Tuberculosis–Cancer Parallels in Immune Response Regulation


Abstract
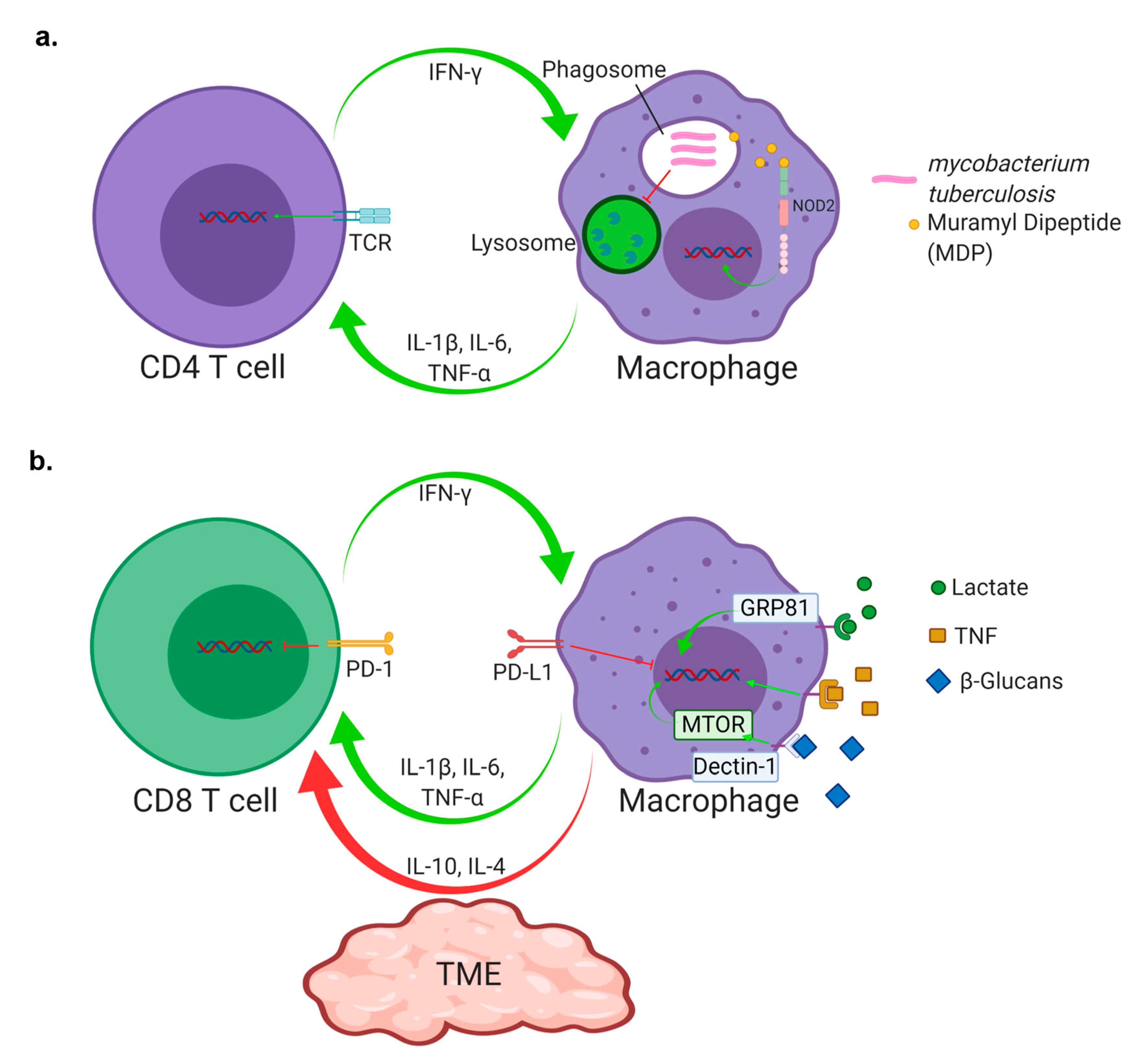
1. Introduction
2. Macrophages: Potent Modulators of Protection from the Innate Immune System through Trained Innate Immunity
3. Innate Immune-Based Immunotherapies for Cancer and TB
4. Checkpoint Inhibition
5. T Cell Infiltration and Homing
6. T Cell Antigen Exposure and Exhaustion
7. Discussion
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BCG | Bacille Calmetter Guerine |
| CTLA-4 | Cytotoxic T Lymphocyte Associated protein 4 |
| HNSCC | Head and Neck Squamous Cell Carcinoma |
| MDP | Muramyl Dipeptide |
| NETs | Neutrophil Extracellular Traps |
| NO | Nitric Oxide |
| PD-1 | Programmed Death Protein 1 |
| PD-L1 | Programmed Death Protein Ligand 1 |
| TAM | Tumor Associated Macrophage |
| TB | Tuberculosis |
| TIM-3 | T cell Immunoglobulin and Mucin Domain Containing-3 |
| TME | Tumor Microenvironment |
| Mtb | Mycobacterium tuberculosis |
References
- Moreira, L.; Wang, J.I.E.; Tsenova-Berkova, L.; Hellmann, W.; Freedman, V.H.; Kaplan, G. Sequestration of Mycobacterium tuberculosis in tight vacuoles in vivo in Lung Macrophages of Mice Infected by the Respiratory Route. Infect. Immun. 1997, 65, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Simmons, J.D.; Stein, C.M.; Seshadri, C.; Campo, M.; Alter, G.; Fortune, S.; Schurr, E.; Wallis, R.S.; Churchyard, G.; Mayanja-Kizza, H.; et al. Immunological mechanisms of human resistance to persistent Mycobacterium tuberculosis infection. Nat. Rev. Immunol. 2018, 18, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Ng, V.H.; Cox, J.S.; Sousa, A.O.; MacMicking, J.D.; McKinney, J.D. Role of KatG catalase-peroxidase in mycobacterial pathogenisis: Countering the phagocyte oxidative burst. Mol. Microbiol. 2004, 52, 1291–1302. [Google Scholar] [CrossRef] [PubMed]
- Bryk, R.; Griffin, P.; Nathan, C. Peroxynitrite reductase activity of bacterial peroxiredoxins. Nature 2000, 407, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Bryk, R.; Lima, C.D.; Erdjument-Bromage, H.; Tempst, P.; Nathan, C. Metabolic enzymes of mycobacteria linked to antioxidant defense by a thioredoxin-like protein. Science 2002, 295, 1073–1077. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Bryk, R.; Shi, S.; Erdjument-Bromage, H.; Tempst, P.; Nathan, C. Mycobacterium tuberculosis appears to lack α-ketoglutarate dehydrogenase and encodes pyruvate dehydrogenase in widely separated genes. Mol. Microbiol. 2005, 57, 859–868. [Google Scholar] [CrossRef]
- Weissbach, H.; Etienne, F.; Hoshi, T.; Heinemann, S.H.; Lowther, W.T.; Matthews, B.; St. John, G.; Nathan, C.; Brot, N. Peptide methionine sulfoxide reductase: Structure, mechanism of action, and biological function. Arch. Biochem. Biophys. 2002, 397, 172–178. [Google Scholar] [CrossRef]
