Clinical Aspects of Janus Kinase (JAK) Inhibitors in the Cardiovascular System in Patients with Rheumatoid Arthritis



Abstract
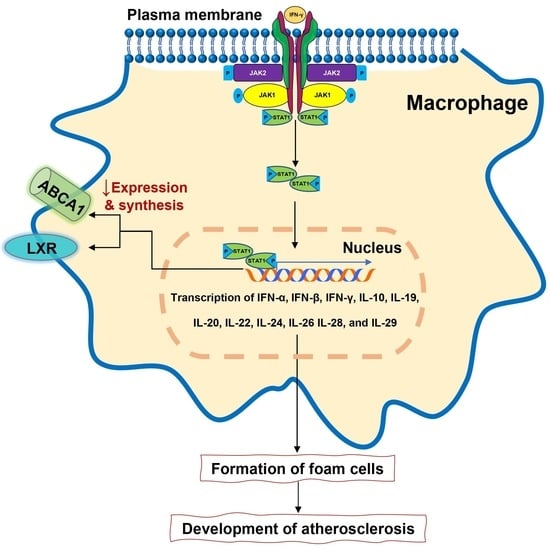
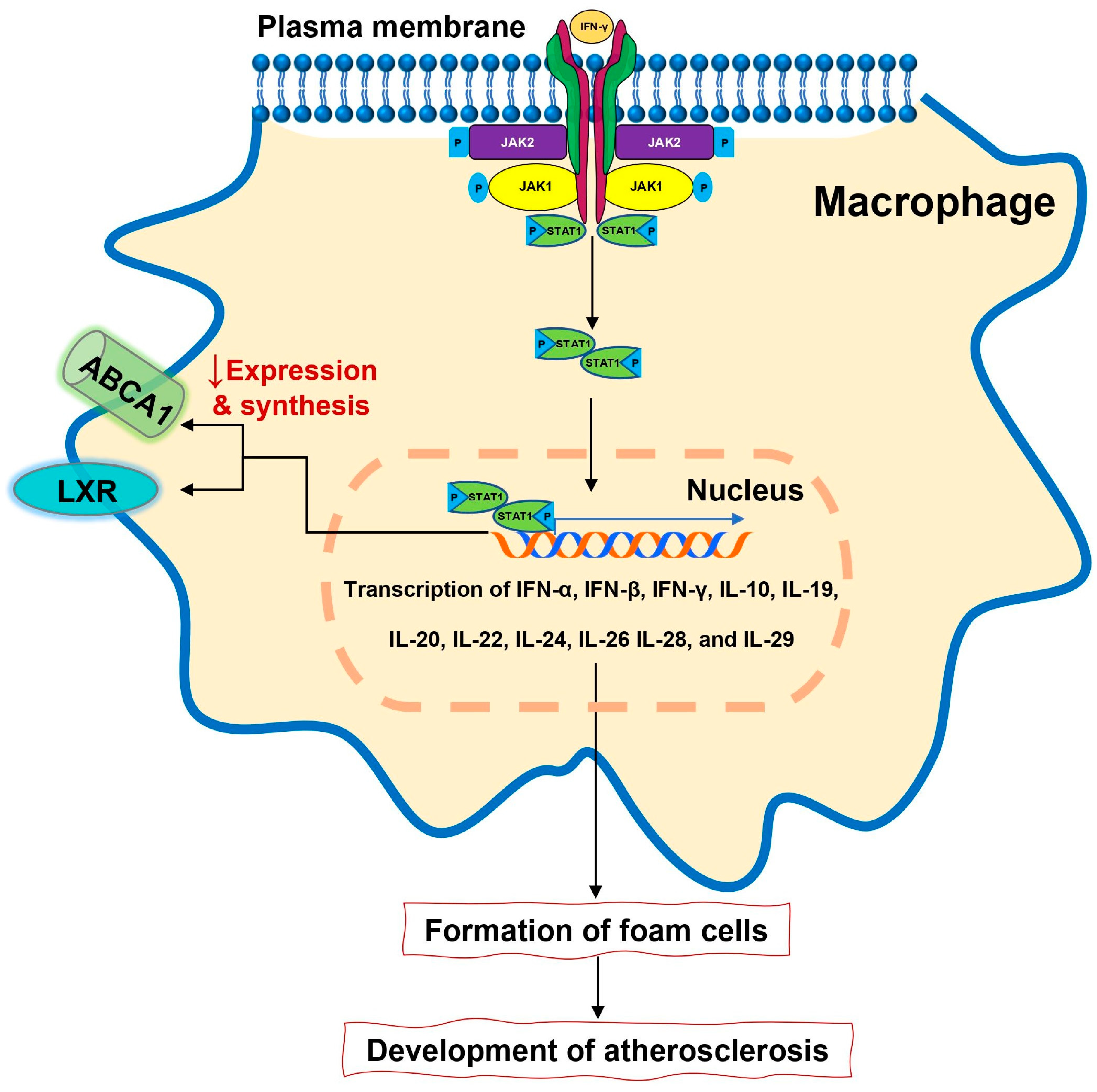
1. Introduction
2. JAK-STAT Pathway
3. The Role of JAK/STAT Pathway in Immunity and Autoimmunity
4. JAK Inhibitors and Rheumatoid Arthritis
5. Safety Issues of JAK Inhibitors
5.1. Heart Failure
5.2. Lipid Profile
5.2.1. JAK Inhibitors and Lipid Profile
5.2.2. Thromboembolic Events
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| RA | Rheumatoid arthritis |
| DMARDs | Disease-modifying antirheumatic drugs |
| MTX | Methotrexate |
| JAK | Janus kinases |
| STAT | Signal transducers and activators of transcription |
| IFN | Interferon |
| TNF | Tumor necrosis factor |
| IL | Interleukin |
| SOCS | Suppressor of cytokine signaling |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| MACE | Major adverse cardiovascular events |
| LDL | Low-density lipoproteins |
| HDL | High-density lipoprotein |
| ABCA1 | ATP-binding cassette transporter |
| VTE | Venous thromboembolic event |
| TF | Tissue factor |
References
- Smolen, J.S.; Aletaha, D.; McInnes, I.B. Rheumatoid arthritis. Lancet 2016, 388, 2023–2038. [Google Scholar] [CrossRef]
- Kotyla, P.J.; Sliwinska-Kotyla, B.; Kucharz, E.J. Treatment with infliximab may contribute to the development of peripheral neuropathy among the patients with rheumatoid arthritis. Clin. Rheumatol. 2007, 26, 1595–1596. [Google Scholar] [CrossRef] [PubMed]
- Kotyla, P.J. Bimodal function of anti-tnf treatment: Shall we be concerned about anti-tnf treatment in patients with rheumatoid arthritis and heart failure? Int. J. Mol. Sci. 2018, 19, 1739. [Google Scholar] [CrossRef] [PubMed]
- Sartori, N.S.; de Andrade, N.P.B.; da Silva Chakr, R.M. Incidence of tuberculosis in patients receiving anti-tnf therapy for rheumatic diseases: A systematic review. Clin. Rheumatol. 2020, 39, 1439–1447. [Google Scholar] [CrossRef]
- Angelini, J.; Talotta, R.; Roncato, R.; Fornasier, G.; Barbiero, G.; Dal Cin, L.; Brancati, S.; Scaglione, F. Jak-inhibitors for the treatment of rheumatoid arthritis: A focus on the present and an outlook on the future. Biomolecules 2020, 10, 1002. [Google Scholar] [CrossRef]
- Jamilloux, Y.; El Jammal, T.; Vuitton, L.; Gerfaud-Valentin, M.; Kerever, S.; Sève, P. Jak inhibitors for the treatment of autoimmune and inflammatory diseases. Autoimmun. Rev. 2019, 18, 102390. [Google Scholar] [CrossRef]
- Singh, S.; Singh, S. Jak-stat inhibitors: Immersing therapeutic approach for management of rheumatoid arthritis. Int. Immunopharmacol. 2020, 86, 106731. [Google Scholar] [CrossRef]
- Xu, P.; Shen, P.; Yu, B.; Xu, X.; Ge, R.; Cheng, X.; Chen, Q.; Bian, J.; Li, Z.; Wang, J. Janus kinases (jaks): The efficient therapeutic targets for autoimmune diseases and myeloproliferative disorders. Eur. J. Med. Chem. 2020, 192, 112155. [Google Scholar] [CrossRef]
- El Jammal, T.; Gerfaud-Valentin, M.; Sève, P.; Jamilloux, Y. Inhibition of jak/stat signaling in rheumatologic disorders: The expanding spectrum. Jt. Bone Spine 2020, 87, 119–129. [Google Scholar] [CrossRef]
- Kotyla, P.J. Are janus kinase inhibitors superior over classic biologic agents in ra patients? Biomed. Res. Int. 2018, 2018, 7492904. [Google Scholar] [CrossRef]
