Ketones Elicit Distinct Alterations in Adipose Mitochondrial Bioenergetics


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
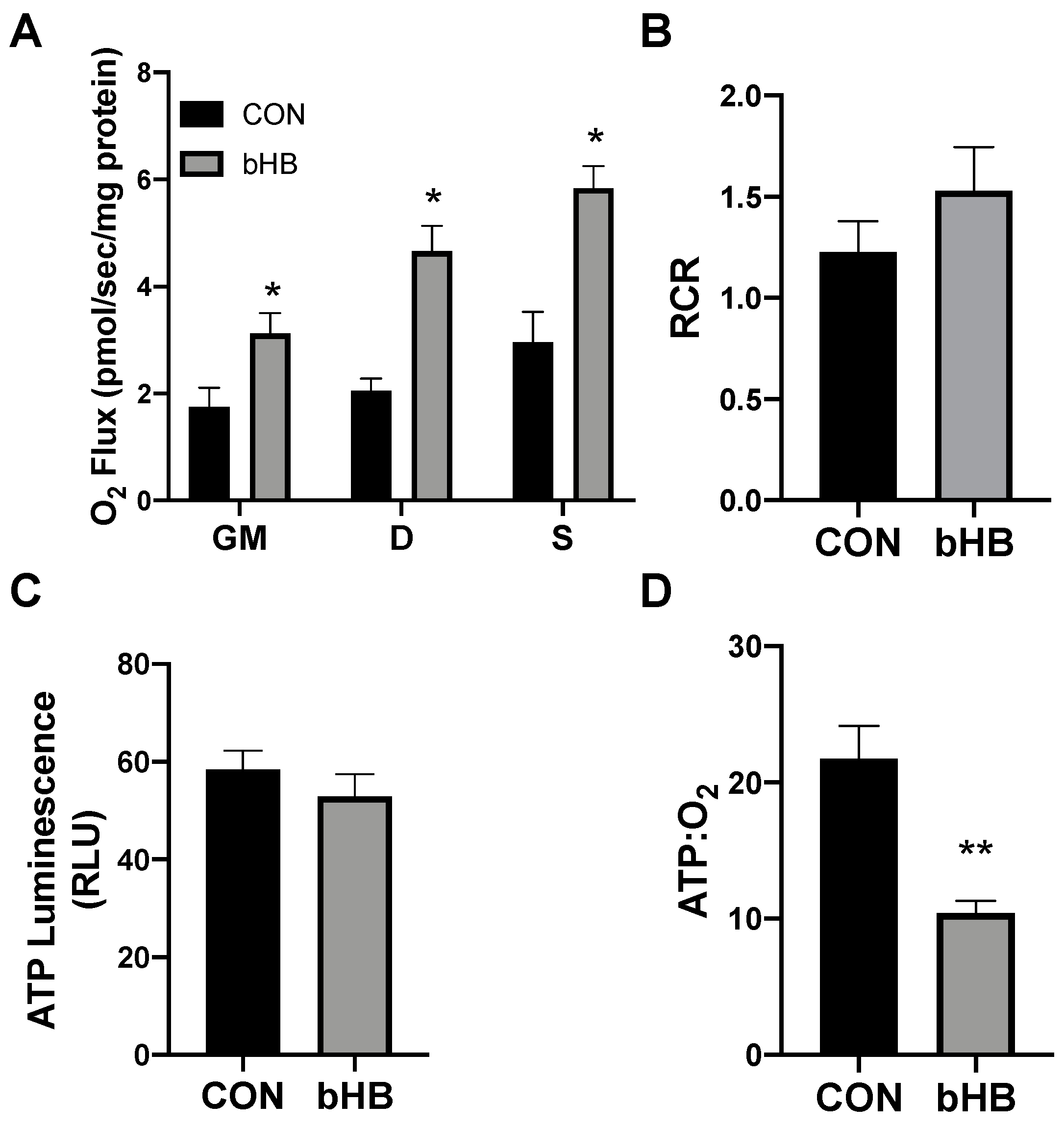
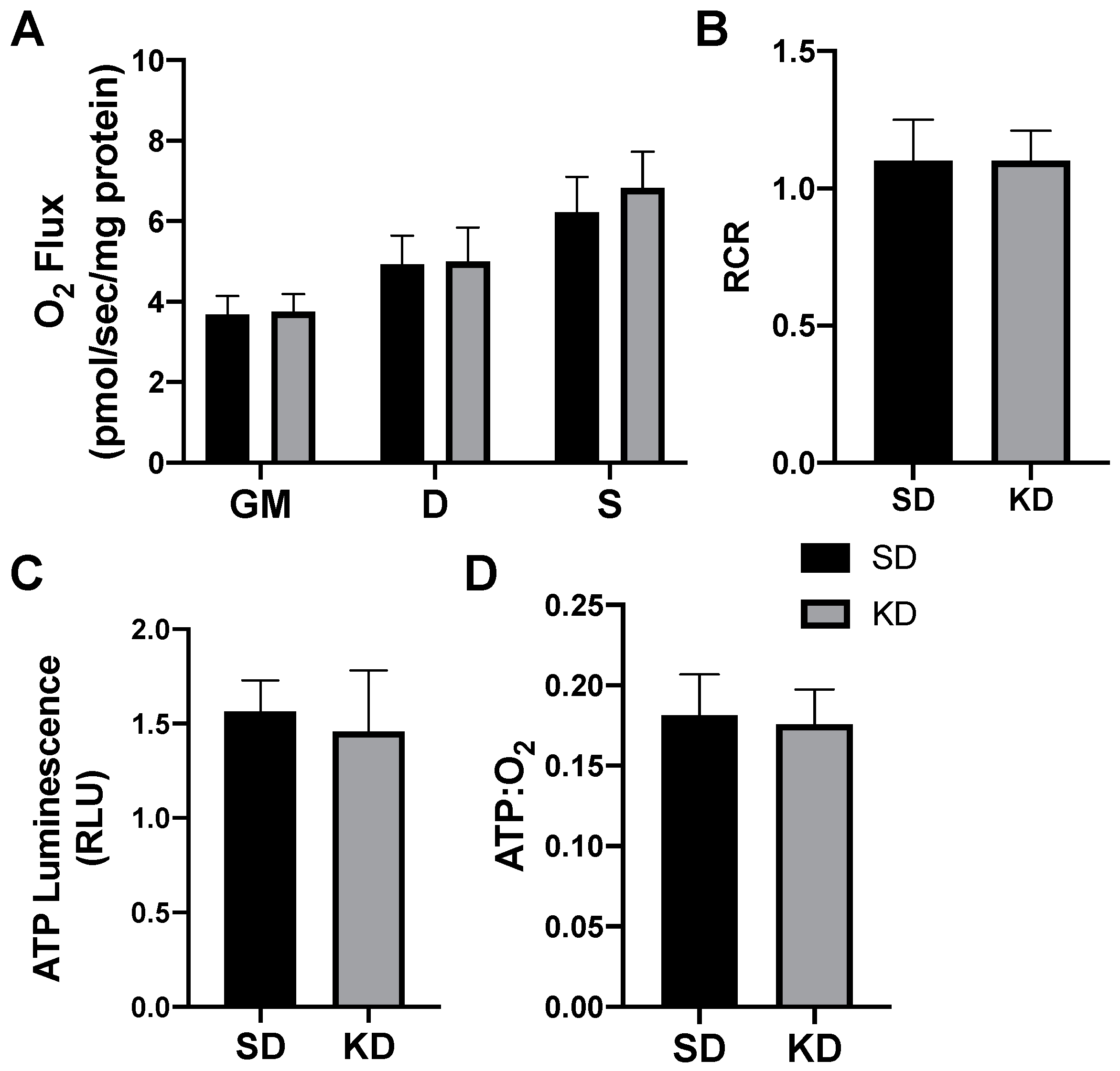
2.1. β-Hydroxybutyrate Alters Mitochondrial Respiration in Cultured Adipocytes
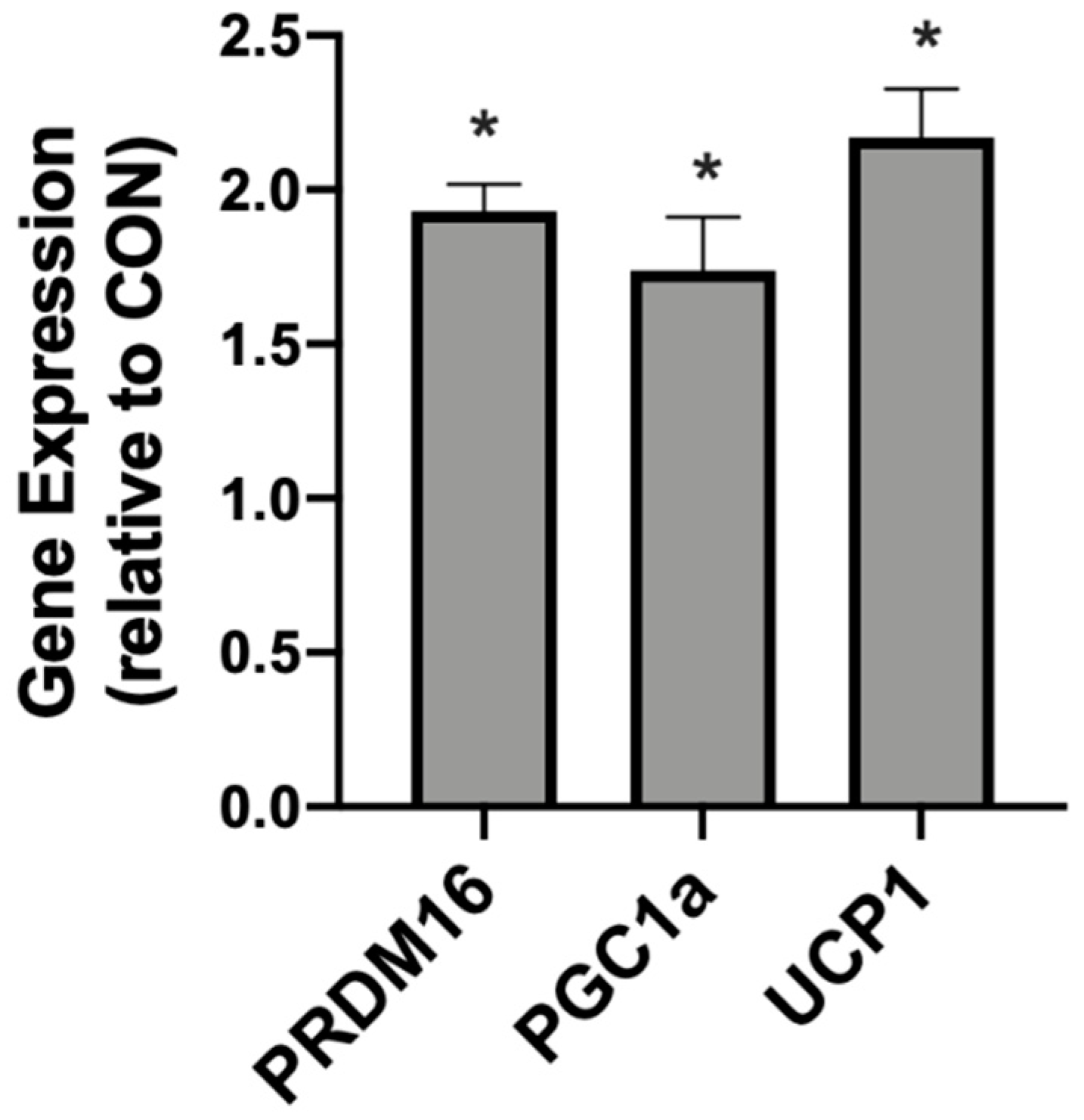
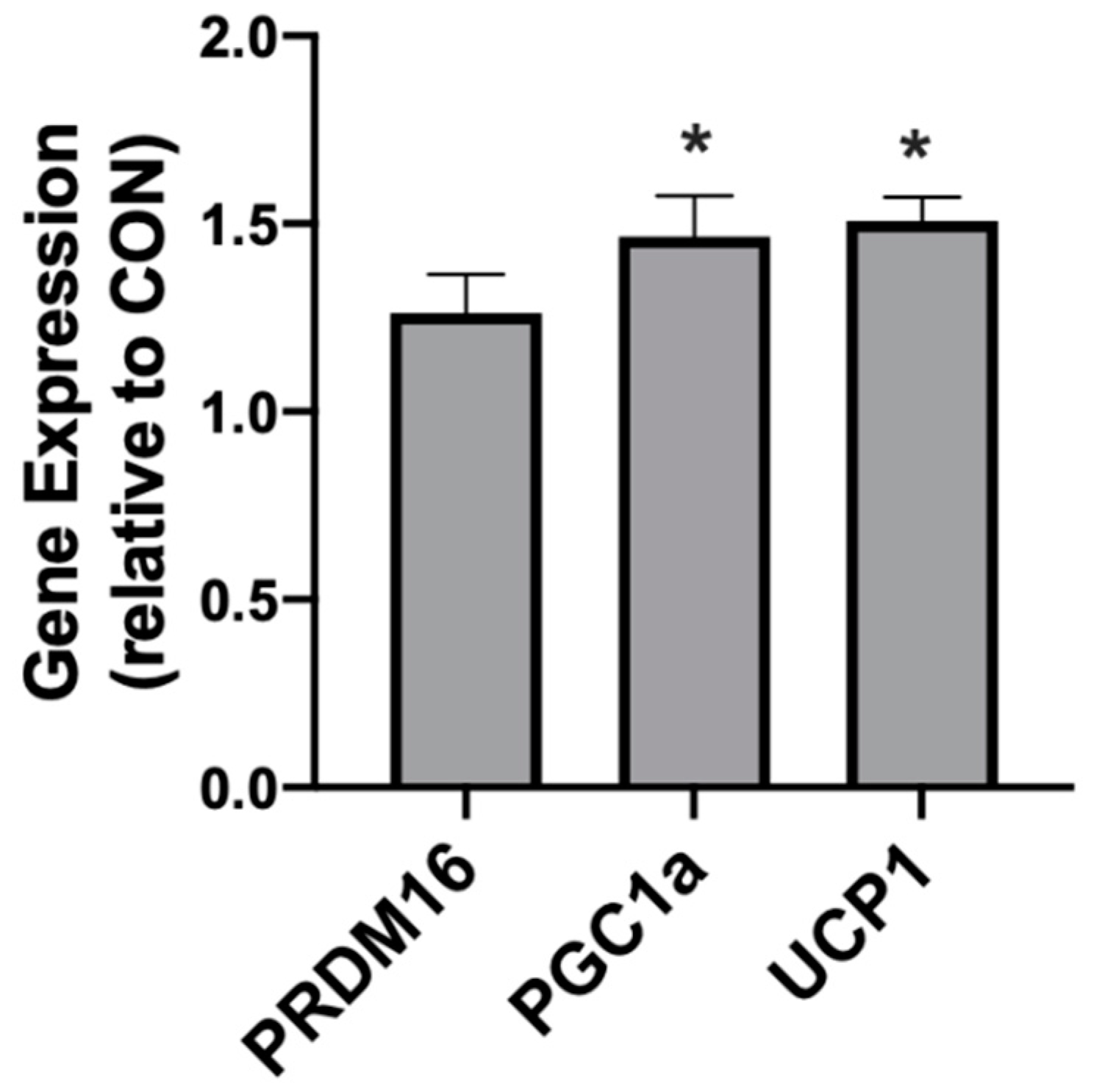
2.2. β-Hydroxybutyrate Changes Adipocyte Gene Expression
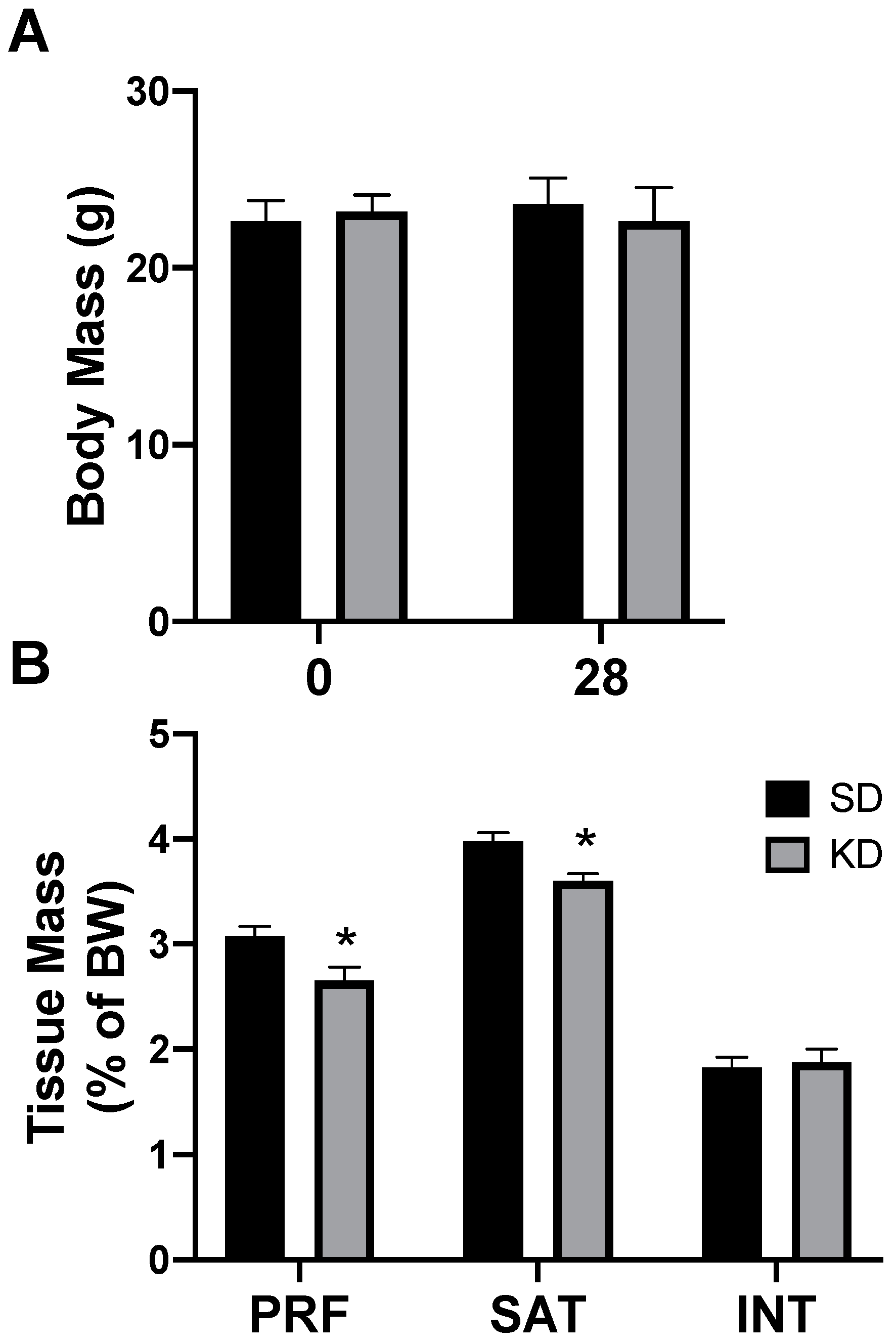
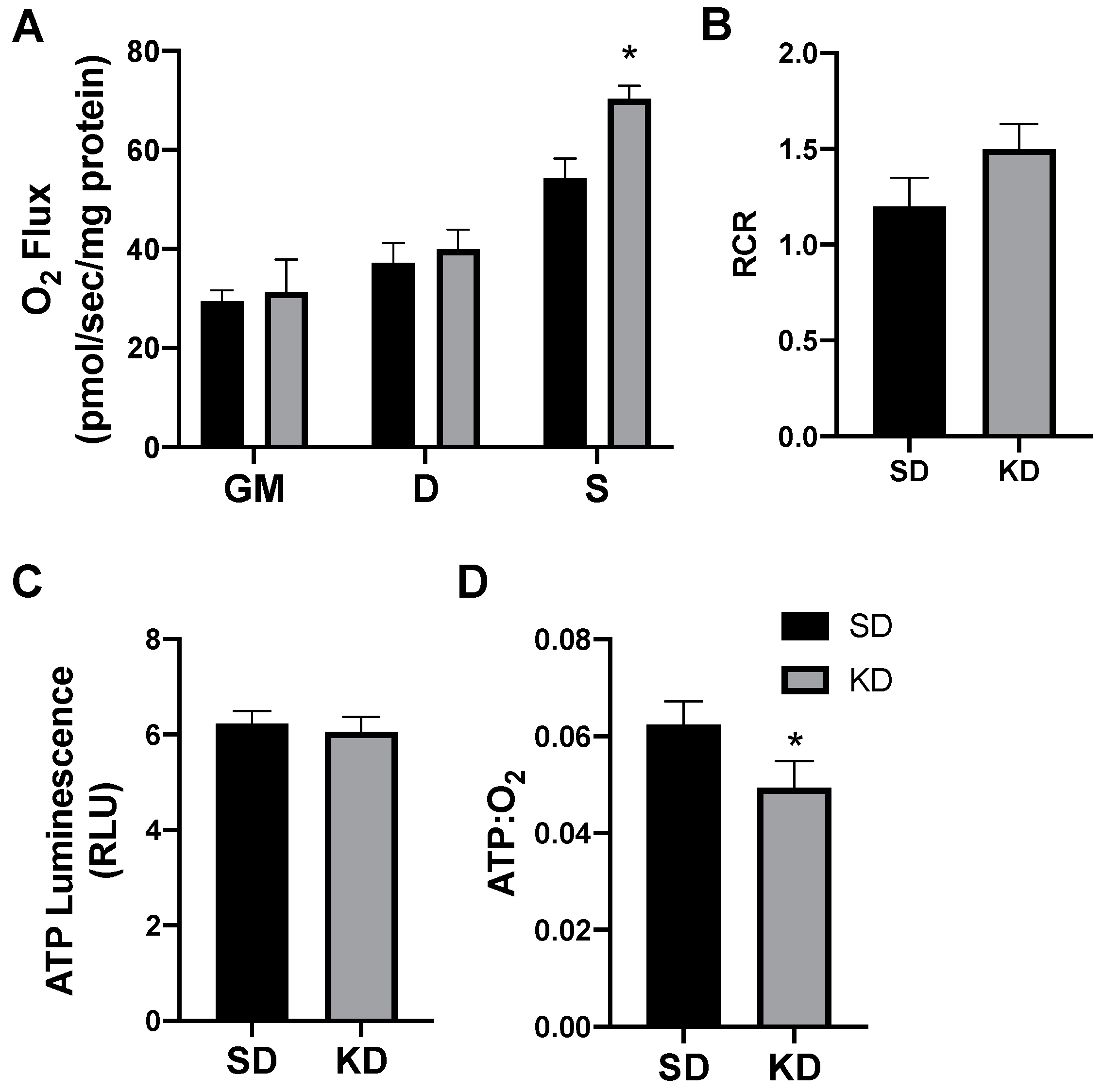
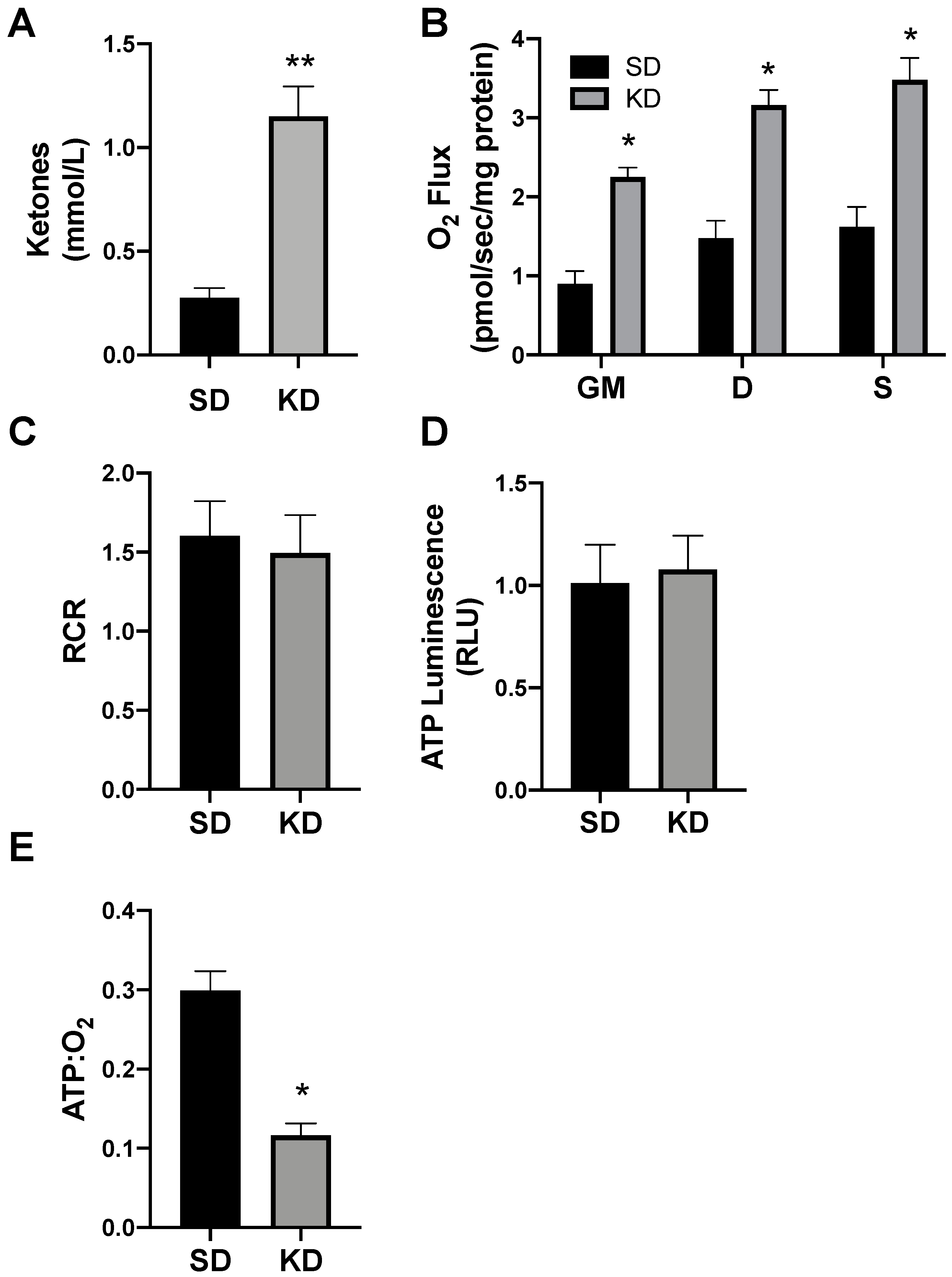
2.3. A Ketogenic Diet Alters Adipose Mitochondrial Bioenergetics
2.4. A Ketogenic Diet Alters Adipose Tissue Genetics in a Depot-Specific Manner
2.5. Shifts in Ketone-Induced Mitochondrial Bioenergetics Are Conserved in Human Adipose
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Animals
4.3. Mitochondrial Respiration
4.4. Tissue Homogenization
4.5. ATP Measurements
4.5.1. Cell Culture
4.5.2. Tissue Samples
4.6. RT-qPCR
4.7. Human Diet
4.8. Human Fat Biopsies
4.9. Statistical Methods
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hales, C.M.; Carroll, M.D.; Fryar, C.D.; Ogden, C.L. Prevalence of obesity and severe obesity among adults: United States, 2017–2018. NCHS Data Brief 2020, 360, 1–8. [Google Scholar]
