Insight into the Folding and Dimerization Mechanisms of the N-Terminal Domain from Human TDP-43


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
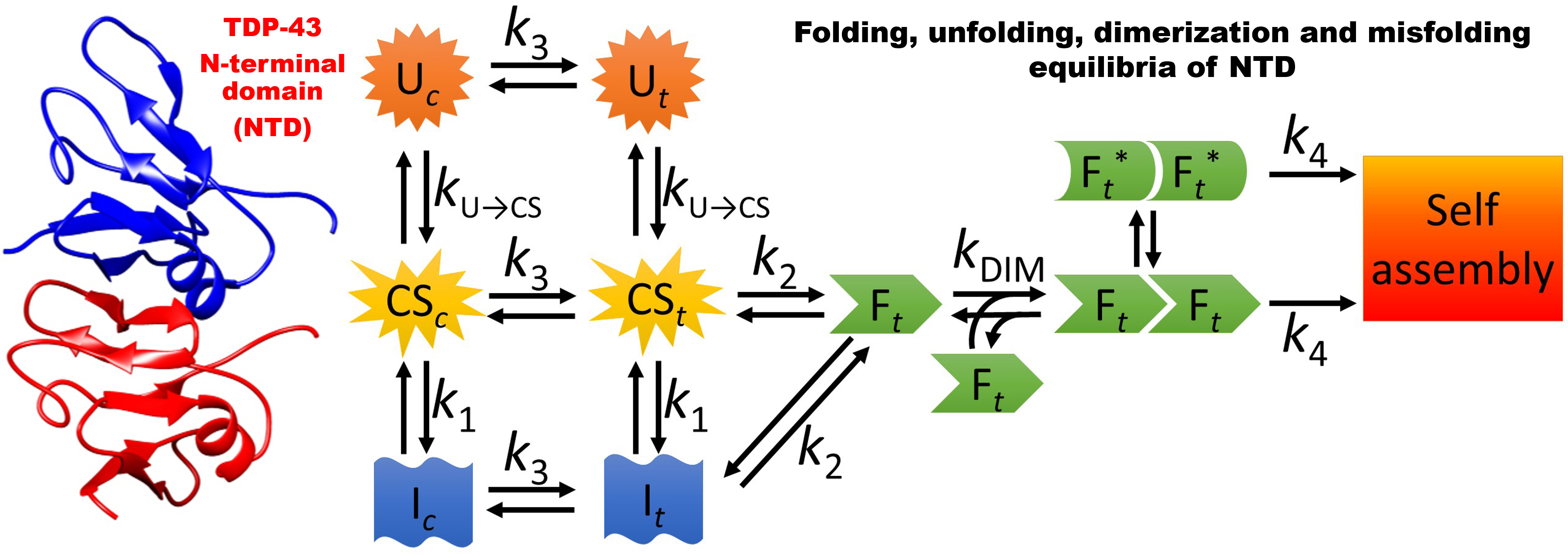
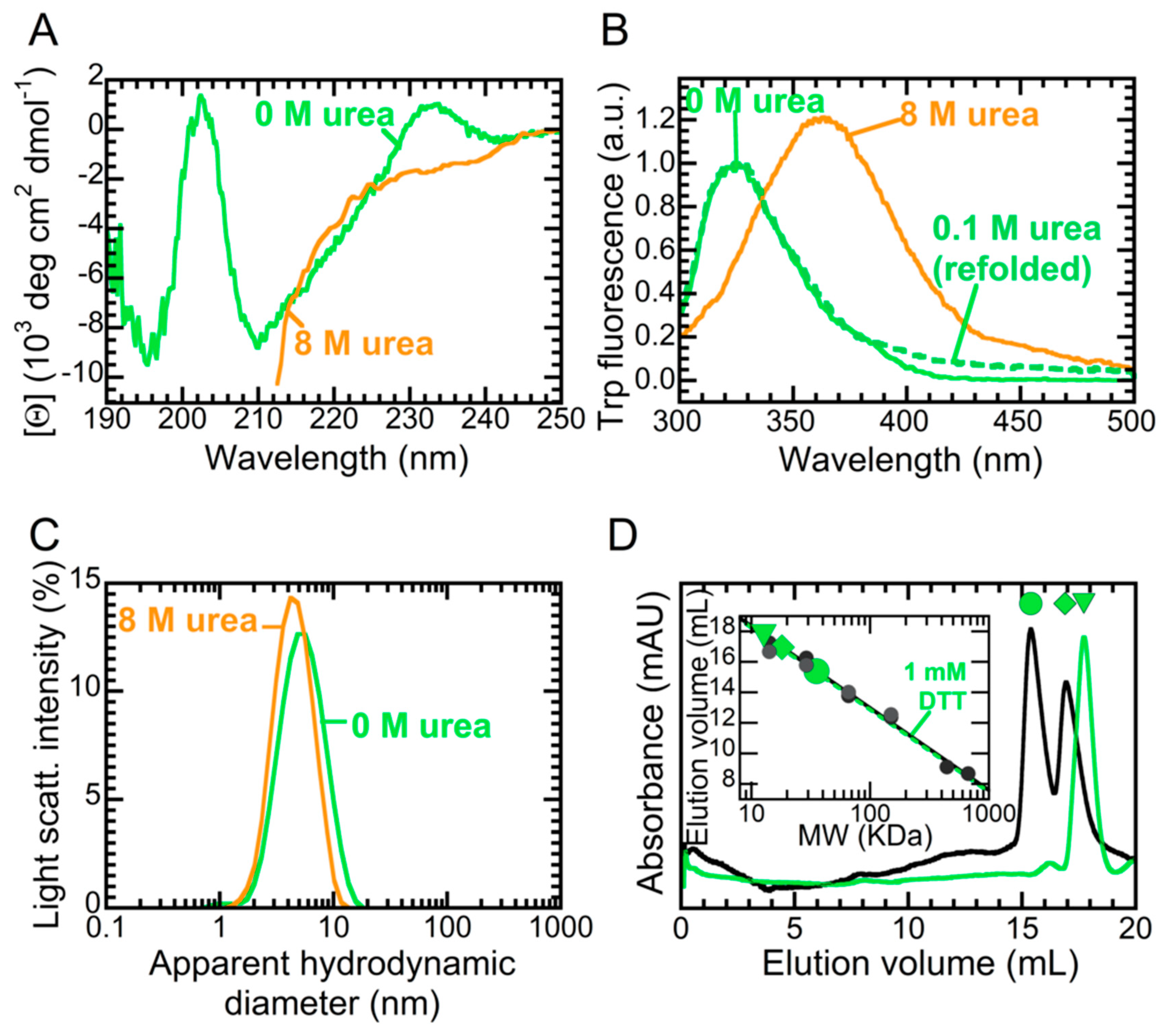
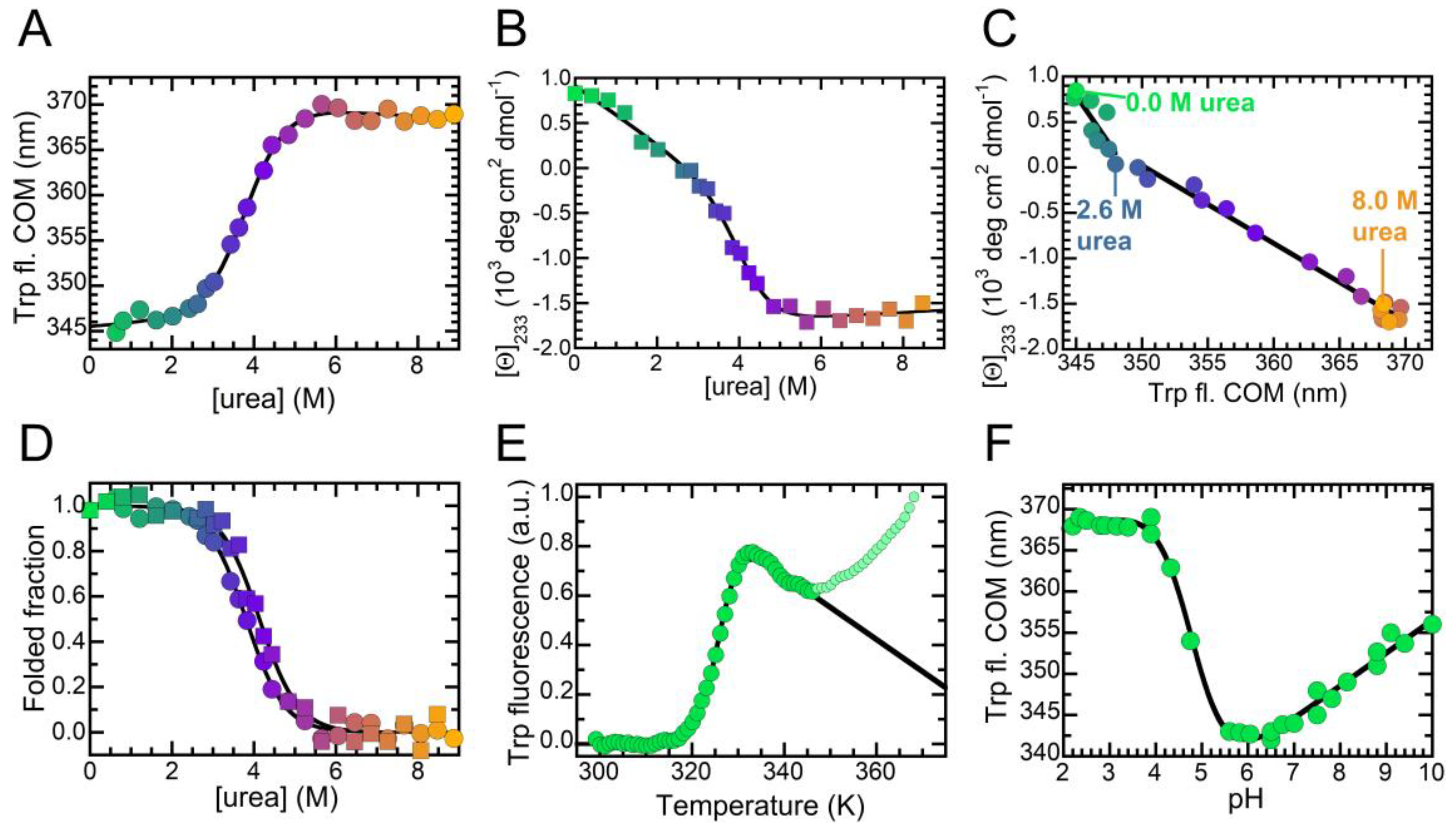
2.1. NTD is a Folded Dimer
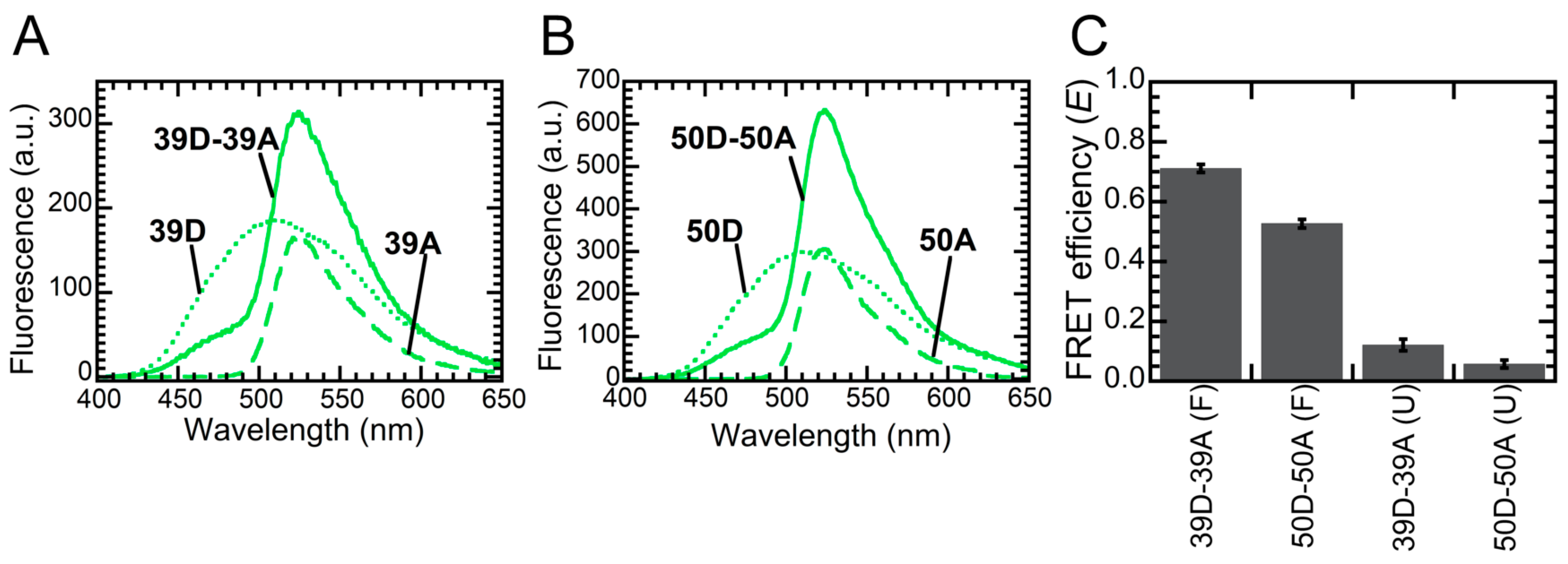
2.2. FRET Reveals That Monomers Interact Head-to-Tail in the NTD Dimer
2.3. The Folded State of NTD Is Susceptible to Small Amounts of Denaturant
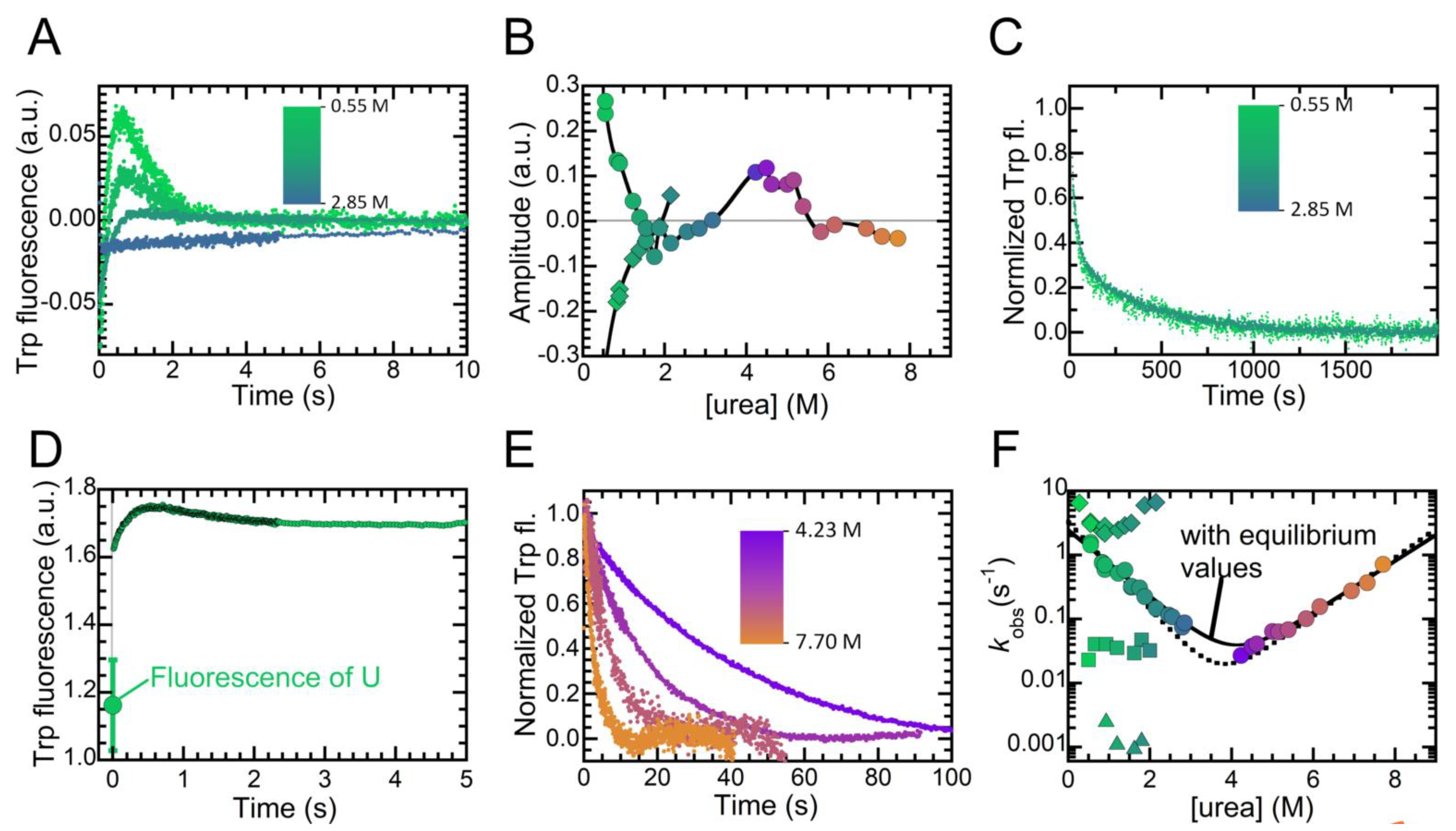
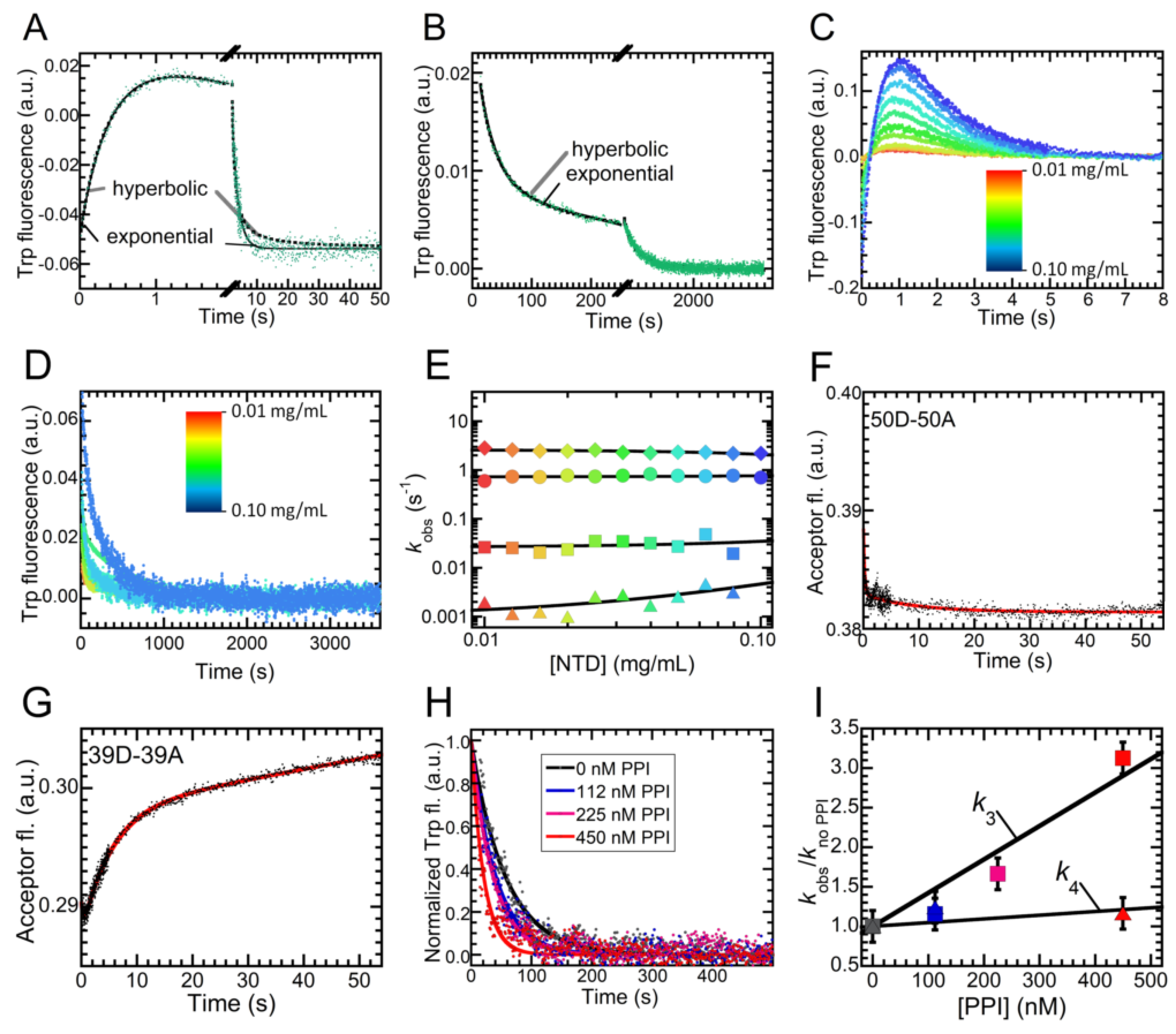
2.4. Folding Kinetics of NTD Reveal Four Exponential Phases and a High Energy Intermediate State
2.5. Folding of NTD Proceeds through a Hydrophobic Collapse Followed First by Structural Rearrangements, then by Dimerization
3. Discussion
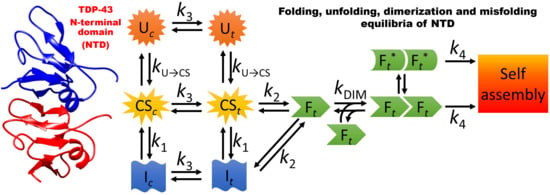
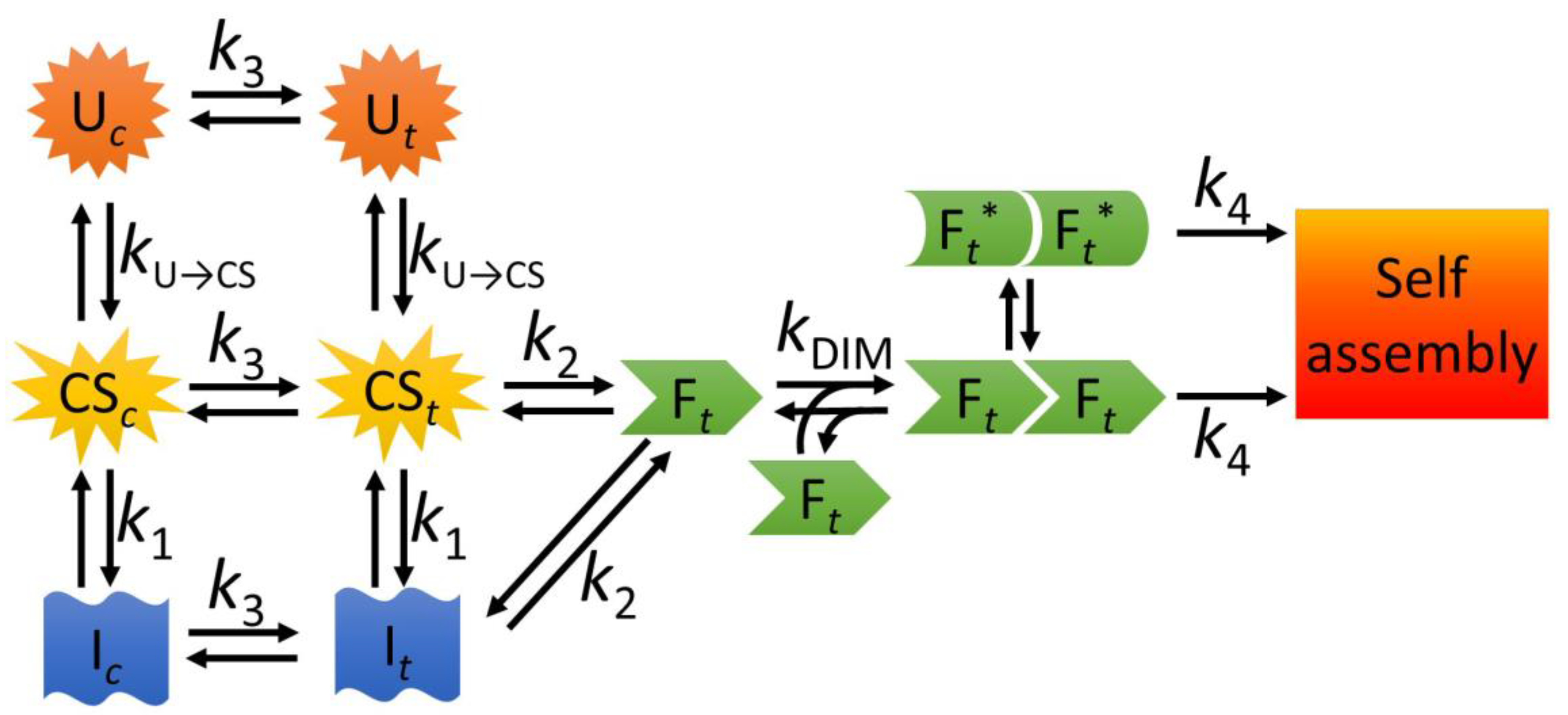
3.1. A Model for the Folding and Dimerization of NTD
3.2. The Role of Partially Folded Conformations in Misfolding
3.3. NTD as a Highly Plastic Protein Domain
4. Materials and Methods
4.1. Gene Cloning, Mutagenesis, Expression and Purification
4.2. Far-UV Circular Dichroism Spectroscopy
4.3. Fluorescence Spectroscopy
4.4. Dynamic Light Scattering
4.5. Analytical Gel Filtration
4.6. Labelling
4.7. Förster Resonance Energy Transfer (FRET)
4.8. Equilibrium Unfolding
4.9. Differential Scanning Fluorimetry
4.10. Acid-Induced Denaturation
4.11. Unfolding/Refolding Kinetics
4.12. Dimerization Kinetics Using FRET
4.13. Proline Isomerization
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 1,5-IAEDANS | 5-((((2-iodoacetyl)amino)ethyl)amino)naphthalene-1-sulfonic acid |
| 6-IAF | 6-iodoacetamidofluorescein |
| AD | Alzheimer’s Disease |
| ALS | Amyotrophic Lateral Sclerosis |
| β2m | β2-microglobulin |
| CD | Circular Dichroism |
| COM | Center Of spectral Mass |
| CS | Collapsed State |
| CTD | C-terminal Domain |
| DLS | Dynamic Light Scattering |
| DMF | Dimethylformamide |
| DMSO | Dimethyl sulfoxide |
| DSF | Differential Scanning Fluorimetry |
| DTT | Dithiothreitol |
| fALS | Familial Amyotrophic Lateral Sclerosis |