- Colangeli, R.; Haq, A.; Arcus, V.L.; Summers, E.; Magliozzo, R.S.; McBride, A.; Mitra, A.K.; Radjainia, M.; Khajo, A.; Jacobs, W.R.; et al. The multifunctional histone-like protein Lsr2 protects mycobacteria against reactive oxygen intermediates. Proc. Natl. Acad. Sci. USA 2009, 106, 4414–4418. [Google Scholar] [CrossRef]
- Boshoff, H.I.M.; Reed, M.B.; Barry, C.E.; Mizrahi, V. DnaE2 polymerase contributes to in vivo survival and the emergence of drug resistance in Mycobacterium tuberculosis. Cell 2003, 113, 183–193. [Google Scholar] [CrossRef]
- van der Wel, N.; Hava, D.; Houben, D.; Fluitsma, D.; van Zon, M.; Pierson, J.; Brenner, M.; Peters, P.J.M. M. tuberculosis and M. leprae Translocate from the Phagolysosome to the Cytosol in Myeloid Cells. Cell 2007, 129, 1287–1298. [Google Scholar] [CrossRef]
- Goren, M.B.; Hart, P.D.; Yount, M.R.; Armstrong, J.A. Prevention of phagosome-lysosome fusion in cultured macrophages by sulfatides of Mycobacterium tuberculosis. Proc. Nat. Acad. Sci. USA 1976, 73, 2510–2514. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.G.; Cardona, P.J.; Kim, M.J.; Allain, S.; Altare, F. Foamy macrophages and the progression of the human tuberculosis granuloma. Nat. Immunol. 2009, 10, 943–948. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Elias, E.J.; Timm, J.; Botha, T.; Wai-Tsing, C.; Gomez, J.E.; Mckinney, J.D. Replication dynamics of Mycobacterium tuberculosis in chronically infected mice. Infect. Immun. 2005, 73, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Alatas, F.; Alatas, Ö.; Metintas, M.; Özarslan, A.; Erginel, S.; Yildirim, H. Vascular endothelial growth factor levels in active pulmonary tuberculosis. Chest 2004, 125, 2156–2159. [Google Scholar] [CrossRef]
- Cáceres, N.; Tapia, G.; Ojanguren, I.; Altare, F.; Gil, O.; Pinto, S.; Vilaplana, C.; Cardona, P.J. Evolution of foamy macrophages in the pulmonary granulomas of experimental tuberculosis models. Tuberculosis 2009, 89, 175–182. [Google Scholar] [CrossRef]
- Peyron, P.; Vaubourgeix, J.; Poquet, Y.; Levillain, F.; Botanch, C.; Bardou, F.; Daffé, M.; Emile, J.F.; Marchou, B.; Cardona, P.J.; et al. Foamy macrophages from tuberculous patients’ granulomas constitute a nutrient-rich reservoir for M. tuberculosis persistence. PLoS Pathog. 2008, 4, 1–14. [Google Scholar] [CrossRef]
- Ulrichs, T.; Kosmiadi, G.A.; Trusov, V.; Jörg, S.; Pradl, L.; Titukhina, M.; Mishenko, V.; Gushina, N.; Kaufmann, S.H.E. Human tuberculous granulomas induce peripheral lymphoid follicle-like structures to orchestrate local host defence in the lung. J. Pathol. 2004, 204, 217–228. [Google Scholar] [CrossRef]
- Kaplan, G.; Post, F.A.; Moreira, A.L.; Wainwright, H.; Kreiswirth, B.N.; Melike, T.; Mathema, B.; Ramaswamy, S.V.; Walther, G.; Steyn, L.M.; et al. Mycobacterium tuberculosis growth at the cavity surface: A microenvironment with failed immunity. Infect. Immun. 2003, 71, 7099–7108. [Google Scholar] [CrossRef]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef]
