Airborne Particulate Matter (PM10) Inhibits Apoptosis through PI3K/AKT/FoxO3a Pathway in Lung Epithelial Cells: The Role of a Second Oxidant Stimulus

 , , , ,
, , , ,  and
and
Abstract
1. Introduction
2. Results
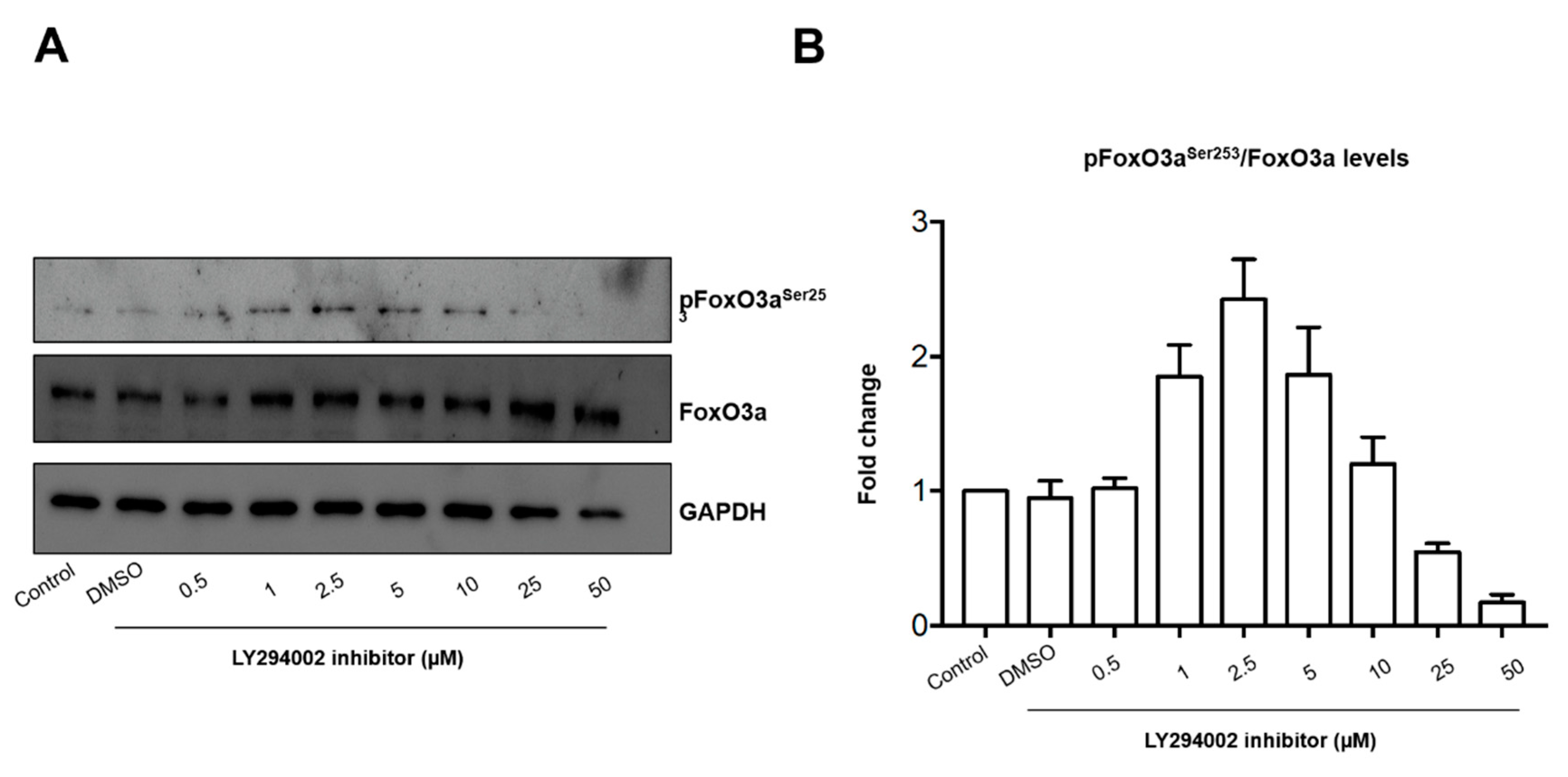
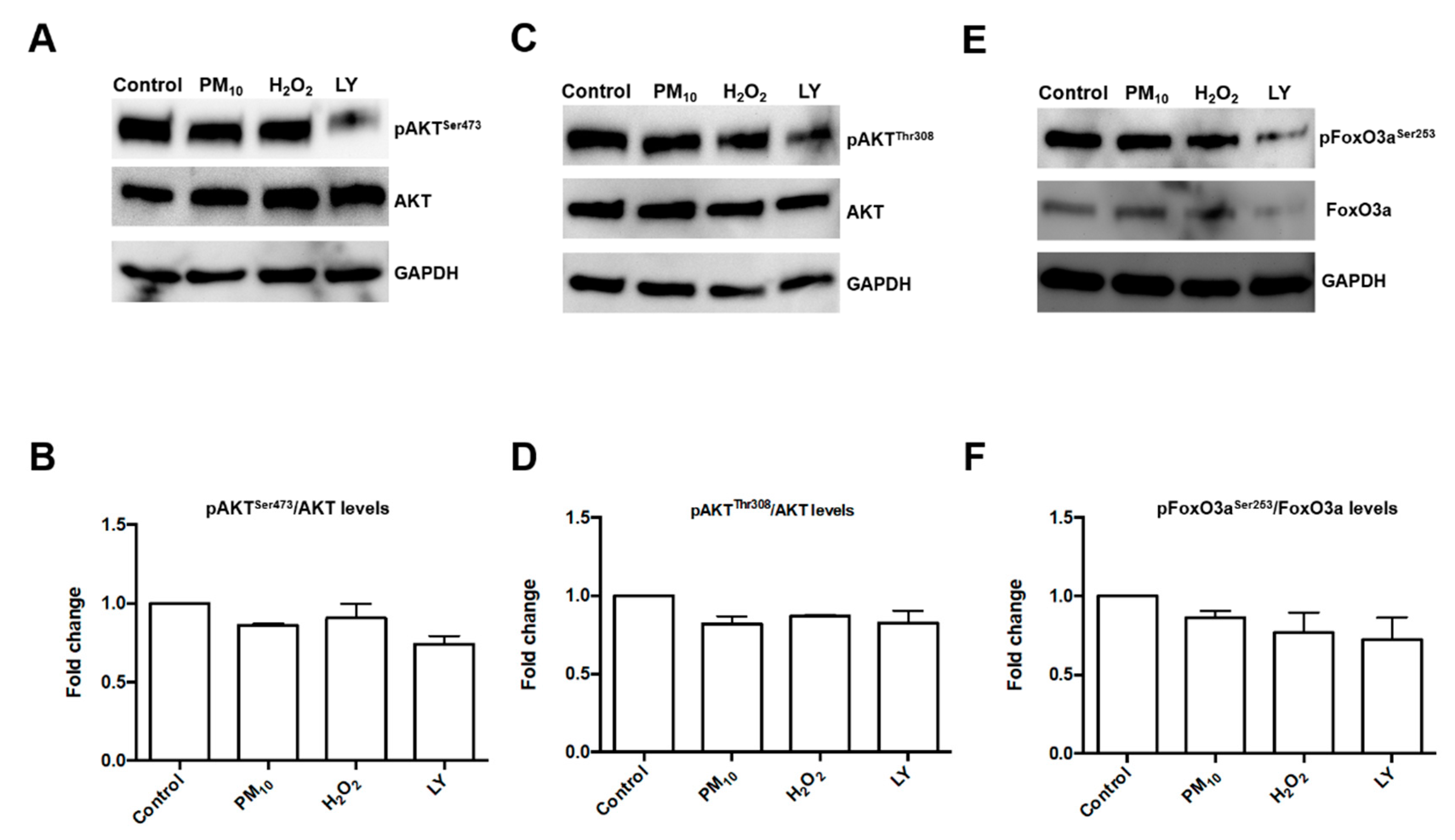
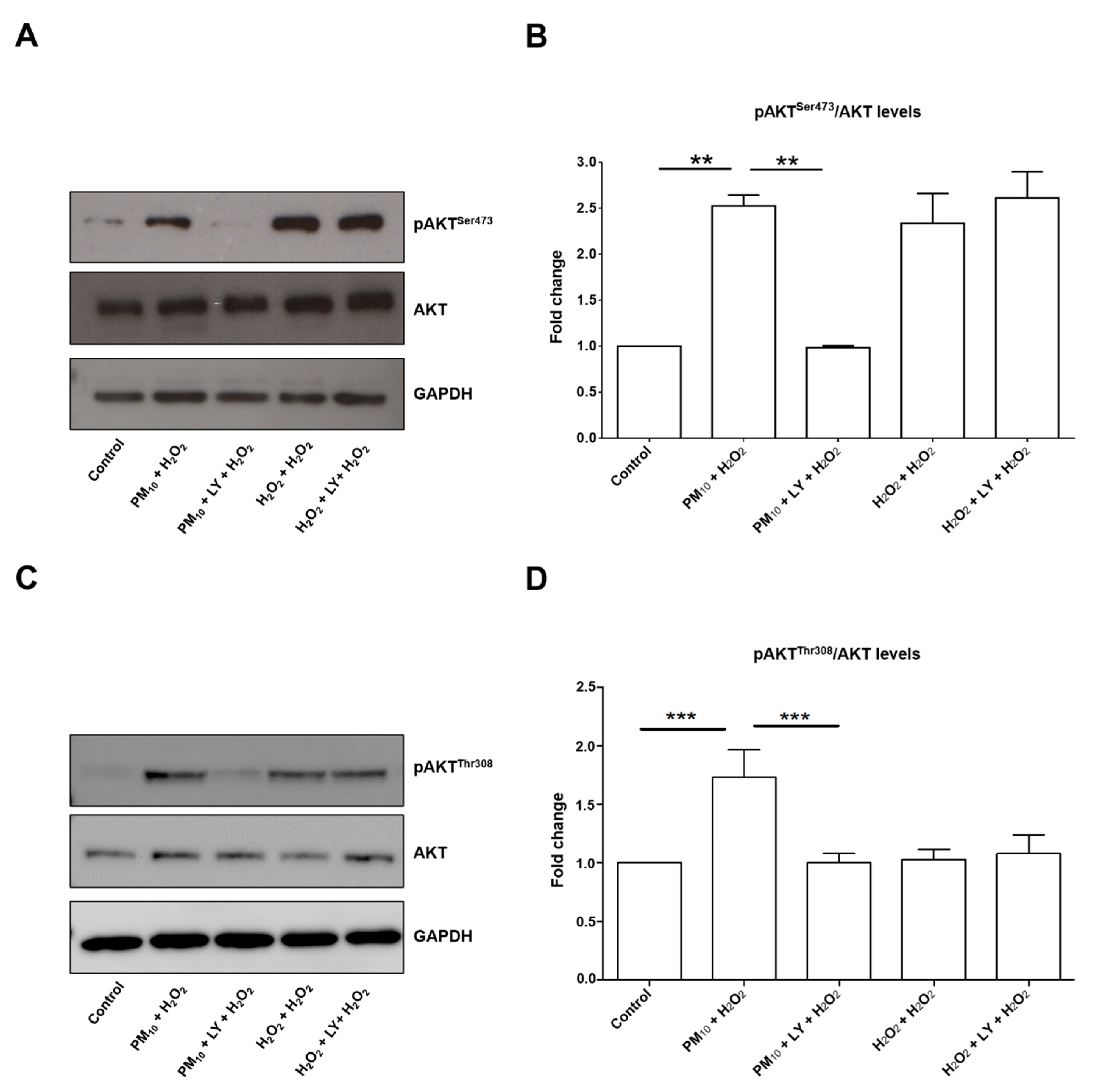
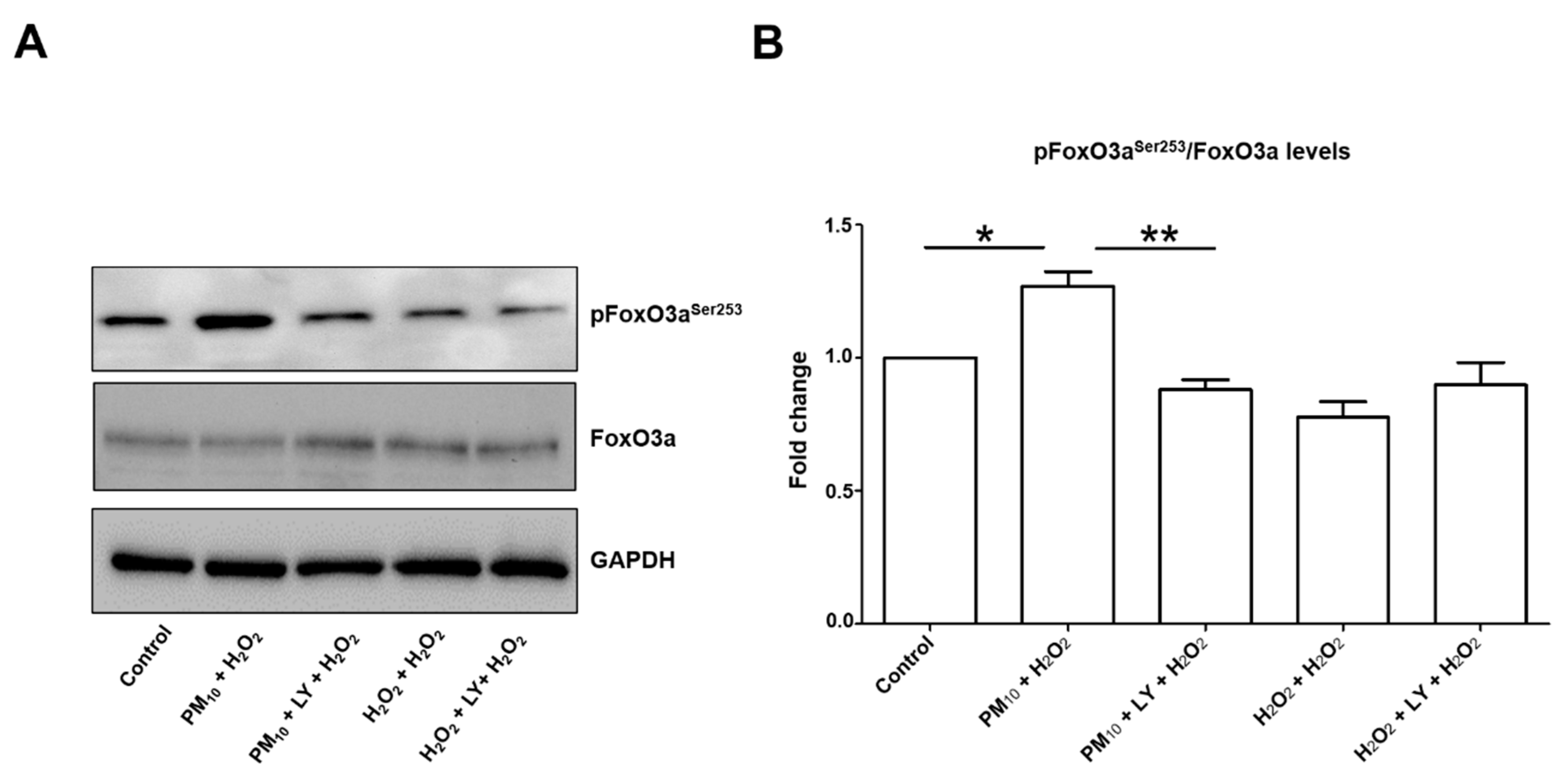
2.1. Pre-Exposure to PM10 Followed by H2O2 Treatment Induced AKT and FoxO3 Phosphorylation through PI3K Activation
2.2. Pre-Exposure to PM10 Decreased Catalase and p27kip1 Protein through PI3K Activation
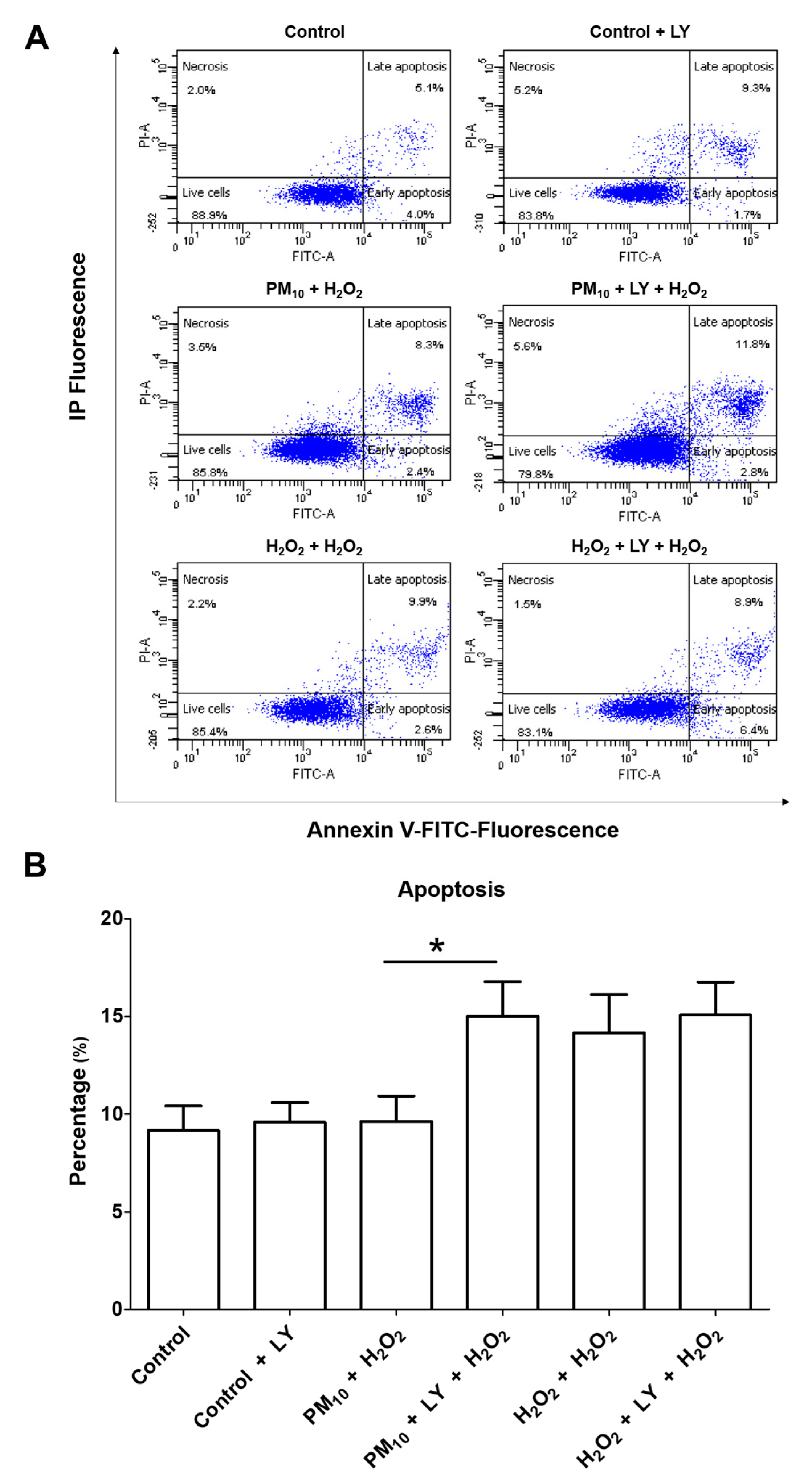
2.3. Inhibition of Apoptosis via PI3K/AKT/FoxO3a by Pre-Exposure to PM10 Followed by H2O2 Treatment
3. Discussion
4. Materials and Methods
4.1. PM10 Sampling
4.2. Lung Epithelial A549 Cell Culture
4.3. PM10 Exposure, H2O2 Treatment, and LY294002 Inhibitor in Cell Cultures
4.4. Cellular Viability
4.5. Determination of Protein Levels
4.6. Apoptosis Measurement
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- IARC. Outdoor Air Pollution. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; IARC: Lyon, France, 2016; Volume 109, pp. 9–444. [Google Scholar]
- Chen, X.; Zhang, L.W.; Huang, J.J.; Song, F.J.; Zhang, L.P.; Qian, Z.M.; Trevathan, E.; Mao, H.J.; Han, B.; Vaughn, M.; et al. Long-term exposure to urban air pollution and lung cancer mortality: A 12-year cohort study in northern china. Sci. Total Environ. 2016, 571, 855–861. [Google Scholar] [CrossRef] [PubMed]
- Pope, C.A.; Burnett, R.T.; Thun, M.J.; Calle, E.E.; Krewski, D.; Ito, K.; Thurston, G.D. Lung cancer, cardiopulmonary mortality, and long-term exposure to fine particulate air pollution. Jama 2002, 287, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Loomis, D.; Grosse, Y.; Lauby-Secretan, B.; El Ghissassi, F.; Bouvard, V.; Benbrahim-Tallaa, L.; Guha, N.; Baan, R.; Mattock, H.; Straif, K.; et al. The carcinogenicity of outdoor air pollution. Lancet Oncol. 2013, 14, 1262–1263. [Google Scholar] [CrossRef]