| F | Folded State |
| F* | Native-like State |
| FRET | Förster Resonance Energy Transfer |
| FTLD-U | Ubiquitin-positive FrontoTemporal Lobar Degeneration |
| HD | Huntington’s Disease |
| HT | High Tension |
| I | Intermediate State |
| MES | 2-(N-morpholino)ethanesulfonic acid |
| NES | Nuclear Export Signal |
| NLS | Nuclear Localization Signal |
| NMR | Nuclear Magnetic Resonance |
| NOE | Nuclear Overhauser Effect |
| NTD | N-terminal Domain |
| PCR | Polymerase Chain Reaction |
| PD | Parkinson’s Disease |
| PDB | Protein Data Bank |
| PF | Partially Folded State |
| PPI | Peptidyl Prolyl Isomerase |
| RRM1 | RNA Recognition Motif 1 |
| RRM2 | RNA Recognition Motif 2 |
| SH3 | Src Homology 3 |
| TDP-43 | TAR DNA-binding Protein 43 |
| U | Unfolded State |
References
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Chanson, J.B.; Echaniz-Laguna, A.; Vogel, T.; Mohr, M.; Benoilid, A.; Kaltenbach, G.; Kiesmann, M. TDP43-positive intraneuronal inclusions in a patient with motor neuron disease and Parkinson’s disease. Neurodegener. Dis. 2010, 7, 260–264. [Google Scholar] [CrossRef]
- Rohn, T.T. Caspase-cleaved TAR DNA-binding protein-43 is a major pathological finding in Alzheimer’s disease. Brain Res. 2008, 1228, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Schwab, C.; Arai, T.; Hasegawa, M.; Yu, S.; McGeer, P.L. Colocalization of transactivation-responsive DNA-binding protein 43 and huntingtin in inclusions of Huntington disease. J. Neuropathol. Exp. Neurol. 2008, 67, 1159–1165. [Google Scholar] [CrossRef]
- Polymenidou, M.; Cleveland, D.W. The seeds of neurodegeneration: Prion-like spreading in ALS. Cell 2011, 147, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Ravits, J.M.; La Spada, A.R. ALS motor phenotype heterogeneity, focality, and spread: Deconstructing motor neuron degeneration. Neurology 2009, 73, 805–811. [Google Scholar] [CrossRef]
- Mackenzie, I.R.; Neumann, M.; Bigio, E.H.; Cairns, N.J.; Alafuzoff, I.; Kril, J.; Kovacs, G.G.; Ghetti, B.; Halliday, G.; Holm, I.E.; et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: An update. Acta Neuropathol. 2010, 119, 1–4. [Google Scholar] [CrossRef]