- Pauken, K.E.; Sammons, M.A.; Odorizzi, P.M.; Manne, S.; Godec, J.; Khan, O.; Drake, A.M.; Chen, Z.; Sen, D.R.; Kurachi, M.; et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 2016, 354, 1160–1165. [Google Scholar] [CrossRef]
- Johnson, P.; Challis, R.; Chowdhury, F.; Gao, Y.; Harvey, M.; Geldart, T.; Kerr, P.; Chan, C.; Smith, A.; Steven, N.; et al. Clinical and biological effects of an agonist anti-CD40 antibody a cancer research UK phase I study. Clin. Cancer Res. 2015, 21, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Segal, N.H.; Gada, P.; Senzer, N.; Gargano, M.A.; Patchen, M.L.; Saltz, L.B. A Phase II Efficacy and Safety, Open-Label, Multicenter Study of Imprime PGG Injection in Combination with Cetuximab in Patients with Stage IV KRAS-Mutant Colorectal Cancer. Clin. Colorectal Cancer 2016, 15, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.F.; Kong, L.Y.; Jordan, J.; Conrad, C.; Madden, T.; Fokt, I.; Priebe, W.; Heimberger, A.B. A novel small molecule inhibitor of signal transducers and activators of transcription 3 reverses immune tolerance in malignant glioma patients. Cancer Res. 2007, 67, 9630–9636. [Google Scholar] [CrossRef]
- Yuhas, Y.; Berent, E.; Ovadiah, H.; Azoulay, I.; Ashkenazi, S. Rifampin augments cytokine-induced nitric oxide production in human alveolar epithelial cells. Antimicrob. Agents Chemother. 2006, 50, 396–398. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Novakovic, B.; Habibi, E.; Wang, S.-Y.; Arts, R.J.W.; Davar, R.; Megchelenbrink, W.; Kim, B.; Kuznetsova, T.; Kox, M.; Zwaag, J.; et al. β-Glucan Reverses the Epigenetic State of LPS-Induced Immunological Tolerance. Cell 2016, 167, 1354–1368. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.A.; Liao, W.; Sarkar, A.; Kim, M.V.; Bivona, M.R.; Liu, K.; Pamer, E.G.; Li, M.O. The cellular and molecular origin of tumor-associated macrophages. Science 2014, 344, 921–925. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, Y. Tumor-associated macrophages, potential targets for cancer treatment. Biomark. Res. 2017, 5, 1–6. [Google Scholar] [CrossRef]
- Qian, B.Z.; Pollard, J.W. Macrophage Diversity Enhances Tumor Progression and Metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef]
- Zwerling, A.; Behr, M.A.; Verma, A.; Brewer, T.F.; Menzies, D.; Pai, M. The BCG world atlas: A database of global BCG vaccination policies and practices. PLoS Med. 2011, 8. [Google Scholar] [CrossRef]
- Shelley, M.D.; Wilt, T.J.; Court, J.; Coles, B.; Kynaston, H.; Mason, M.D. Intravesical bacillus Calmette-Guérin is superior to mitomycin C in reducing tumour recurrence in high-risk superficial bladder cancer: A meta-analysis of randomized trials. Urol. Oncol. 2004, 485–490. [Google Scholar] [CrossRef]
- Morales, A.; Eidinger, D.; Bruce, A.W. Intracavitary Bacillus Calmette-Guerin in the Treatment of Superficial Bladder Tumors. J. Urol. 1976, 116, 180–183. [Google Scholar] [CrossRef]