- Chirino, Y.I.; Sanchez-Perez, Y.; Osornio-Vargas, A.R.; Morales-Barcenas, R.; Gutierrez-Ruiz, M.C.; Segura-Garcia, Y.; Rosas, I.; Pedraza-Chaverri, J.; Garcia-Cuellar, C.M. Pm(10) impairs the antioxidant defense system and exacerbates oxidative stress driven cell death. Toxicol. Lett. 2010, 193, 209–216. [Google Scholar] [CrossRef]
- Sanchez-Perez, Y.; Chirino, Y.I.; Osornio-Vargas, A.R.; Morales-Barcenas, R.; Gutierrez-Ruiz, C.; Vazquez-Lopez, I.; Garcia-Cuellar, C.M. DNA damage response of a549 cells treated with particulate matter (pm10) of urban air pollutants. Cancer Lett. 2009, 278, 192–200. [Google Scholar] [CrossRef]
- Quezada-Maldonado, E.M.; Sanchez-Perez, Y.; Chirino, Y.I.; Vaca-Paniagua, F.; Garcia-Cuellar, C.M. miRNAs deregulation in lung cells exposed to airborne particulate matter (pm10) is associated with pathways deregulated in lung tumors. Environ. Pollut. 2018, 241, 351–358. [Google Scholar] [CrossRef]
- Santibanez-Andrade, M.; Sanchez-Perez, Y.; Chirino, Y.I.; Morales-Barcenas, R.; Herrera, L.A.; Garcia-Cuellar, C.M. Airborne particulate matter induces mitotic slippage and chromosomal missegregation through disruption of the spindle assembly checkpoint (sac). Chemosphere 2019, 235, 794–804. [Google Scholar] [CrossRef]
- Sanchez-Perez, Y.; Chirino, Y.I.; Osornio-Vargas, A.R.; Herrera, L.A.; Morales-Barcenas, R.; Lopez-Saavedra, A.; Gonzalez-Ramirez, I.; Miranda, J.; Garcia-Cuellar, C.M. Cytoplasmic p21(cip1/waf1), erk1/2 activation, and cytoskeletal remodeling are associated with the senescence-like phenotype after airborne particulate matter (pm(10)) exposure in lung cells. Toxicol. Lett. 2014, 225, 12–19. [Google Scholar] [CrossRef]