- Tan, R.H.; Kril, J.J.; Fatima, M.; McGeachie, A.; McCann, H.; Shepherd, C.; Forrest, S.L.; Affleck, A.; Kwok, J.B.; Hodges, J.R.; et al. TDP-43 proteinopathies: Pathological identification of brain regions differentiating clinical phenotypes. Brain 2015, 138, 3110–3322. [Google Scholar] [CrossRef]
- Mompean, M.; Romano, V.; Pantoja-Uceda, D.; Stuani, C.; Baralle, F.E.; Buratti, E.; Laurents, D.V. Point mutations in the N-terminal domain of transactive response DNA-binding protein 43 kDa (TDP-43) compromise its stability, dimerization, and functions. J. Biol. Chem. 2017, 292, 11992–12006. [Google Scholar] [CrossRef]
- Afroz, T.; Hock, E.M.; Ernst, P.; Foglieni, C.; Jambeau, M.; Gilhespy, L.A.B.; Laferriere, F.; Maniecka, Z.; Pluckthun, A.; Mittl, P.; et al. ALS-linked protein TDP-43 antagonizes its pathologic aggregation. Nat. Comm. 2017, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.L.; Xue, W.; Hong, J.Y.; Zhang, J.T.; Li, M.J.; Yu, S.N.; He, J.H.; Hu, H.Y. The N-terminal dimerization is required for TDP-43 splicing activity. Sci. Rep. 2017, 7, 6196. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Conicella, A.E.; Schmidt, H.B.; Martin, E.W.; Rhoads, S.N.; Reeb, A.N.; Nourse, A.; Ramirez Montero, D.; Ryan, V.H.; Rohatgi, R.; et al. A single N-terminal phosphomimic disrupts TDP-43 polymerization, phase separation, and RNA splicing. EMBO J. 2018, 37. [Google Scholar] [CrossRef] [PubMed]
- Kuo, P.H.; Doudeva, L.G.; Wang, Y.T.; Shen, C.K.; Yuan, H.S. Structural insights into TDP-43 in nucleic-acid binding and domain interactions. Nucleic Acids Res. 2009, 37, 1799–1808. [Google Scholar] [CrossRef] [PubMed]
- Kuo, P.H.; Chiang, C.H.; Wang, Y.T.; Doudeva, L.G.; Yuan, H.S. The crystal structure of TDP-43 RRM1-DNA complex reveals the specific recognition for UG- and TG-rich nucleic acids. Nucleic Acids Res. 2014, 42, 4712–4722. [Google Scholar] [CrossRef] [PubMed]