- Flegal, K.M.; Kruszon-Moran, D.; Carroll, M.D.; Fryar, C.D.; Ogden, C.L. Trends in obesity among adults in the United States, 2005 to 2014. JAMA 2016, 315, 2284–2291. [Google Scholar] [CrossRef] [PubMed]
- Despres, J. Abdominal obesity as important component of insulin-resistance syndrome. Nutrition 1993, 9, 452–459. [Google Scholar] [PubMed]
- Bikman, B.T. A role for sphingolipids in the pathophysiology of obesity-induced inflammation. Cell. Mol. Life Sci. 2012, 69, 2135–2146. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Page, A.J.; Hatzinikolas, G.; Chen, M.X.; Wittert, G.A.; Heilbronn, L.K. Intermittent Fasting Improves Glucose Tolerance and Promotes Adipose Tissue Remodeling in Male Mice Fed a High-Fat Diet. Endocrinology 2019, 160, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Calle, E.E.; Kaaks, R. Overweight, obesity and cancer: Epidemiological evidence and proposed mechanisms. Nat. Rev. Cancer 2004, 4, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Farmer, S.R.; Boss, O. Recruitment of brown adipose tissue as a therapy for obesity-associated diseases. Front. Endocrinol. 2012, 3, 14. [Google Scholar]
- Cinti, S. The role of brown adipose tissue in human obesity. Nutr. Metab. Cardiovasc. Dis. 2006, 16, 569–574. [Google Scholar] [CrossRef]
- Dallon, B.W.; Parker, B.A.; Hodson, A.E.; Tippetts, T.S.; Harrison, M.E.; Appiah, M.M.A.; Witt, J.E.; Gibbs, J.L.; Gray, H.M.; Sant, T.M.; et al. Insulin selectively reduces mitochondrial uncoupling in brown adipose tissue in mice. Biochem. J. 2018, 475, 561–569. [Google Scholar] [CrossRef]