- Reyes-Zarate, E.; Sanchez-Perez, Y.; Gutierrez-Ruiz, M.C.; Chirino, Y.I.; Osornio-Vargas, A.R.; Morales-Barcenas, R.; Souza-Arroyo, V.; Garcia-Cuellar, C.M. Atmospheric particulate matter (pm10) exposure-induced cell cycle arrest and apoptosis evasion through stat3 activation via pkczeta and src kinases in lung cells. Environ. Pollut. 2016, 214, 646–656. [Google Scholar] [CrossRef]
- Bowman, T.; Garcia, R.; Turkson, J.; Jove, R. Stats in oncogenesis. Oncogene 2000, 19, 2474–2488. [Google Scholar] [CrossRef]
- Spitzner, M.; Ebner, R.; Wolff, H.A.; Ghadimi, B.M.; Wienands, J.; Grade, M. Stat3: A novel molecular mediator of resistance to chemoradiotherapy. Cancers 2014, 6, 1986–2011. [Google Scholar] [CrossRef] [PubMed]
- Gharbi, S.I.; Zvelebil, M.J.; Shuttleworth, S.J.; Hancox, T.; Saghir, N.; Timms, J.F.; Waterfield, M.D. Exploring the specificity of the pi3k family inhibitor ly294002. Biochem. J. 2007, 404, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mtor, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-kinase akt pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Calnan, D.R.; Brunet, A. The foxo code. Oncogene 2008, 27, 2276–2288. [Google Scholar] [CrossRef]
- Wu, D.; Liang, M.; Dang, H.; Fang, F.; Xu, F.; Liu, C. Hydrogen protects against hyperoxia-induced apoptosis in type ii alveolar epithelial cells via activation of pi3k/akt/foxo3a signaling pathway. Biochem. Biophys. Res. Commun. 2018, 495, 1620–1627. [Google Scholar] [CrossRef]
- Brown, A.K.; Webb, A.E. Regulation of foxo factors in mammalian cells. Curr. Top. Dev. Biol. 2018, 127, 165–192. [Google Scholar]
- Lloyd, R.V.; Erickson, L.A.; Jin, L.; Kulig, E.; Qian, X.; Cheville, J.C.; Scheithauer, B.W. P27kip1: A multifunctional cyclin-dependent kinase inhibitor with prognostic significance in human cancers. Am. J. Pathol. 1999, 154, 313–323. [Google Scholar] [CrossRef]