- Lukavsky, P.J.; Daujotyte, D.; Tollervey, J.R.; Ule, J.; Stuani, C.; Buratti, E.; Baralle, F.E.; Damberger, F.F.; Allain, F.H. Molecular basis of UG-rich RNA recognition by the human splicing factor TDP-43. Nat. Struct. Mol. Biol. 2013, 20, 1443–1449. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.H.; Grauffel, C.; Wu, L.S.; Kuo, P.H.; Doudeva, L.G.; Lim, C.; Shen, C.K.; Yuan, H.S. Structural analysis of disease-related TDP-43 D169G mutation: Linking enhanced stability and caspase cleavage efficiency to protein accumulation. Sci. Rep. 2016, 6, 21581. [Google Scholar] [CrossRef]
- Jiang, L.L.; Zhao, J.; Yin, X.F.; He, W.T.; Yang, H.; Che, M.X.; Hu, H.Y. Two mutations G335D and Q343R within the amyloidogenic core region of TDP-43 influence its aggregation and inclusion formation. Sci. Rep. 2016, 6, 23928. [Google Scholar] [CrossRef]
- Lim, L.; Wei, Y.; Lu, Y.; Song, J. ALS-Causing Mutations Significantly Perturb the Self-Assembly and Interaction with Nucleic Acid of the Intrinsically Disordered Prion-Like Domain of TDP-43. PLoS Biol. 2016, 14, e1002338. [Google Scholar] [CrossRef]
- Conicella, A.E.; Zerze, G.H.; Mittal, J.; Fawzi, N.L. ALS Mutations Disrupt Phase Separation Mediated by alpha-Helical Structure in the TDP-43 Low-Complexity C-Terminal Domain. Structure 2016, 24, 1537–1549. [Google Scholar] [CrossRef]
- Li, H.R.; Chen, T.C.; Hsiao, C.L.; Shi, L.; Chou, C.Y.; Huang, J.R. The physical forces mediating self-association and phase-separation in the C-terminal domain of TDP-43. Biochim. Biophys. Acta Proteins Proteom. 2018, 1866, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Xu, Y.F.; Dickey, C.A.; Buratti, E.; Baralle, F.; Bailey, R.; Pickering-Brown, S.; Dickson, D.; Petrucelli, L. Progranulin mediates caspase-dependent cleavage of TAR DNA binding protein-43. J. Neurosci. 2007, 27, 10530–10534. [Google Scholar] [CrossRef]
- Gregory, R.I.; Yan, K.P.; Amuthan, G.; Chendrimada, T.; Doratotaj, B.; Cooch, N.; Shiekhattar, R. The Microprocessor complex mediates the genesis of microRNAs. Nature 2004, 432, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef] [PubMed]