- Ghoreschi, K.; Laurence, A.; O’Shea, J.J. Janus kinases in immune cell signaling. Immunol. Rev. 2009, 228, 273–287. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, M.; McVicar, D.W.; Johnston, J.A.; Blake, T.B.; Chen, Y.-Q.; Lal, B.K.; Lloyd, A.R.; Kelvin, D.J.; Staples, J.E.; Ortaldo, J.R. Molecular cloning of l-jak, a janus family protein-tyrosine kinase expressed in natural killer cells and activated leukocytes. Proc. Natl. Acad. Sci. USA 1994, 91, 6374–6378. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Murray, P.J. Cytokine signaling modules in inflammatory responses. Immunity 2008, 28, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Villarino, A.V.; Kanno, Y.; O’Shea, J.J. Mechanisms and consequences of jak–stat signaling in the immune system. Nat. Immunol. 2017, 18, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Levy, D.E. Comparative evolutionary genomics of the stat family of transcription factors. JAK-STAT 2012, 1, 23–33. [Google Scholar] [CrossRef]
- Zouein, F.A.; Duhé, R.J.; Booz, G.W. Jaks go nuclear: Emerging role of nuclear jak1 and jak2 in gene expression and cell growth. Growth Factors 2011, 29, 245–252. [Google Scholar] [CrossRef]
- Schwartz, D.M.; Bonelli, M.; Gadina, M.; O’shea, J.J. Type i/ii cytokines, jaks, and new strategies for treating autoimmune diseases. Nat. Rev. Rheumatol. 2016, 12, 25. [Google Scholar] [CrossRef]
- Waickman, A.T.; Park, J.-Y.; Park, J.-H. The common γ-chain cytokine receptor: Tricks-and-treats for t cells. Cell. Mol. Life Sci. 2016, 73, 253–269. [Google Scholar] [CrossRef]
- Nicola, N.A.; Babon, J.J. Leukemia inhibitory factor (lif). Cytokine Growth Factor Rev. 2015, 26, 533–544. [Google Scholar] [CrossRef]
- Boulanger, M.J.; Bankovich, A.J.; Kortemme, T.; Baker, D.; Garcia, K.C. Convergent mechanisms for recognition of divergent cytokines by the shared signaling receptor gp130. Mol. Cell 2003, 12, 577–589. [Google Scholar] [CrossRef]
- Murakami, M.; Kamimura, D.; Hirano, T. Pleiotropy and specificity: Insights from the interleukin 6 family of cytokines. Immunity 2019, 50, 812–831. [Google Scholar] [CrossRef] [PubMed]
- Collison, L.W.; Delgoffe, G.M.; Guy, C.S.; Vignali, K.M.; Chaturvedi, V.; Fairweather, D.; Satoskar, A.R.; Garcia, K.C.; Hunter, C.A.; Drake, C.G. The composition and signaling of the il-35 receptor are unconventional. Nat. Immunol. 2012, 13, 290. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Huang, W.; Costa, M.M.; Martin, S.A.; Secombes, C.J. Two copies of the genes encoding the subunits of putative interleukin (il)-4/il-13 receptors, il-4ralpha, il-13ralpha1 and il-13ralpha2, have been identified in rainbow trout (oncorhynchus mykiss) and have complex patterns of expression and modulation. Immunogenetics 2011, 63, 235–253. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Schwartz, D.M.; Villarino, A.V.; Gadina, M.; McInnes, I.B.; Laurence, A. The jak-stat pathway: Impact on human disease and therapeutic intervention. Annu. Rev. Med. 2015, 66, 311–328. [Google Scholar] [CrossRef]
- Esch, A.; Masiarz, A.; Mossner, S.; Moll, J.M.; Grötzinger, J.; Schröder, J.; Scheller, J.; Floss, D.M. Deciphering site 3 interactions of interleukin 12 and interleukin 23 with their cognate murine and human receptors. J. Biol. Chem. 2020, 295, 10478–10492. [Google Scholar] [CrossRef]
- Reddy, V.; Cohen, S. Jak inhibitors: What is new? Curr. Rheumatol. Rep. 2020, 22, 50. [Google Scholar] [CrossRef]
- Tokumasa, N.; Suto, A.; Kagami, S.-i.; Furuta, S.; Hirose, K.; Watanabe, N.; Saito, Y.; Shimoda, K.; Iwamoto, I.; Nakajima, H. Expression of tyk2 in dendritic cells is required for il-12, il-23, and ifn-γ production and the induction of th1 cell differentiation. Blood J. Am. Soc. Hematol. 2007, 110, 553–560. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Holland, S.M.; Staudt, L.M. Jaks and stats in immunity, immunodeficiency, and cancer. N. Engl. J. Med. 2013, 368, 161–170. [Google Scholar] [CrossRef]