- Li, N.; Hao, M.; Phalen, R.F.; Hinds, W.C.; Nel, A.E. Particulate air pollutants and asthma. A paradigm for the role of oxidative stress in pm-induced adverse health effects. Clin. Immunol. 2003, 109, 250–265. [Google Scholar] [CrossRef]
- Zhai, C.; Lv, J.; Wang, K.; Li, Q.; Qu, Y. Hsp70 silencing aggravates apoptosis induced by hypoxia/reoxygenation in vitro. Exp. Ther. Med. 2019, 18, 1013–1020. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Li, S.; Jia, W.; Lv, G.; Song, C.; Kang, C.; Zhang, Q. Effects of diesel exhaust particles on microrna-21 in human bronchial epithelial cells and potential carcinogenic mechanisms. Mol. Med. Rep. 2015, 12, 2329–2335. [Google Scholar] [CrossRef] [PubMed]
- Glorieux, C.; Zamocky, M.; Sandoval, J.M.; Verrax, J.; Calderon, P.B. Regulation of catalase expression in healthy and cancerous cells. Free Radic. Biol. Med. 2015, 87, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Ganguli, G.; Mukherjee, U.; Sonawane, A. Peroxisomes and oxidative stress: Their implications in the modulation of cellular immunity during mycobacterial infection. Front. Microbiol. 2019, 10, 1121. [Google Scholar] [CrossRef]
- Hosakote, Y.M.; Komaravelli, N.; Mautemps, N.; Liu, T.; Garofalo, R.P.; Casola, A. Antioxidant mimetics modulate oxidative stress and cellular signaling in airway epithelial cells infected with respiratory syncytial virus. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L991–L1000. [Google Scholar] [CrossRef]
- Horne, G.A.; Kinstrie, R.; Copland, M. Novel drug therapies in myeloid leukemia. Pharm. Pat. Anal. 2015, 4, 187–205. [Google Scholar] [CrossRef]
- Kato, M.; Yuan, H.; Xu, Z.G.; Lanting, L.; Li, S.L.; Wang, M.; Hu, M.C.; Reddy, M.A.; Natarajan, R. Role of the akt/foxo3a pathway in tgf-beta1-mediated mesangial cell dysfunction: A novel mechanism related to diabetic kidney disease. J. Am. Soc. Nephrol. JASN 2006, 17, 3325–3335. [Google Scholar] [CrossRef]