- CDC The role of BCG vaccine in the prevention and control of tuberculosis in the United States. A joint statement by the Advisory Council for the Elimination of Tuberculosis and the Advisory Committee on Immunization Practices. MMWR. Recomm. Rep. 1996, 45, 1–18. [Google Scholar]
- Abubakar, I.; Pimpin, L.; Ariti, C.; Beynon, R.; Mangtani, P.; Sterne, J.; Fine, P.; Smith, P.; Lipman, M.; Elliman, D.; et al. Systematic review and meta-analysis of the current evidence on the duration of protection by bacillus Calmette-Guérin vaccination against tuberculosis. Health Technol. Assess. 2013, 17, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Crevel, R. Van Seminars in Immunology BCG-induced protection: Effects on innate immune memory. Semin. Immunol. 2014, 26, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Bickett, T.E.; McLean, J.; Creissen, E.; Hagan, C.; Izzo, L.; Izzo, A.; Silva, F.; Izzo Angelo, A. Characterizing the BCG Induced Macrophage and Neutrophil Mechanisms for Defense Against Mycobacterium tuberculosis. Front. Immunol. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Kleinnijenhuis, J.; Quintin, J.; Preijers, F.; Joosten, L.A.B.; Ifrim, D.C.; Saeed, S.; Jacobs, C.; van Loenhout, J.; de Jong, D.; Stunnenberg, H.G.; et al. Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc. Natl. Acad. Sci. USA 2012, 109, 17537–17542. [Google Scholar] [CrossRef] [PubMed]
- Inohara, N.; Ogura, Y.; Fontalba, A.; Gutierrez, O.; Pons, F.; Crespo, J.; Fukase, K.; Inamura, S.; Kusumoto, S.; Hashimoto, M.; et al. Host Recognition of Bacterial Muramyl Dipeptide Mediated through NOD2. J. Biol. Chem. 2003, 278, 5509–5513. [Google Scholar] [CrossRef]
- Kaufmann, E.; Sanz, J.; Dunn, J.L.; Robbins, C.S.; Barreiro, L.B.; Divangahi, M.; Kaufmann, E.; Sanz, J.; Dunn, J.L.; Khan, N.; et al. BCG Educates Hematopoietic Stem Cells to Generate Protective Innate Immunity against Article BCG Educates Hematopoietic Stem Cells to Generate Protective Innate Immunity against Tuberculosis. Cell 2017, 176–190. [Google Scholar] [CrossRef]
- Kleinnijenhuis, J.; Quintin, J.; Preijers, F.; Joosten, L.A.B.; Jacobs, C.; Xavier, R.J.; van der Meer, J.W.M.; van Crevel, R.; Netea, M.G. BCG-induced trained immunity in NK cells: Role for non-specific protection to infection. Clin. Immunol. 2014, 155, 213–219. [Google Scholar] [CrossRef]
- Netea, M.G.; Joosten, L.A.B.; van der Meer, J.W.M. Hypothesis: Stimulation of trained immunity as adjunctive immunotherapy in cancer. J. Leukoc. Biol. 2017, 102, 1323–1332. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Carmi, Y.; Dotan, S.; Rider, P.; Kaplanov, I.; White, M.R.; Baron, R.; Abutbul, S.; Huszar, M.; Dinarello, C.A.; Apte, R.N.; et al. The role of IL-1β in the early tumor cell-induced angiogenic response. J. Immunol. 2013, 190, 3500–3509. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Recht, L.; Strober, S. The Promise of Targeting Macrophages in Cancer Therapy. Clin. Cancer Res. 2017, 23, 3241–3250. [Google Scholar] [CrossRef] [PubMed]
