.; Howard, B.H.; Nakatani, Y. The Transcriptional Coactivators p300 and CBP Are Histone Acetyltransferases. Cell 1996, 87, 953–959. [Google Scholar] [CrossRef]
- Coulibaly, A.; Haas, A.; Steinmann, S.; Jakobs, A.; Schmidt, T.J.; Klempnauer, K.-H. The natural anti-tumor compound Celastrol targets a Myb-C/EBPβ-p300 transcriptional module implicated in myeloid gene expression. PLoS ONE 2018, 13, e0190934. [Google Scholar] [CrossRef]
- Uttarkar, S.; Piontek, T.; Dukare, S.; Schomburg, C.; Schlenke, P.; Berdel, W.E.; Müller-Tidow, C.; Schmidt, T.J.; Klempnauer, K.-H. Small-Molecule Disruption of the Myb/p300 Cooperation Targets Acute Myeloid Leukemia Cells. Mol. Cancer Ther. 2016, 15, 2905–2915. [Google Scholar] [CrossRef]
- Denis, C.M.; Langelaan, D.N.; Kirlin, A.C.; Chitayat, S.; Munro, K.; Spencer, H.L.; LeBrun, D.P.; Smith, S.P. Functional redundancy between the transcriptional activation domains of E2A is mediated by binding to the KIX domain of CBP/p300. Nucleic Acids Res. 2014, 42, 7370–7382. [Google Scholar] [CrossRef] [PubMed]
- Uttarkar, S.; Dassé, E.; Coulibaly, A.; Steinmann, S.; Jakobs, A.; Schomburg, C.; Trentmann, A.; Jose, J.; Schlenke, P.; Berdel, W.E.; et al. Targeting acute myeloid leukemia with a small molecule inhibitor of the Myb/p300 interaction. Blood 2016, 127, 1173–1182. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tsai, S.Q.; Zheng, Z.; Nguyen, N.T.; Liebers, M.; Topkar, V.V.; Thapar, V.; Wyvekens, N.; Khayter, C.; Iafrate, A.J.; Le, L.P.; et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015, 33, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Van Essen, D.; Engist, B.; Natoli, G.; Saccani, S. Two Modes of Transcriptional Activation at Native Promoters by NF-κB p65. PLoS Biol. 2009, 7, e1000073. [Google Scholar] [CrossRef] [PubMed]
- Thakore, P.I.; Black, J.B.; Hilton, I.B.; Gersbach, C.A. Editing the epigenome: Technologies for programmable transcription and epigenetic modulation. Nat. Methods 2016, 13, 127–137. [Google Scholar] [CrossRef]
- Wang, W.; Qin, J.-J.; Voruganti, S.; Nag, S.; Zhou, J.; Zhang, R. Polycomb Group (PcG) Proteins and Human Cancers: Multifaceted Functions and Therapeutic Implications. Med. Res. Rev. 2015, 35, 1220–1267. [Google Scholar] [CrossRef]
- Lecoq, L.; Raiola, L.; Chabot, P.R.; Cyr, N.; Arseneault, G.; Legault, P.; Omichinski, J.G. Structural characterization of interactions between transactivation domain 1 of the p65 subunit of NF-κB and transcription regulatory factors. Nucleic Acids Res. 2017, 45, 5564–5576. [Google Scholar] [CrossRef]
- Huang, Z.-Q.; Li, J.; Sachs, L.M.; Cole, P.A.; Wong, J. A role for cofactor-cofactor and cofactor-histone interactions in targeting p300, SWI/SNF and Mediator for transcription. EMBO J. 2003, 22, 2146–2155. [Google Scholar] [CrossRef]
- Haas, M.; Siegert, M.; Schürmann, A.; Sodeik, B.; Wolfes, H. c-Myb protein interacts with Rcd-1, a component of the CCR4 transcription mediator complex. Biochemistry 2004, 43, 8152–8159. [Google Scholar] [CrossRef]
- Fukasawa, R.; Iida, S.; Tsutsui, T.; Hirose, Y.; Ohkuma, Y. Mediator complex cooperatively regulates transcription of retinoic acid target genes with Polycomb Repressive Complex 2 during neuronal differentiation. J. Biochem. 2015, 158, 373–384. [Google Scholar] [CrossRef]
- Englert, N.A.; Luo, G.; Goldstein, J.A.; Surapureddi, S. Epigenetic modification of histone 3 lysine 27: Mediator subunit MED25 is required for the dissociation of polycomb repressive complex 2 from the promoter of cytochrome P450 2C9. J. Biol. Chem. 2015, 290, 2264–2278. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, L.; Ferrari, R.; Vashisht, A.A.; Wohlschlegel, J.A.; Kurdistani, S.K.; Carey, M. Polycomb repressive complex 1 (PRC1) disassembles RNA polymerase II preinitiation complexes. J. Biol. Chem. 2012, 287, 35784–35794. [Google Scholar] [CrossRef] [PubMed]
- Oakes, B.L.; Nadler, D.C.; Flamholz, A.; Fellmann, C.; Staahl, B.T.; Doudna, J.A.; Savage, D.F. Profiling of engineering hotspots identifies an allosteric CRISPR-Cas9 switch. Nat. Biotechnol. 2016, 34, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Greber, D.; El-Baba, M.D.; Fussenegger, M. Intronically encoded siRNAs improve dynamic range of mammalian gene regulation systems and toggle switch. Nucleic Acids Res. 2008, 36, e101. [Google Scholar] [CrossRef] [PubMed]