- Taylor, S.; Lam, M.; Pararasa, C.; Brown, J.E.; Carmichael, A.R.; Griffiths, H.R. Evaluating the evidence for targeting foxo3a in breast cancer: A systematic review. Cancer Cell Int. 2015, 15, 1. [Google Scholar] [CrossRef]
- Vlahos, C.J.; Matter, W.F.; Hui, K.Y.; Brown, R.F. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4h-1-benzopyran-4-one (ly294002). J. Biol. Chem. 1994, 269, 5241–5248. [Google Scholar]
- Sun, X.; Meng, L.; Qiao, W.; Yang, R.; Gao, Q.; Peng, Y.; Bian, Z. Vascular endothelial growth factor a/vascular endothelial growth factor receptor 2 axis promotes human dental pulp stem cell migration via the fak/pi3k/akt and p38 mapk signalling pathways. Int. Endod. J. 2019, 52, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Arcaro, A.; Wymann, M.P. Wortmannin is a potent phosphatidylinositol 3-kinase inhibitor: The role of phosphatidylinositol 3,4,5-trisphosphate in neutrophil responses. Biochem. J. 1993, 296 Pt 2, 297–301. [Google Scholar] [CrossRef]
- Yano, H.; Nakanishi, S.; Kimura, K.; Hanai, N.; Saitoh, Y.; Fukui, Y.; Nonomura, Y.; Matsuda, Y. Inhibition of histamine secretion by wortmannin through the blockade of phosphatidylinositol 3-kinase in rbl-2h3 cells. J. Biol. Chem. 1993, 268, 25846–25856. [Google Scholar] [PubMed]
- Brunn, G.J.; Williams, J.; Sabers, C.; Wiederrecht, G.; Lawrence, J.C., Jr.; Abraham, R.T. Direct inhibition of the signaling functions of the mammalian target of rapamycin by the phosphoinositide 3-kinase inhibitors, wortmannin and ly294002. EMBO J. 1996, 15, 5256–5267. [Google Scholar] [CrossRef] [PubMed]
- Davies, S.P.; Reddy, H.; Caivano, M.; Cohen, P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 2000, 351, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, M.D.; Black, J.; Futer, O.; Swenson, L.; Hare, B.; Fleming, M.; Saxena, K. Pim-1 ligand-bound structures reveal the mechanism of serine/threonine kinase inhibition by ly294002. J. Biol. Chem. 2005, 280, 13728–13734. [Google Scholar] [CrossRef]