- Ayala, Y.M.; Misteli, T.; Baralle, F.E. TDP-43 regulates retinoblastoma protein phosphorylation through the repression of cyclin-dependent kinase 6 expression. Proc. Natl. Acad. Sci. USA 2008, 105, 3785–3789. [Google Scholar] [CrossRef]
- Ayala, Y.M.; Pantano, S.; D’Ambrogio, A.; Buratti, E.; Brindisi, A.; Marchetti, C.; Romano, M.; Baralle, F.E. Human, Drosophila, and C.elegans TDP43: Nucleic acid binding properties and splicing regulatory function. J. Mol. Biol. 2005, 348, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Lagier-Tourenne, C.; Polymenidou, M.; Cleveland, D.W. TDP-43 and FUS/TLS: Emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet. 2010, 19, R46–R64. [Google Scholar] [CrossRef]
- Casafont, I.; Bengoechea, R.; Tapia, O.; Berciano, M.T.; Lafarga, M. TDP-43 localizes in mRNA transcription and processing sites in mammalian neurons. J. Struct. Biol. 2009, 167, 235–241. [Google Scholar] [CrossRef]
- Sephton, C.F.; Good, S.K.; Atkin, S.; Dewey, C.M.; Mayer, P.; Herz, J.; Yu, G. TDP-43 is a developmentally regulated protein essential for early embryonic development. J. Biol. Chem. 2010, 285, 6826–6834. [Google Scholar] [CrossRef]
- Chang, C.K.; Wu, T.H.; Wu, C.Y.; Chiang, M.H.; Toh, E.K.; Hsu, Y.C.; Lin, K.F.; Liao, Y.H.; Huang, T.H.; Huang, J.J. The N-terminus of TDP-43 promotes its oligomerization and enhances DNA binding affinity. Biochem. Biophys. Res. Commun. 2012, 425, 219–224. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Caulfield, T.; Xu, Y.F.; Gendron, T.F.; Hubbard, J.; Stetler, C.; Sasaguri, H.; Whitelaw, E.C.; Cai, S.; Lee, W.C.; et al. The dual functions of the extreme N-terminus of TDP-43 in regulating its biological activity and inclusion formation. Hum. Mol. Genet. 2013, 22, 3112–3122. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Lim, L.Z.; Wei, Y.; Song, J. TDP-43 N terminus encodes a novel ubiquitin-like fold and its unfolded form in equilibrium that can be shifted by binding to ssDNA. Proc. Natl. Acad. Sci. USA 2014, 111, 18619–18624. [Google Scholar] [CrossRef] [PubMed]
- Mompean, M.; Romano, V.; Pantoja-Uceda, D.; Stuani, C.; Baralle, F.E.; Buratti, E.; Laurents, D.V. The TDP-43 N-terminal domain structure at high resolution. FEBS J. 2016, 283, 1242–1260. [Google Scholar] [CrossRef]