- Stanton, B.C.; Siciliano, V.; Ghodasara, A.; Wroblewska, L.; Clancy, K.; Trefzer, A.C.; Chesnut, J.D.; Weiss, R.; Voigt, C.A. Systematic transfer of prokaryotic sensors and circuits to mammalian cells. ACS Synth. Biol. 2014, 3, 880–891. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, M.; Strittmatter, T.; Fussenegger, M. Light-Controlled Mammalian Cells and Their Therapeutic Applications in Synthetic Biology. Adv. Sci. Lett. 2018, 1800952. [Google Scholar] [CrossRef] [PubMed]
- Inobe, T.; Nukina, N. Rapamycin-induced oligomer formation system of FRB-FKBP fusion proteins. J. Biosci. Bioeng. 2016, 122, 40–46. [Google Scholar] [CrossRef]
- Rivera, V.M.; Clackson, T.; Natesan, S.; Pollock, R.; Amara, J.F.; Keenan, T.; Magari, S.R.; Phillips, T.; Courage, N.L.; Cerasoli, F.; et al. A humanized system for pharmacologic control of gene expression. Nat. Med. 1996, 2, 1028–1032. [Google Scholar] [CrossRef]
- DeRose, R.; Miyamoto, T.; Inoue, T. Manipulating signaling at will: Chemically-inducible dimerization (CID) techniques resolve problems in cell biology. Pflug. Arch. 2013, 465, 409–417. [Google Scholar] [CrossRef]
- Cascão, R.; Fonseca, J.E.; Moita, L.F. Celastrol: A Spectrum of Treatment Opportunities in Chronic Diseases. Front. Med. 2017, 4, 69. [Google Scholar] [CrossRef]
- Venkatesha, S.H.; Dudics, S.; Astry, B.; Moudgil, K.D. Control of autoimmune inflammation by celastrol, a natural triterpenoid. Pathog. Dis. 2016, 74, ftw059. [Google Scholar] [CrossRef] [PubMed]
- Ju, S.M.; Youn, G.S.; Cho, Y.S.; Choi, S.Y.; Park, J. Celastrol ameliorates cytokine toxicity and pro-inflammatory immune responses by suppressing NF-κB activation in RINm5F beta cells. BMB Rep. 2015, 48, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Liu, D.; Zhang, Y.; Qian, Y.; Zhang, H.; Guo, S.; Sunagawa, M.; Hisamitsu, T.; Liu, Y. Celastrol inhibits lipopolysaccharide-stimulated rheumatoid fibroblast-like synoviocyte invasion through suppression of TLR4/NF-κB-mediated matrix metalloproteinase-9 expression. PLoS ONE 2013, 8, e68905. [Google Scholar] [CrossRef] [PubMed]
- Raja, S.M.; Clubb, R.J.; Ortega-Cava, C.; Williams, S.H.; Bailey, T.A.; Duan, L.; Zhao, X.; Reddi, A.L.; Nyong, A.M.; Natarajan, A.; et al. Anticancer activity of Celastrol in combination with ErbB2-targeted therapeutics for treatment of ErbB2-overexpressing breast cancers. Cancer Biol. Ther. 2011, 11, 263–276. [Google Scholar] [CrossRef]
- Yang, H.; Chen, D.; Cui, Q.C.; Yuan, X.; Dou, Q.P. Celastrol, a triterpene extracted from the Chinese “Thunder of God Vine,” is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res. 2006, 66, 4758–4765. [Google Scholar] [CrossRef]
- Cleren, C.; Calingasan, N.Y.; Chen, J.; Beal, M.F. Celastrol protects against MPTP- and 3-nitropropionic acid-induced neurotoxicity. J. Neurochem. 2005, 94, 995–1004. [Google Scholar] [CrossRef]
- Konieczny, J.; Jantas, D.; Lenda, T.; Domin, H.; Czarnecka, A.; Kuter, K.; Śmiałowska, M.; Lasoń, W.; Lorenc-Koci, E. Lack of neuroprotective effect of celastrol under conditions of proteasome inhibition by lactacystin in in vitro and in vivo studies: Implications for Parkinson’s disease. Neurotox. Res. 2014, 26, 255–273. [Google Scholar] [CrossRef]
- Tekel, S.J.; Barrett, C.; Vargas, D.; Haynes, K.A. Design, Construction, and Validation of Histone-Binding Effectors in Vitro and in Cells. Biochemistry 2018, 57, 4707–4716. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, O.; Blake Joseph Wall, J.; Zheng, M.; Zhou, Y.; Wang, L.; Vaseghi, H.R.; Qian, L.; Liu, J. Systematic comparison of 2A peptides for cloning multi-genes in a polycistronic vector. Sci. Rep. 2017, 7, 2193. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barrett, C.M.; McCracken, R.; Elmer, J.; Haynes, K.A. Components from the Human c-myb Transcriptional Regulation System Reactivate Epigenetically Repressed Transgenes. Int. J. Mol. Sci. 2020, 21, 530. https://doi.org/10.3390/ijms21020530
Barrett CM, McCracken R, Elmer J, Haynes KA. Components from the Human c-myb Transcriptional Regulation System Reactivate Epigenetically Repressed Transgenes. International Journal of Molecular Sciences. 2020; 21(2):530. https://doi.org/10.3390/ijms21020530
Chicago/Turabian StyleBarrett, Cassandra M., Reilly McCracken, Jacob Elmer, and Karmella A. Haynes. 2020. "Components from the Human c-myb Transcriptional Regulation System Reactivate Epigenetically Repressed Transgenes" International Journal of Molecular Sciences 21, no. 2: 530. https://doi.org/10.3390/ijms21020530
APA StyleBarrett, C. M., McCracken, R., Elmer, J., & Haynes, K. A. (2020). Components from the Human c-myb Transcriptional Regulation System Reactivate Epigenetically Repressed Transgenes. International Journal of Molecular Sciences, 21(2), 530. https://doi.org/10.3390/ijms21020530