- Wang, Y.; Kuramitsu, Y.; Baron, B.; Kitagawa, T.; Tokuda, K.; Akada, J.; Maehara, S.I.; Maehara, Y.; Nakamura, K. Pi3k inhibitor ly294002, as opposed to wortmannin, enhances akt phosphorylation in gemcitabine-resistant pancreatic cancer cells. Int. J. Oncol. 2017, 50, 606–612. [Google Scholar] [CrossRef]
- Yan, W.; Zhang, M.; Yu, Y.; Yi, X.; Guo, T.; Hu, H.; Sun, Q.; Chen, M.; Xiong, H.; Chen, L. Blockade of voltage-gated potassium channels ameliorates diabetes-associated cognitive dysfunction in vivo and in vitro. Exp. Neurol. 2019, 320, 112988. [Google Scholar] [CrossRef]
- Pham-Huy, L.A.; He, H.; Pham-Huy, C. Free radicals, antioxidants in disease and health. Int. J. Biomed. Sci. IJBS 2008, 4, 89–96. [Google Scholar]
- Liu, S.T.; Chan, G.K.; Li, W. Editorial: Non-cell cycle functions of cell cycle regulators. Front. Cell Dev. Biol. 2019, 7, 122. [Google Scholar] [CrossRef]
- Deiuliis, J.A.; Kampfrath, T.; Zhong, J.; Oghumu, S.; Maiseyeu, A.; Chen, L.C.; Sun, Q.; Satoskar, A.R.; Rajagopalan, S. Pulmonary t cell activation in response to chronic particulate air pollution. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L399–L409. [Google Scholar] [CrossRef] [PubMed]
- Miyata, R.; Bai, N.; Vincent, R.; Sin, D.D.; Van Eeden, S.F. Novel properties of statins: Suppression of the systemic and bone marrow responses induced by exposure to ambient particulate matter (pm(10)) air pollution. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L492–L499. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, P.E.; Ho, T.R.; Mann, E.H.; Kelly, F.J.; Sehlstedt, M.; Pourazar, J.; Dove, R.E.; Sandstrom, T.; Mudway, I.S.; Hawrylowicz, C.M. Urban particulate matter stimulation of human dendritic cells enhances priming of naive cd8 t lymphocytes. Immunology 2018, 153, 502–512. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, D.P.; Puig, K.L.; Gorr, M.W.; Wold, L.E.; Combs, C.K. A pilot study to assess effects of long-term inhalation of airborne particulate matter on early alzheimer-like changes in the mouse brain. PLoS ONE 2015, 10, e0127102. [Google Scholar] [CrossRef] [PubMed]
- Krawczyk, A.; Nowak, D.; Nowak, P.J.; Padula, G.; Kwiatkowska, S. Elevated exhalation of hydrogen peroxide in patients with non-small cell lung cancer is not affected by chemotherapy. Redox Rep. Commun. Free Radic. Res. 2017, 22, 308–314. [Google Scholar] [CrossRef][Green Version]
- Wei, L.; Zhu, S.; Wang, J.; Liu, J. Activation of the phosphatidylinositol 3-kinase/akt signaling pathway during porcine circovirus type 2 infection facilitates cell survival and viral replication. J. Virol. 2012, 86, 13589–13597. [Google Scholar] [CrossRef]
- Hopkins, B.D.; Pauli, C.; Du, X.; Wang, D.G.; Li, X.; Wu, D.; Amadiume, S.C.; Goncalves, M.D.; Hodakoski, C.; Lundquist, M.R.; et al. Suppression of insulin feedback enhances the efficacy of pi3k inhibitors. Nature 2018, 560, 499–503. [Google Scholar] [CrossRef]
- Alfaro-Moreno, E.; Martinez, L.; Garcia-Cuellar, C.; Bonner, J.C.; Murray, J.C.; Rosas, I.; Rosales, S.P.; Osornio-Vargas, A.R. Biologic effects induced in vitro by pm10 from three different zones of mexico city. Environ. Health Perspect. 2002, 110, 715–720. [Google Scholar] [CrossRef]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Girish, V.; Vijayalakshmi, A. Affordable image analysis using nih image/imagej. Indian J. Cancer 2004, 41, 47. [Google Scholar]
- Gorczyca, W.; Melamed, M.R.; Darzynkiewicz, Z. Analysis of apoptosis by flow cytometry. Methods Mol. Biol. 1998, 91, 217–238. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | LY294002 | PM10+H2O2 | PM10+LY+H2O2 | H2O2+H2O2 | H2O2+LY+H2O2 |