- Bloemena, E.; Gall, H.; Ransom, J.; Pomato, N.; Murray, J.; Bos, E.; Scheper, R.; Meijer, C.; Hanna, M.; Vermorken, J. Delayed-Type Hypersensitivity Reactions to Tumor-associated Antigens in Colon Carcinoma Patients Immunized with an Autologous Tumor Cell/Bacillus Calmette-Guérin Vaccine. Am. Assoc. Cancer Res. 1992, 53, 456–459. [Google Scholar]
- Thommen, D.S.; Schumacher, T.N. T Cell Dysfunction in Cancer. Cancer Cell 2018, 33, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Sun, E.; Lei, M.; Li, L.; Gao, J.; Nian, X.; Wang, L. BCG-induced formation of neutrophil extracellular traps play an important role in bladder cancer treatment. Clin. Immunol. 2019, 201, 4–14. [Google Scholar] [CrossRef]
- Thiel, T.; Ryk, C.; Chatzakos, V.; Hallén Grufman, K.; Bavand-Chobot, N.; Flygare, J.; Wiklund, N.P.; De Verdier, P.J. Secondary stimulation from Bacillus Calmette-Guérin induced macrophages induce nitric oxide independent cell-death in bladder cancer cells. Cancer Lett. 2014, 348, 119–125. [Google Scholar] [CrossRef]
- Jallad, S.; Thomas, P.; Newport, M.J.; Kern, F. Baseline cytokine profiles of tuberculin-specific CD4+ T cells in non-muscle-invasive bladder cancer may predict outcomes of BCG immunotherapy. Cancer Immunol. Res. 2018, 6, 1212–1219. [Google Scholar] [CrossRef]
- Rao, V.; Dhar, N.; Shakila, H.; Singh, R.; Khera, A.; Jain, R.; Naseema, M.; Paramasivan, C.N.; Narayanan, P.R.; Ramanathan, V.D.; et al. Increased expression of Mycobacterium tuberculosis 19 kDa lipoprotein obliterates the protective efficacy of BCG by polarizing host immune responses to the Th2 subtype. Scand. J. Immunol. 2005, 61, 410–417. [Google Scholar] [CrossRef]
- Pichler, R.; Gruenbacher, G.; Culig, Z.; Brunner, A.; Fuchs, D.; Fritz, J.; Gander, H.; Rahm, A.; Thurnher, M. Intratumoral Th2 predisposition combines with an increased Th1 functional phenotype in clinical response to intravesical BCG in bladder cancer. Cancer Immunol. Immunother. 2017, 66, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.T.; Knops, A.; Swendseid, B.; Martinez-Outschoom, U.; Harshyne, L.; Philp, N.; Rodeck, U.; Luginbuhl, A.; Cognetti, D.; Johnson, J.; et al. Prognostic Significance of Tumor-Associated Macrophage Content in Head and Neck Squamous Cell Carcinoma: A Meta-Analysis. Front. Oncol. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nardin, A.; Lefebvre, M.; Labroquere, K.; Faure, O.; Abastado, J. Liposomal Muramyl Tripeptide Phosphatidylethanolamine: Targeting and Activating Macrophages for Adjuvant Treatment of Osteosarcoma. Curr. Cancer Drug Targets 2006, 6, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Bekkering, S.; Arts, R.J.W.; Novakovic, B.; Kourtzelis, I.; van der Heijden, C.D.C.C.; Li, Y.; Popa, C.D.; ter Horst, R.; van Tuijl, J.; Netea-Maier, R.T.; et al. Metabolic Induction of Trained Immunity through the Mevalonate Pathway. Cell 2018, 172, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Arts, R.J.W.; Plantinga, T.S.; Tuit, S.; Ulas, T.; Heinhuis, B.; Tesselaar, M.; Sloot, Y.; Adema, G.J.; Joosten, L.A.B.; Smit, J.W.A.; et al. Transcriptional and metabolic reprogramming induce an inflammatory phenotype in non-medullary thyroid carcinoma-induced macrophages. Oncoimmunology 2016, 5, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sia, J.K.; Bizzell, E.; Madan-Lala, R.; Rengarajan, J. Engaging the CD40-CD40L pathway augments T-helper cell responses and improves control of Mycobacterium tuberculosis infection. PLoS Pathog. 2017, 13, 1–22. [Google Scholar] [CrossRef]
- Harling, K.; Adankwah, E.; Guler, A.; Afum-Adjei Awuah, A.; Adu-Amoah, L.; Mayatepek, E.; Owusu-Dabo, E.; Nausch, N.; Jacobsen, M. Constitutive STAT3 phosphorylation and IL-6/IL-10 co-expression are associated with impaired T-cell function in tuberculosis patients. Cell. Mol. Immunol. 2018, 16, 175–187. [Google Scholar] [CrossRef]
- Genoula, M.; Franco, J.L.M.; Dupont, M.; Kviatcovsky, D.; Milillo, A.; Schierloh, P.; Moraña, E.J.; Poggi, S.; Palmero, D.; Mata-Espinosa, D.; et al. Formation of foamy macrophages by tuberculous pleural effusions is triggered by the interleukin-10/signal transducer and activator of transcription 3 axis through ACAT upregulation. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Bogdan, C. Nitric oxide and the immune response. Nat. Immunol. 2001, 2, 907–916. [Google Scholar] [CrossRef]
- Gordon, S.; Taylor, P.R. Monocyte and Macrophage Heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Dyck, L.; Mills, K.H.G. Immune checkpoints and their inhibition in cancer and infectious diseases. Eur. J. Immunol. 2017, 47, 765–779. [Google Scholar] [CrossRef] [PubMed]
- Pauken, K.E.; Wherry, E.J. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015, 36, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Jurado, J.O.; Alvarez, I.B.; Pasquinelli, V.; Martínez, G.J.; Quiroga, M.F.; Abbate, E.; Musella, R.M.; Chuluyan, H.E.; Verónica, E.; Jurado, J.O.; et al. Programmed Death (PD)-1:PD-Ligand 1/PD-Ligand 2 Pathway Inhibits T Cell Effector Functions during Human Tuberculosis. J. Immunol. 2008, 181, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Mohan, A.; Dey, A.B.; Mitra, D.K. Inhibiting the Programmed Death 1 Pathway Rescues Mycobacterium tuberculosis—Specific Interferon γ—Producing T Cells From Apoptosis in Patients With Pulmonary Tuberculosis. J. Infect. Dis. 2013, 208, 603–615. [Google Scholar] [CrossRef]
- Musvosvi, M.; Penn-nicholson, A.; Andersen, P.; Scriba, T.J.; Urdahl, K.B.; Moguche, A.O.; Musvosvi, M.; Penn-nicholson, A.; Plumlee, C.R.; Hoff, S.T.; et al. Antigen Availability Shapes T Cell Differentiation and Function during Tuberculosis. Cell Host Microbe 2017, 21, 695–706. [Google Scholar] [CrossRef]
- Khan, N.; Vidyarthi, A.; Amir, M.; Mushtaq, K.; Agrewala, J.N. T-cell exhaustion in tuberculosis: Pitfalls and prospects. Crit. Rev. Microbiol. 2017, 43, 133–141. [Google Scholar] [CrossRef]