- Holland, S.M.; DeLeo, F.R.; Elloumi, H.Z.; Hsu, A.P.; Uzel, G.; Brodsky, N.; Freeman, A.F.; Demidowich, A.; Davis, J.; Turner, M.L. Stat3 mutations in the hyper-ige syndrome. N. Engl. J. Med. 2007, 357, 1608–1619. [Google Scholar] [CrossRef]
- Cho, J.H.; Gregersen, P.K. Genomics and the multifactorial nature of human autoimmune disease. N. Engl. J. Med. 2011, 365, 1612–1623. [Google Scholar] [CrossRef]
- Remmers, E.F.; Cosan, F.; Kirino, Y.; Ombrello, M.J.; Abaci, N.; Satorius, C.; Le, J.M.; Yang, B.; Korman, B.D.; Cakiris, A. Genome-wide association study identifies variants in the mhc class i, il10, and il23r-il12rb2 regions associated with behcet’s disease. Nat. Genet. 2010, 42, 698–702. [Google Scholar] [CrossRef] [PubMed]
- Duerr, R.H.; Taylor, K.D.; Brant, S.R.; Rioux, J.D.; Silverberg, M.S.; Daly, M.J.; Steinhart, A.H.; Abraham, C.; Regueiro, M.; Griffiths, A. A genome-wide association study identifies il23r as an inflammatory bowel disease gene. Science (N.Y.) 2006, 314, 1461–1463. [Google Scholar] [CrossRef] [PubMed]
- Remmers, E.F.; Plenge, R.M.; Lee, A.T.; Graham, R.R.; Hom, G.; Behrens, T.W.; De Bakker, P.I.; Le, J.M.; Lee, H.-S.; Batliwalla, F. Stat4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N. Engl. J. Med. 2007, 357, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Alunno, A.; Padjen, I.; Fanouriakis, A.; Boumpas, D.T. Pathogenic and therapeutic relevance of jak/stat signaling in systemic lupus erythematosus: Integration of distinct inflammatory pathways and the prospect of their inhibition with an oral agent. Cells 2019, 8, 898. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, S.; Jouanguy, E.; Al-Hajjar, S.; Fieschi, C.; Al-Mohsen, I.Z.; Al-Jumaah, S.; Yang, K.; Chapgier, A.; Eidenschenk, C.; Eid, P. Impaired response to interferon-α/β and lethal viral disease in human stat1 deficiency. Nat. Genet. 2003, 33, 388–391. [Google Scholar] [CrossRef]
- Chapgier, A.; Kong, X.-F.; Boisson-Dupuis, S.; Jouanguy, E.; Averbuch, D.; Feinberg, J.; Zhang, S.-Y.; Bustamante, J.; Vogt, G.; Lejeune, J. A partial form of recessive stat1 deficiency in humans. J. Clin. Investig. 2009, 119, 1502–1514. [Google Scholar] [CrossRef]
- Baechler, E.C.; Batliwalla, F.M.; Karypis, G.; Gaffney, P.M.; Ortmann, W.A.; Espe, K.J.; Shark, K.B.; Grande, W.J.; Hughes, K.M.; Kapur, V.; et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. USA 2003, 100, 2610–2615. [Google Scholar] [CrossRef]
- Barrat, F.J.; Crow, M.K.; Ivashkiv, L.B. Interferon target-gene expression and epigenomic signatures in health and disease. Nat. Immunol. 2019, 20, 1574–1583. [Google Scholar] [CrossRef]
- Gallay, L.; Mouchiroud, G.; Chazaud, B. Interferon-signature in idiopathic inflammatory myopathies. Curr. Opin. Rheumatol. 2019, 31, 634–642. [Google Scholar] [CrossRef]
- Marketos, N.; Cinoku, I.; Rapti, A.; Mavragani, C.P. Type i interferon signature in sjögren’s syndrome: Pathophysiological and clinical implications. Clin. Exp. Rheumatol. 2019, 37 (Suppl. 118), 185–191. [Google Scholar]
- Rönnblom, L.; Eloranta, M.L. The interferon signature in autoimmune diseases. Curr. Opin. Rheumatol. 2013, 25, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Zhao, M.; Chang, C.; Wu, H.; Lu, Q. Type i interferons in the pathogenesis and treatment of autoimmune diseases. Clin. Rev. Allergy Immunol. 2020, 59, 248–272. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, T.; Masaki, T.; Hirai, T.; Doi, S.; Kuratsune, M.; Arihiro, K.; Kohno, N.; Yorioka, N. Activation of signal transducer and activator of transcription 3 correlates with cell proliferation and renal injury in human glomerulonephritis. Nephrol. Dial. Transplant. 2008, 23, 3418–3426. [Google Scholar] [CrossRef] [PubMed]