- Tsoi, P.S.; Choi, K.J.; Leonard, P.G.; Sizovs, A.; Moosa, M.M.; MacKenzie, K.R.; Ferreon, J.C.; Ferreon, A.C.M. The N-Terminal Domain of ALS-Linked TDP-43 Assembles without Misfolding. Angew. Chem. Int. Ed. Engl. 2017, 56, 12590–12593. [Google Scholar] [CrossRef]
- Micsonai, A.; Wien, F.; Kernya, L.; Lee, Y.H.; Goto, Y.; Refregiers, M.; Kardos, J. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl. Acad. Sci. USA 2015, 112, E3095–E3103. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, D.K.; Grimshaw, S.B.; Receveur, V.; Dobson, C.M.; Jones, J.A.; Smith, L.J. Hydrodynamic radii of native and denatured proteins measured by pulse field gradient NMR techniques. Biochemistry 1999, 38, 16424–16431. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.M.; Bolen, D.W. Unfolding free energy changes determined by the linear extrapolation method. 1. Unfolding of phenylmethanesulfonyl alpha-chymotrypsin using different denaturants. Biochemistry 1988, 27, 8063–8068. [Google Scholar] [CrossRef]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef]
- Sun, Y.; Chakrabartty, A. Phase to Phase with TDP-43. Biochemistry 2017, 56, 809–823. [Google Scholar] [CrossRef]
- Travaglini-Allocatelli, C.; Gianni, S.; Dubey, V.K.; Borgia, A.; Di Matteo, A.; Bonivento, D.; Cutruzzola, F.; Bren, K.L.; Brunori, M. An obligatory intermediate in the folding pathway of cytochrome c552 from Hydrogenobacter thermophilus. J. Biol. Chem. 2005, 280, 25729–25734. [Google Scholar] [CrossRef]
- Neudecker, P.; Robustelli, P.; Cavalli, A.; Walsh, P.; Lundstrom, P.; Zarrine-Afsar, A.; Sharpe, S.; Vendruscolo, M.; Kay, L.E. Structure of an intermediate state in protein folding and aggregation. Science 2012, 336, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Krobath, H.; Estacio, S.G.; Faisca, P.F.; Shakhnovich, E.I. Identification of a conserved aggregation-prone intermediate state in the folding pathways of Spc-SH3 amyloidogenic variants. J. Mol. Biol. 2012, 422, 705–722. [Google Scholar] [CrossRef] [PubMed]