- Barber, D.L.; Mayer-Barber, K.D.; Feng, C.G.; Sharpe, A.H.; Sher, A. CD4 T Cells Promote Rather than Control Tuberculosis in the Absence of PD-1–Mediated Inhibition. J. Immunol. 2011, 186, 1598–1607. [Google Scholar] [CrossRef]
- Tousif, S.; Singh, Y.; Prasad, D.V.R.; Sharma, P.; van Kaer, L.; Das, G. T cells from programmed death-1 deficient mice respond poorly to mycobacterium tuberculosis infection. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Reiley, W.W.; Shafiani, S.; Wittmer, S.T.; Tucker-Heard, G.; Moon, J.J.; Jenkins, M.K.; Urdahl, K.B.; Winslow, G.M.; Woodland, D.L. Distinct functions of antigen-specific CD4 T cells during murine Mycobacterium tuberculosis infection. Proc. Natl. Acad. Sci. USA 2010, 107, 19408–19413. [Google Scholar] [CrossRef]
- Ferris, R.L.; Blumenschein, G.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef] [PubMed]
- Seiwert, T.Y.; Burtness, B.; Mehra, R.; Weiss, J.; Berger, R.; Eder, J.P.; Heath, K.; McClanahan, T.; Lunceford, J.; Gause, C.; et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): An open-label, multicentre, phase 1b trial. Lancet Oncol. 2016, 17, 956–965. [Google Scholar] [CrossRef]
- Jayaraman, P.; Jacques, M.K.; Zhu, C.; Steblenko, K.M.; Stowell, B.L.; Madi, A.; Anderson, A.C.; Kuchroo, V.K.; Behar, S.M. TIM3 Mediates T Cell Exhaustion during Mycobacterium tuberculosis Infection. PLoS Pathog. 2016, 12, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Pe, S.; Sierra-Madero, J.; Torre-Bouscoulet, L.; Addo, M.M. Tim-3 blocking rescue macrophage and T cell function against Mycobacterium tuberculosis infection in HIV patients. J. Int. AIDS Soc. 2015, 1–10. [Google Scholar]
- Shayan, G.; Srivastava, R.; Li, J.; Schmitt, N.; Kane, L.P.; Ferris, R.L. Adaptive resistance to anti-PD1 therapy by tim-3 upregulation is mediated by the PI3k-akt pathway in head and neck cancer. Oncoimmunology 2017, 6, 1–11. [Google Scholar] [CrossRef]
- Fesnak, A.D.; June, C.H.; Levine, B.L. Engineered T cells: The promise and challenges of cancer immunotherapy. Nat. Rev. Cancer 2016, 16, 566–581. [Google Scholar] [CrossRef]
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guérin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti–PD-1 treatment. Proc. Natl. Acad. Sci. USA 2018, 115, E4041–E4050. [Google Scholar] [CrossRef]
- Whilding, L.M.; Halim, L.; Draper, B.; Parente-pereira, A.C.; Zabinski, T.; Davies, D.M.; Maher, J. CAR T-Cells Targeting the Integrin αvβ6 and Co-Expressing the Chemokine Receptor CXCR2 Demonstrate Enhanced Homing and Efficacy against Several Solid Malignancies. Cancers 2019, 11, 674. [Google Scholar] [CrossRef]
- Tang, H.; Wang, Y.; Chlewicki, L.K.; Zhang, Y.; Guo, J.; Liang, W.; Wang, J.; Wang, X.; Fu, Y.X. Facilitating T Cell Infiltration in Tumor Microenvironment Overcomes Resistance to PD-L1 Blockade. Cancer Cell 2016, 29, 285–296. [Google Scholar] [CrossRef]