- Justiz Vaillant, A.A.; Qurie, A. Interleukin. In Statpearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2020. [Google Scholar]
- Rochman, Y.; Spolski, R.; Leonard, W.J. New insights into the regulation of t cells by γ c family cytokines. Nat. Rev. Immunol. 2009, 9, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.; Kishimoto, T. Il-6: Regulator of treg/th17 balance. Eur. J. Immunol. 2010, 40, 1830–1835. [Google Scholar] [CrossRef]
- Fragoulis, G.E.; McInnes, I.B.; Siebert, S. Jak-inhibitors. New players in the field of immune-mediated diseases, beyond rheumatoid arthritis. Rheumatology (Oxford) 2019, 58, i43–i54. [Google Scholar] [CrossRef]
- Emery, P.; Pope, J.E.; Kruger, K.; Lippe, R.; DeMasi, R.; Lula, S.; Kola, B. Efficacy of monotherapy with biologics and jak inhibitors for the treatment of rheumatoid arthritis: A systematic review. Adv. Ther. 2018, 35, 1535–1563. [Google Scholar] [CrossRef]
- Kerschbaumer, A.; Sepriano, A.; Smolen, J.S.; van der Heijde, D.; Dougados, M.; van Vollenhoven, R.; McInnes, I.B.; Bijlsma, J.W.J.; Burmester, G.R.; de Wit, M.; et al. Efficacy of pharmacological treatment in rheumatoid arthritis: A systematic literature research informing the 2019 update of the eular recommendations for management of rheumatoid arthritis. Ann. Rheum. Dis. 2020, 79, 744–759. [Google Scholar] [CrossRef]
- Taylor, P.C.; Keystone, E.C.; van der Heijde, D.; Weinblatt, M.E.; del Carmen Morales, L.; Reyes Gonzaga, J.; Yakushin, S.; Ishii, T.; Emoto, K.; Beattie, S. Baricitinib versus placebo or adalimumab in rheumatoid arthritis. N. Engl. J. Med. 2017, 376, 652–662. [Google Scholar] [CrossRef]
- Fleischmann, R.; Pangan, A.L.; Song, I.H.; Mysler, E.; Bessette, L.; Peterfy, C.; Durez, P.; Ostor, A.J.; Li, Y.; Zhou, Y. Upadacitinib versus placebo or adalimumab in patients with rheumatoid arthritis and an inadequate response to methotrexate: Results of a phase iii, double-blind, randomized controlled trial. Arthritis Rheumatol. 2019, 71, 1788–1800. [Google Scholar] [CrossRef]
- Fleischmann, R.; Mysler, E.; Hall, S.; Kivitz, A.J.; Moots, R.J.; Luo, Z.; DeMasi, R.; Soma, K.; Zhang, R.; Takiya, L. Efficacy and safety of tofacitinib monotherapy, tofacitinib with methotrexate, and adalimumab with methotrexate in patients with rheumatoid arthritis (oral strategy): A phase 3b/4, double-blind, head-to-head, randomised controlled trial. Lancet 2017, 390, 457–468. [Google Scholar] [CrossRef]
- Wollenhaupt, J.; Lee, E.-B.; Curtis, J.R.; Silverfield, J.; Terry, K.; Soma, K.; Mojcik, C.; DeMasi, R.; Strengholt, S.; Kwok, K. Safety and efficacy of tofacitinib for up to 9.5 years in the treatment of rheumatoid arthritis: Final results of a global, open-label, long-term extension study. Arthritis Res. Ther. 2019, 21, 89. [Google Scholar] [CrossRef] [PubMed]
- Keystone, E.C.; Genovese, M.C.; Schlichting, D.E.; de la Torre, I.; Beattie, S.D.; Rooney, T.P.; Taylor, P.C. Safety and efficacy of baricitinib through 128 weeks in an open-label, longterm extension study in patients with rheumatoid arthritis. J. Rheumatol. 2018, 45, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.C.; Weinblatt, M.E.; Burmester, G.R.; Rooney, T.P.; Witt, S.; Walls, C.D.; Issa, M.; Salinas, C.A.; Saifan, C.; Zhang, X. Cardiovascular safety during treatment with baricitinib in rheumatoid arthritis. Arthritis Rheumatol. 2019, 71, 1042–1055. [Google Scholar] [CrossRef] [PubMed]
- El Jammal, T.; Sève, P.; Gerfaud-Valentin, M.; Jamilloux, Y. State of the art: Approved and emerging jak inhibitors for rheumatoid arthritis. Expert Opin. Pharmacother. 2020, 1–14. [Google Scholar] [CrossRef]