- Rosenbaum, M.; Leibel, R.L. Adaptive thermogenesis in humans. Int. J. Obes. 2010, 34, S47–S55. [Google Scholar] [CrossRef]
- Choe, S.S.; Huh, J.Y.; Hwang, I.J.; Kim, J.I.; Kim, J.B. Adipose tissue remodeling: Its role in energy metabolism and metabolic disorders. Front. Endocrinol. 2016, 7, 30. [Google Scholar] [CrossRef] [PubMed]
- Bartelt, A.; Heeren, J. Adipose tissue browning and metabolic health. Nat. Rev. Endocrinol. 2014, 10, 24. [Google Scholar] [CrossRef] [PubMed]
- Gantner, M.L.; Hazen, B.C.; Conkright, J.; Kralli, A. GADD45γ regulates the thermogenic capacity of brown adipose tissue. Proc. Natl. Acad. Sci. USA 2014, 111, 11870–11875. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.A.; Walton, C.M.; Carr, S.T.; Andrus, J.L.; Cheung, E.C.; Duplisea, M.J.; Wilson, E.K.; Draney, C.; Lathen, D.R.; Kenner, K.B. β-Hydroxybutyrate Elicits Favorable Mitochondrial Changes in Skeletal Muscle. Int. J. Mol. Sci. 2018, 19, 2247. [Google Scholar] [CrossRef]
- Youm, Y.-H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome–mediated inflammatory disease. Nat. Med. 2015, 21, 263. [Google Scholar] [CrossRef]