- Honda, R.P.; Xu, M.; Yamaguchi, K.; Roder, H.; Kuwata, K. A Native-like intermediate serves as a branching point between the folding and aggregation pathways of the mouse prion protein. Structure 2015, 23, 1735–1742. [Google Scholar] [CrossRef]
- Sen Mojumdar, S.Z.N.S.; Dee, D.R.; Rouleau, L.; Anand, U.; Garen, C.; Woodside, M.T. Partially native intermediates mediate misfolding of SOD1 in single-molecule folding trajectories. Nat. Commun. 2017, 8, 1881. [Google Scholar] [CrossRef]
- Heidarsson, P.O.; Naqvi, M.M.; Otazo, M.R.; Mossa, A.; Kragelund, B.B.; Cecconi, C. Direct single-molecule observation of calcium-dependent misfolding in human neuronal calcium sensor-1. Proc. Natl. Acad. Sci. USA 2014, 111, 13069–13074. [Google Scholar] [CrossRef] [PubMed]
- Del Poggetto, E.; Chiti, F.; Bemporad, F. The Folding process of Human Profilin-1, a novel protein associated with familial amyotrophic lateral sclerosis. Sci. Rep. 2015, 5, 12332. [Google Scholar] [CrossRef]
- Del Poggetto, E.; Toto, A.; Aloise, C.; Di Piro, F.; Gori, L.; Malatesta, F.; Gianni, S.; Chiti, F.; Bemporad, F. Stability of an aggregation-prone partially folded state of human profilin-1 correlates with aggregation propensity. J. Biol. Chem. 2018, 293, 10303–10313. [Google Scholar] [CrossRef]
- Mackness, B.C.; Tran, M.T.; McClain, S.P.; Matthews, C.R.; Zitzewitz, J.A. Folding of the RNA recognition motif (RRM) domains of the amyotrophic lateral sclerosis (ALS)-linked protein TDP-43 reveals an intermediate state. J. Biol. Chem. 2014, 289, 8264–8276. [Google Scholar] [CrossRef]
- Stoppini, M.; Bellotti, V. Systemic amyloidosis: Lessons from beta2-microglobulin. J. Biol. Chem. 2015, 290, 9951–9958. [Google Scholar] [CrossRef]
- Chiti, F.; De Lorenzi, E.; Grossi, S.; Mangione, P.; Giorgetti, S.; Caccialanza, G.; Dobson, C.M.; Merlini, G.; Ramponi, G.; Bellotti, V. A partially structured species of beta 2-microglobulin is significantly populated under physiological conditions and involved in fibrillogenesis. J. Biol. Chem. 2001, 276, 46714–46721. [Google Scholar] [CrossRef]