- Spolski, R.; Gromer, D.; Leonard, W.J. The γ c family of cytokines: Fine-tuning signals from il-2 and il-21 in the regulation of the immune response. F1000Research 2017, 6, 1872. [Google Scholar] [CrossRef]
- Fukuzawa, J.; Booz, G.W.; Hunt, R.A.; Shimizu, N.; Karoor, V.; Baker, K.M.; Dostal, D.E. Cardiotrophin-1 increases angiotensinogen mrna in rat cardiac myocytes through stat3: An autocrine loop for hypertrophy. Hypertension 2000, 35, 1191–1196. [Google Scholar] [CrossRef]
- Hirota, H.; Chen, J.; Betz, U.A.; Rajewsky, K.; Gu, Y.; Ross, J., Jr.; Müller, W.; Chien, K.R. Loss of a gp130 cardiac muscle cell survival pathway is a critical event in the onset of heart failure during biomechanical stress. Cell 1999, 97, 189–198. [Google Scholar] [CrossRef]
- Li, H.; Cen, K.; Sun, W.; Feng, B. Predictive value of blood interleukin-6 level in patients with acute coronary syndrome: A meta-analysis. Immunol. Investig. 2020, 1–13. [Google Scholar] [CrossRef]
- Xu, L.; Yan, J.; Zhang, F.; Zhou, C.; Fan, T.; Chen, X.; Cui, X.; Zhou, H.; Liang, Y. Use of inflammatory biomarkers and real-time cardiac catheterisation to evaluate the left ventricular diastolic function in patients with diastolic heart failure. Heart Lung Circ. 2020. [Google Scholar] [CrossRef]
- Ridker, P.M.; Libby, P.; MacFadyen, J.G.; Thuren, T.; Ballantyne, C.; Fonseca, F.; Koenig, W.; Shimokawa, H.; Everett, B.M.; Glynn, R.J. Modulation of the interleukin-6 signalling pathway and incidence rates of atherosclerotic events and all-cause mortality: Analyses from the canakinumab anti-inflammatory thrombosis outcomes study (cantos). Eur. Heart J. 2018, 39, 3499–3507. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; MacFadyen, J.G.; Glynn, R.J.; Bradwin, G.; Hasan, A.A.; Rifai, N. Comparison of interleukin-6, c-reactive protein, and low-density lipoprotein cholesterol as biomarkers of residual risk in contemporary practice: Secondary analyses from the cardiovascular inflammation reduction trial. Eur. Heart J. 2020. [Google Scholar] [CrossRef]
- Kanda, T.; Takahashi, T. Interleukin-6 and cardiovascular diseases. Jpn. Heart J. 2004, 45, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, A.S.; Martinsson, A.; Wretlind, B.; Ahnve, S. Il-6 levels in acute and post myocardial infarction: Their relation to crp levels, infarction size, left ventricular systolic function, and heart failure. Eur. J. Intern. Med. 2004, 15, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, B.; Xie, Y.; Xia, N.; Li, M.; Gao, G. Statin rosuvastatin inhibits apoptosis of human coronary artery endothelial cells through upregulation of the jak2/stat3 signaling pathway. Mol. Med. Rep. 2020, 22, 2052–2062. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Yu, J.; Xie, P.; Maimaitili, Y.; Wang, J.; Yang, L.; Ma, H.; Zhang, X.; Yang, Y.; Zheng, H. Sevoflurane postconditioning protects the myocardium against ischemia/reperfusion injury via activation of the jak2–stat3 pathway. PeerJ 2017, 5, e3196. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Hu, X.; Guo, X.; Zhang, B.; Xu, W.; Jiang, H. Promoting effects of il-23 on myocardial ischemia and reperfusion are associated with increased expression of il-17a and upregulation of the jak2-stat3 signaling pathway. Mol. Med. Rep. 2017, 16, 9309–9316. [Google Scholar] [CrossRef]
- Liao, Y.H.; Xia, N.; Zhou, S.F.; Tang, T.T.; Yan, X.X.; Lv, B.J.; Nie, S.F.; Wang, J.; Iwakura, Y.; Xiao, H.; et al. Interleukin-17a contributes to myocardial ischemia/reperfusion injury by regulating cardiomyocyte apoptosis and neutrophil infiltration. J. Am. Coll. Cardiol. 2012, 59, 420–429. [Google Scholar] [CrossRef]