- Frey, S.; Geffroy, G.; Desquiret-Dumas, V.; Gueguen, N.; Bris, C.; Belal, S.; Amati-Bonneau, P.; Chevrollier, A.; Barth, M.; Henrion, D.; et al. The addition of ketone bodies alleviates mitochondrial dysfunction by restoring complex I assembly in a MELAS cellular model. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 284–291. [Google Scholar] [CrossRef]
- Kim, D.Y.; Rho, J.M. The ketogenic diet and epilepsy. Curr. Opin. Clin. Nutr. Metab. Care 2008, 11, 113–120. [Google Scholar] [CrossRef]
- Yuen, A.W.C.; Walcutt, I.A.; Sander, J.W. An acidosis-sparing ketogenic (ASK) diet to improve efficacy and reduce adverse effects in the treatment of refractory epilepsy. Epilepsy Behav. 2017, 74, 15–21. [Google Scholar] [CrossRef]
- Kim, D.Y.; Davis, L.M.; Sullivan, P.G.; Maalouf, M.; Simeone, T.A.; van Brederode, J.; Rho, J.M. Ketone bodies are protective against oxidative stress in neocortical neurons. J. Neurochem. 2007, 101, 1316–1326. [Google Scholar] [CrossRef]
- Myette-Cote, E.; Neudorf, H.; Rafiei, H.; Clarke, K.; Little, J.P. Prior ingestion of exogenous ketone monoester attenuates the glycaemic response to an oral glucose tolerance test in healthy young individuals. J. Physiol. 2018, 596, 1385–1395. [Google Scholar] [CrossRef]
- Seyfried, B.T.; Kiebish, M.; Marsh, J.; Mukherjee, P. Targeting energy metabolism in brain cancer through calorie restriction and the ketogenic diet. J. Cancer Res. Ther. 2009, 5, S7–S15. [Google Scholar] [CrossRef]