|---|---|---|---|---|---|
| 100 | 95.23 (SD ± 15.2) | 108.8 (SD ± 7.7) | 103.7 (SD ± 3.2) | 83.3 (SD ± 3.2) | 82 (SD ± 7.9) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Cuellar, C.M.; Chirino, Y.I.; Morales-Bárcenas, R.; Soto-Reyes, E.; Quintana-Belmares, R.; Santibáñez-Andrade, M.; Sánchez-Pérez, Y. Airborne Particulate Matter (PM10) Inhibits Apoptosis through PI3K/AKT/FoxO3a Pathway in Lung Epithelial Cells: The Role of a Second Oxidant Stimulus. Int. J. Mol. Sci. 2020, 21, 473. https://doi.org/10.3390/ijms21020473
García-Cuellar CM, Chirino YI, Morales-Bárcenas R, Soto-Reyes E, Quintana-Belmares R, Santibáñez-Andrade M, Sánchez-Pérez Y. Airborne Particulate Matter (PM10) Inhibits Apoptosis through PI3K/AKT/FoxO3a Pathway in Lung Epithelial Cells: The Role of a Second Oxidant Stimulus. International Journal of Molecular Sciences. 2020; 21(2):473. https://doi.org/10.3390/ijms21020473
Chicago/Turabian StyleGarcía-Cuellar, Claudia M., Yolanda I. Chirino, Rocío Morales-Bárcenas, Ernesto Soto-Reyes, Raúl Quintana-Belmares, Miguel Santibáñez-Andrade, and Yesennia Sánchez-Pérez. 2020. "Airborne Particulate Matter (PM10) Inhibits Apoptosis through PI3K/AKT/FoxO3a Pathway in Lung Epithelial Cells: The Role of a Second Oxidant Stimulus" International Journal of Molecular Sciences 21, no. 2: 473. https://doi.org/10.3390/ijms21020473
APA StyleGarcía-Cuellar, C. M., Chirino, Y. I., Morales-Bárcenas, R., Soto-Reyes, E., Quintana-Belmares, R., Santibáñez-Andrade, M., & Sánchez-Pérez, Y. (2020). Airborne Particulate Matter (PM10) Inhibits Apoptosis through PI3K/AKT/FoxO3a Pathway in Lung Epithelial Cells: The Role of a Second Oxidant Stimulus. International Journal of Molecular Sciences, 21(2), 473. https://doi.org/10.3390/ijms21020473