- Charles-Schoeman, C.; Wicker, P.; Gonzalez-Gay, M.A.; Boy, M.; Zuckerman, A.; Soma, K.; Geier, J.; Kwok, K.; Riese, R. Cardiovascular safety findings in patients with rheumatoid arthritis treated with tofacitinib, an oral janus kinase inhibitor. Semin. Arthritis Rheum. 2016, 46, 261–271. [Google Scholar] [CrossRef]
- Xie, W.; Huang, Y.; Xiao, S.; Sun, X.; Fan, Y.; Zhang, Z. Impact of janus kinase inhibitors on risk of cardiovascular events in patients with rheumatoid arthritis: Systematic review and meta-analysis of randomised controlled trials. Ann. Rheum. Dis. 2019, 78, 1048–1054. [Google Scholar] [CrossRef]
- Charles-Schoeman, C.; DeMasi, R.; Valdez, H.; Soma, K.; Hwang, L.J.; Boy, M.G.; Biswas, P.; McInnes, I.B. Risk factors for major adverse cardiovascular events in phase iii and long-term extension studies of tofacitinib in patients with rheumatoid arthritis. Arthritis Rheumatol. 2019, 71, 1450–1459. [Google Scholar] [CrossRef] [PubMed]
- Choy, E.; Sattar, N. Interpreting lipid levels in the context of high-grade inflammatory states with a focus on rheumatoid arthritis: A challenge to conventional cardiovascular risk actions. Ann. Rheum. Dis. 2009, 68, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Johnsson, H.; Panarelli, M.; Cameron, A.; Sattar, N. Analysis and modelling of cholesterol and high-density lipoprotein cholesterol changes across the range of c-reactive protein levels in clinical practice as an aid to better understanding of inflammation–lipid interactions. Ann. Rheum. Dis. 2014, 73, 1495–1499. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J.; Peters, M.J.; McInnes, I.B.; Sattar, N. Changes in lipid levels with inflammation and therapy in ra: A maturing paradigm. Nat. Rev. Rheumatol. 2013, 9, 513. [Google Scholar] [CrossRef]
- Souto, A.; Salgado, E.; Maneiro, J.R.; Mera, A.; Carmona, L.; Gómez-Reino, J.J. Lipid profile changes in patients with chronic inflammatory arthritis treated with biologic agents and tofacitinib in randomized clinical trials: A systematic review and meta-analysis. Arthritis Rheumatol. 2015, 67, 117–127. [Google Scholar] [CrossRef]
- Smolen, J.S.; Beaulieu, A.; Rubbert-Roth, A.; Ramos-Remus, C.; Rovensky, J.; Alecock, E.; Woodworth, T.; Alten, R. Effect of interleukin-6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (option study): A double-blind, placebo-controlled, randomised trial. Lancet 2008, 371, 987–997. [Google Scholar] [CrossRef]
- Singh, S.; Fumery, M.; Singh, A.G.; Singh, N.; Prokop, L.J.; Dulai, P.S.; Sandborn, W.J.; Curtis, J.R. Comparative risk of cardiovascular events with biologic and synthetic disease-modifying antirheumatic drugs in patients with rheumatoid arthritis: A systematic review and meta-analysis. Arthritis Care Res. 2020, 72, 561–576. [Google Scholar] [CrossRef]
- Castagné, B.; Viprey, M.; Martin, J.; Schott, A.-M.; Cucherat, M.; Soubrier, M. Cardiovascular safety of tocilizumab: A systematic review and network meta-analysis. PLoS ONE 2019, 14, e0220178. [Google Scholar] [CrossRef]
- Ferraz-Amaro, I.; Hernández-Hernández, M.V.; Tejera-Segura, B.; Delgado-Frías, E.; Macía-Díaz, M.; Machado, J.D.; Diaz-González, F. Effect of il-6 receptor blockade on proprotein convertase subtilisin/kexin type-9 and cholesterol efflux capacity in rheumatoid arthritis patients. Horm. Metab. Res. 2019, 51, 200–209. [Google Scholar] [CrossRef]
- Strang, A.C.; Bisoendial, R.J.; Kootte, R.S.; Schulte, D.M.; Dallinga-Thie, G.M.; Levels, J.H.; Kok, M.; Vos, K.; Tas, S.W.; Tietge, U.J. Pro-atherogenic lipid changes and decreased hepatic ldl receptor expression by tocilizumab in rheumatoid arthritis. Atherosclerosis 2013, 229, 174–181. [Google Scholar] [CrossRef]