- Cahill Jr, G.F. Fuel metabolism in starvation. Annu. Rev. Nutr. 2006, 26, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.M.; Williamson, D.H. Physiological roles of ketone bodies as substrates and signals in mammalian tissues. Physiol. Rev. 1980, 60, 143–187. [Google Scholar] [CrossRef] [PubMed]
- McGarry, J.; Foster, D. Regulation of hepatic fatty acid oxidation and ketone body production. Annu. Rev. Biochem. 1980, 49, 395–420. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Morales, P.; Tapia, E.; Pedraza-Chaverri, J. β-hydroxybutyrate: A signaling metabolite in starvation response? Cell. Signal. 2016, 28, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.R.; Pissios, P.; Otu, H.; Roberson, R.; Xue, B.; Asakura, K.; Furukawa, N.; Marino, F.E.; Liu, F.F.; Kahn, B.B.; et al. A high-fat, ketogenic diet induces a unique metabolic state in mice. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E1724–E1739. [Google Scholar] [CrossRef]
- Hua, Z.G.; Xiong, L.J.; Yan, C.; Wei, D.H.; YingPai, Z.; Qing, Z.Y.; Lin, Q.Z.; Fei, F.R.; Ling, W.Y.; Ren, M.Z. Glucose and Insulin Stimulate Lipogenesis in Porcine Adipocytes: Dissimilar and Identical Regulation Pathway for Key Transcription Factors. Mol. Cells 2016, 39, 797–806. [Google Scholar] [CrossRef]