- Karamanos, T.K.; Pashley, C.L.; Kalverda, A.P.; Thompson, G.S.; Mayzel, M.; Orekhov, V.Y.; Radford, S.E. A population shift between sparsely populated folding intermediates determines amyloidogenicity. J. Am. Chem. Soc. 2016, 138, 6271–6280. [Google Scholar] [CrossRef] [PubMed]
- Vivoli Vega, M.; Nigro, A.; Luti, S.; Capitini, C.; Fani, G.; Gonnelli, L.; Boscaro, F.; Chiti, F. Isolation and characterization of soluble human full-lenghtTDP-43 associated with neurodegeneration. FASEB J. 2019, 10, 10780–10793. [Google Scholar] [CrossRef] [PubMed]
- Szyperski, T.; Mills, J.L.; Perl, D.; Balbach, J. Combined NMR-observation of cold denaturation in supercooled water and heat denaturation enables accurate measurement of deltaC(p) of protein unfolding. Eur. Biophys. J. 2006, 35, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.E.; Fersht, A.R. Folding of chymotrypsin inhibitor 2. 1. Evidence for a two-state transition. Biochemistry 1991, 30, 10428–10435. [Google Scholar] [CrossRef] [PubMed]
- Li, M.Z.; Elledge, S.J. Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat. Meth. 2007, 4, 251–256. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vivoli-Vega, M.; Guri, P.; Chiti, F.; Bemporad, F. Insight into the Folding and Dimerization Mechanisms of the N-Terminal Domain from Human TDP-43. Int. J. Mol. Sci. 2020, 21, 6259. https://doi.org/10.3390/ijms21176259
Vivoli-Vega M, Guri P, Chiti F, Bemporad F. Insight into the Folding and Dimerization Mechanisms of the N-Terminal Domain from Human TDP-43. International Journal of Molecular Sciences. 2020; 21(17):6259. https://doi.org/10.3390/ijms21176259
Chicago/Turabian StyleVivoli-Vega, Mirella, Prandvera Guri, Fabrizio Chiti, and Francesco Bemporad. 2020. "Insight into the Folding and Dimerization Mechanisms of the N-Terminal Domain from Human TDP-43" International Journal of Molecular Sciences 21, no. 17: 6259. https://doi.org/10.3390/ijms21176259
APA StyleVivoli-Vega, M., Guri, P., Chiti, F., & Bemporad, F. (2020). Insight into the Folding and Dimerization Mechanisms of the N-Terminal Domain from Human TDP-43. International Journal of Molecular Sciences, 21(17), 6259. https://doi.org/10.3390/ijms21176259