- Greco, D.; Gualtierotti, R.; Agosti, P.; Adorni, M.P.; Ingegnoli, F.; Rota, M.; Bernini, F.; Meroni, P.L.; Ronda, N. Anti-atherogenic modification of serum lipoprotein function in patients with rheumatoid arthritis after tocilizumab treatment, a pilot study. J. Clin. Med. 2020, 9, 2157. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Baos, S.; Barrasa, J.I.; Gratal, P.; Larrañaga-Vera, A.; Prieto-Potin, I.; Herrero-Beaumont, G.; Largo, R. Tofacitinib restores the inhibition of reverse cholesterol transport induced by inflammation: Understanding the lipid paradox associated with rheumatoid arthritis. Br. J. Pharmacol. 2017, 174, 3018–3031. [Google Scholar] [CrossRef]
- Hodge, J.A.; Kawabata, T.T.; Krishnaswami, S.; Clark, J.D.; Telliez, J.-B.; Dowty, M.E.; Menon, S.; Lamba, M.; Zwillich, S. The mechanism of action of tofacitinib-an oral janus kinase inhibitor for the treatment of rheumatoid arthritis. Clin. Exp. Rheumatol. 2016, 34, 318–328. [Google Scholar] [PubMed]
- Li, Y.; Yuan, L.; Yang, J.; Lei, Y.; Zhang, H.; Xia, L.; Shen, H.; Lu, J. Changes in serum cytokines may predict therapeutic efficacy of tofacitinib in rheumatoid arthritis. Mediat. Inflamm. 2019, 2019. [Google Scholar] [CrossRef] [PubMed]
- Rusinova, I.; Forster, S.; Yu, S.; Kannan, A.; Masse, M.; Cumming, H.; Chapman, R.; Hertzog, P.J. Interferome v2. 0: An updated database of annotated interferon-regulated genes. Nucleic Acids Res. 2012, 41, D1040–D1046. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Xu, R.; He, L.; Xu, T.; Zhang, Z.; Han, D.; Du, J. Comparison of pathogenic mechanisms underlying single and recurrent venous thromboembolism based on gene expression profiling. Ann. Vasc. Surg. 2016, 36, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.J.; Lin, K.C.; Huang, S.Y.; Thomas, P.A.; Wu, Y.H.; Wu, H.C.; Lin, K.H.; Sheu, J.R. Role of a janus kinase 2-dependent signaling pathway in platelet activation. Thromb. Res. 2014, 133, 1088–1096. [Google Scholar] [CrossRef] [PubMed]
- Ayer, M.; Menken, İ.; Yamak, M.; Ayer, F.A.; Kırkızlar, O.; Burak Aktuğlu, M. The impact of mean platelet volume (mpv) and jak-2 mutation on thrombosis in chronic myeloproliferative diseases. Indian J. Hematol. Blood Transfus. Off. J. Indian Soc. Hematol. Blood Transfus. 2017, 33, 181–187. [Google Scholar] [CrossRef]
- Nadir, Y. Heparanase in the coagulation system. Adv. Exp. Med. Biol. 2020, 1221, 771–784. [Google Scholar]
- Charles-Schoeman, C.; Fleischmann, R.; Davignon, J.; Schwartz, H.; Turner, S.M.; Beysen, C.; Milad, M.; Hellerstein, M.K.; Luo, Z.; Kaplan, I.V.; et al. Potential mechanisms leading to the abnormal lipid profile in patients with rheumatoid arthritis versus healthy volunteers and reversal by tofacitinib. Arthritis Rheumatol. (Hoboken N.J.) 2015, 67, 616–625. [Google Scholar] [CrossRef]
- Smolen, J.S.; Genovese, M.C.; Takeuchi, T.; Hyslop, D.L.; Macias, W.L.; Rooney, T.; Chen, L.; Dickson, C.L.; Riddle Camp, J.; Cardillo, T.E.; et al. Safety profile of baricitinib in patients with active rheumatoid arthritis with over 2 years median time in treatment. J. Rheumatol. 2019, 46, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, R.; Meirovitz, A.; Hirshoren, N.; Bulvik, R.; Binder, A.; Rubinstein, A.M.; Elkin, M. Versatile role of heparanase in inflammation. Matrix Biol. J. Int. Soc. Matrix Biol. 2013, 32, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Li, R.W.; Freeman, C.; Yu, D.; Hindmarsh, E.J.; Tymms, K.E.; Parish, C.R.; Smith, P.N. Dramatic regulation of heparanase activity and angiogenesis gene expression in synovium from patients with rheumatoid arthritis. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 2008, 58, 1590–1600. [Google Scholar] [CrossRef] [PubMed]