- Kobayashi, T.; Fujimori, K. Very long-chain-fatty acids enhance adipogenesis through coregulation of Elovl3 and PPARgamma in 3T3-L1 cells. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E1461–E1471. [Google Scholar] [CrossRef]
- Srivastava, S.; Baxa, U.; Niu, G.; Chen, X.; Veech, R.L. A ketogenic diet increases brown adipose tissue mitochondrial proteins and UCP1 levels in mice. IUBMB Life 2013, 65, 58–66. [Google Scholar] [CrossRef]
- Virtanen, K.A.; Lidell, M.E.; Orava, J.; Heglind, M.; Westergren, R.; Niemi, T.; Taittonen, M.; Laine, J.; Savisto, N.J.; Enerback, S.; et al. Functional brown adipose tissue in healthy adults. N. Engl. J. Med. 2009, 360, 1518–1525. [Google Scholar] [CrossRef]
- Vijgen, G.H.; Bouvy, N.D.; Teule, G.J.; Brans, B.; Schrauwen, P.; van Marken Lichtenbelt, W.D. Brown adipose tissue in morbidly obese subjects. PLoS ONE 2011, 6, e17247. [Google Scholar] [CrossRef]
- Kaisanlahti, A.; Glumoff, T. Browning of white fat: Agents and implications for beige adipose tissue to type 2 diabetes. J. Physiol. Biochem. 2019, 75, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ebbeling, C.B.; Feldman, H.A.; Klein, G.L.; Wong, J.M.W.; Bielak, L.; Steltz, S.K.; Luoto, P.K.; Wolfe, R.R.; Wong, W.W.; Ludwig, D.S. Effects of a low carbohydrate diet on energy expenditure during weight loss maintenance: Randomized trial. BMJ 2018, 363, k4583. [Google Scholar] [CrossRef] [PubMed]