- Ghoti, H.; Ackerman, S.; Rivella, S.; Casu, C.; Nadir, Y. Heparanase level and procoagulant activity are increased in thalassemia and attenuated by jak-2 inhibition. Am. J. Pathol. 2020. [Google Scholar] [CrossRef]
- Febvre-James, M.; Lecureur, V.; Fardel, O. Potent repression of c-reactive protein (crp) expression by the jak1/2 inhibitor ruxolitinib in inflammatory human hepatocytes. Inflamm. Res. 2020, 69, 51–62. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Typical Drug Representatives | Mode of Action | Side Effects |
|---|---|---|
| csDMARDs | ||
| Methotrexate | At lower doses (as used in rheumatology) methotrexate inhibits the 5-aminoimidazole-4-carboxamide ribonucleotide transformylase. As a result, it increases extracellular pool of adenosine leading to an overall immunomodulatory activity | Oral ulcers, alopecia, nausea, hepatic and hematologic toxicities, and pneumonitis |
| tsDMARDs | ||
JAK inhibitors
| Inhibition of JAK molecule and subsequently JAK stat pathway resulting in reducing expression of cytokine related genes | Lipid profile disturbances, higher risk of infections, and thromboembolic complications |
| bDMARDs | ||
TNF-α inhibitors
| Inhibit (ameliorate) TNF activity upon targeted cells resulting in blockade of inflammatory response driven by this cytokine. | Infections, latent tuberculosis reactivation, neuropathy development (anectodical data), contraindicated in patients with over or latent heart failure, and risk of malignancy |
IL-6 inhibitors
| Inhibit IL-6 activity upon targeted cells. | Infections and lipid profile disturbances |
B-cell depletion
| Antibody against B-cell (anti CD-20). Depletion of whole lines of B-cells expressing CD-20 molecule. | Infections, infusion-related reactions, hepatitis B infection reactivation, cytokine released syndrome, and progressive multifocal leukoencephalopathy |
Inhibitors of co-stimulation
| CTLA-4 regulates T-cell priming, differentiation, and migration. CTLA-4 ensures homeostasis of regulatory T cells and mediates their immunosuppressive capacity. | serious allergic reactions including anaphylaxis and angioedema, latent tuberculosis reactivation, and higher risk for cancer (i.e., skin cancer) |
| DMARDs: disease-modifying antirheumatic drugs; csDMARDs: conventional synthetic DMARDs; tsDMARDs: targeted synthetic DMARDs; bDMARDs: biological DMARDs; TNF: tumor necrosis factor; IL: interleukin; CD: cluster of differentiation; CTLA: cytotoxic T-lymphocyte-associated protein. | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kotyla, P.J.; Islam, M.A.; Engelmann, M. Clinical Aspects of Janus Kinase (JAK) Inhibitors in the Cardiovascular System in Patients with Rheumatoid Arthritis. Int. J. Mol. Sci. 2020, 21, 7390. https://doi.org/10.3390/ijms21197390
Kotyla PJ, Islam MA, Engelmann M. Clinical Aspects of Janus Kinase (JAK) Inhibitors in the Cardiovascular System in Patients with Rheumatoid Arthritis. International Journal of Molecular Sciences. 2020; 21(19):7390. https://doi.org/10.3390/ijms21197390
Chicago/Turabian StyleKotyla, Przemysław J., Md Asiful Islam, and Małgorzata Engelmann. 2020. "Clinical Aspects of Janus Kinase (JAK) Inhibitors in the Cardiovascular System in Patients with Rheumatoid Arthritis" International Journal of Molecular Sciences 21, no. 19: 7390. https://doi.org/10.3390/ijms21197390
APA StyleKotyla, P. J., Islam, M. A., & Engelmann, M. (2020). Clinical Aspects of Janus Kinase (JAK) Inhibitors in the Cardiovascular System in Patients with Rheumatoid Arthritis. International Journal of Molecular Sciences, 21(19), 7390. https://doi.org/10.3390/ijms21197390