- Hall, K.D.; Chen, K.Y.; Guo, J.; Lam, Y.Y.; Leibel, R.L.; Mayer, L.E.; Reitman, M.L.; Rosenbaum, M.; Smith, S.R.; Walsh, B.T.; et al. Energy expenditure and body composition changes after an isocaloric ketogenic diet in overweight and obese men. Am. J. Clin. Nutr. 2016, 104, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Joslin EP, B.F. A Study of Metabolism in Severe Diabetes; Carnegie Institution of Washington: Washington, DC, USA, 1912. [Google Scholar]
- Nair, K.S.; Halliday, D.; Garrow, J.S. Increased energy expenditure in poorly controlled Type 1 (insulin-dependent) diabetic patients. Diabetologia 1984, 27, 13–16. [Google Scholar] [CrossRef]
- Zauner, C.; Schneeweiss, B.; Kranz, A.; Madl, C.; Ratheiser, K.; Kramer, L.; Roth, E.; Schneider, B.; Lenz, K. Resting energy expenditure in short-term starvation is increased as a result of an increase in serum norepinephrine. Am. J. Clin. Nutr. 2000, 71, 1511–1515. [Google Scholar] [CrossRef]
- Melchior, J.C.; Rigaud, D.; Rozen, R.; Malon, D.; Apfelbaum, M. Energy expenditure economy induced by decrease in lean body mass in anorexia nervosa. Eur. J. Clin. Nutr. 1989, 43, 793–799. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Gong, H.; Sun, L.; Chen, B.; Han, Y.; Pang, J.; Wu, W.; Qi, R.; Zhang, T.-M. Evaluation of candidate reference genes for RT-qPCR studies in three metabolism related tissues of mice after caloric restriction. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walton, C.M.; Jacobsen, S.M.; Dallon, B.W.; Saito, E.R.; Bennett, S.L.H.; Davidson, L.E.; Thomson, D.M.; Hyldahl, R.D.; Bikman, B.T. Ketones Elicit Distinct Alterations in Adipose Mitochondrial Bioenergetics. Int. J. Mol. Sci. 2020, 21, 6255. https://doi.org/10.3390/ijms21176255
Walton CM, Jacobsen SM, Dallon BW, Saito ER, Bennett SLH, Davidson LE, Thomson DM, Hyldahl RD, Bikman BT. Ketones Elicit Distinct Alterations in Adipose Mitochondrial Bioenergetics. International Journal of Molecular Sciences. 2020; 21(17):6255. https://doi.org/10.3390/ijms21176255
Chicago/Turabian StyleWalton, Chase M., Samuel M. Jacobsen, Blake W. Dallon, Erin R. Saito, Shantelle L. H. Bennett, Lance E. Davidson, David M. Thomson, Robert D. Hyldahl, and Benjamin T. Bikman. 2020. "Ketones Elicit Distinct Alterations in Adipose Mitochondrial Bioenergetics" International Journal of Molecular Sciences 21, no. 17: 6255. https://doi.org/10.3390/ijms21176255
APA StyleWalton, C. M., Jacobsen, S. M., Dallon, B. W., Saito, E. R., Bennett, S. L. H., Davidson, L. E., Thomson, D. M., Hyldahl, R. D., & Bikman, B. T. (2020). Ketones Elicit Distinct Alterations in Adipose Mitochondrial Bioenergetics. International Journal of Molecular Sciences, 21(17), 6255. https://doi.org/10.3390/ijms21176255