- Schrager, L.K.; Vekemens, J.; Drager, N.; Lewinsohn, D.M.; Olesen, O.F. The status of tuberculosis vaccine development. Lancet Infect. Dis. 2020, 3099, 1–10. [Google Scholar] [CrossRef]
- Wolf, A.J.; Desvignes, L.; Linas, B.; Banaiee, N.; Tamura, T.; Takatsu, K.; Ernst, J.D. Initiation of the adaptive immune response to Mycobacterium tuberculosis depends on antigen production in the local lymph node, not the lungs. J. Exp. Med. 2008, 205, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Khader, S.A.; Partida-Sanchez, S.; Bell, G.; Jelley-Gibbs, D.M.; Swain, S.; Pearl, J.E.; Ghilardi, N.; Desauvage, F.J.; Lund, F.E.; Cooper, A.M. Interleukin 12p40 is required for dendritic cell migration and T cell priming after Mycobacterium tuberculosis infection. J. Exp. Med. 2006, 203, 1805–1815. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Jankovic, D.; Oler, A.J.; Wei, G.; Sharma, S.; Hu, G.; Guo, L.; Yagi, R.; Yamane, H.; Punkosdy, G.; et al. The Transcription Factor T-bet Is Induced by Multiple Pathways and Prevents an Endogenous Th2 Cell Program during Th1 Cell Responses. Immunity 2012, 37, 660–673. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.L.; Chan, J.; Lin, P.L. Macrophages and control of granulomatous inflammation in tuberculosis. Mucosal Immunol. 2011, 4, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Lighvani, A.A.; Frucht, D.M.; Jankovic, D.; Yamane, H.; Aliberti, J.; Hissong, B.D.; Nguyen, B.V.; Gadina, M.; Sher, A.; Paul, W.E.; et al. T-bet is rapidly induced by interferon-γ in lymphoid and myeloid cells. Proc. Natl. Acad. Sci. USA 2002, 98, 15137–15142. [Google Scholar] [CrossRef] [PubMed]
- Sallin, M.A.; Sakai, S.; Kauffman, K.D.; Young, H.A.; Zhu, J.; Barber, D.L. Th1 Differentiation Drives the Accumulation of Intravascular, Non-protective CD4 T Cells during Tuberculosis. Cell Rep. 2017, 18, 3091–3104. [Google Scholar] [CrossRef]
- Sakai, S.; Kauffman, K.D.; Schenkel, J.M.; Mcberry, C.C.; Mayer-barber, K.D.; Barber, D.L. Control of Mycobacterium tuberculosis Infection by a Subset of Lung Parenchyma—Homing CD4 T Cells. J. Immunol. 2019, 192. [Google Scholar] [CrossRef]
- Cohen, S.B.; Urdahl, K.B. Going beyond gamma for TB protection. Nat. Microbiol. 2018, 3, 1194–1195. [Google Scholar] [CrossRef]
- Kauffman, K.D.; Sallin, M.A.; Sakai, S.; Kamenyeva, O.; Kabat, J.; Weiner, D.; Sutphin, M.; Schimel, D.; Via, L.; Barry, C.E.; et al. Defective positioning in granulomas but not lung-homing limits CD4 T-cell interactions with Mycobacterium tuberculosis-infected macrophages in rhesus macaques. Mucosal Immunol. 2018, 11, 462–473. [Google Scholar] [CrossRef]
- Alspach, E.; Lussier, D.M.; Miceli, A.P.; Kizhvatov, I.; DuPage, M.; Luoma, A.M.; Meng, W.; Lichti, C.F.; Esaulova, E.; Vomund, A.N.; et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature 2019, 574, 696–701. [Google Scholar] [CrossRef]
- Angell, H.; Galon, J. From the immune contexture to the Immunoscore: The role of prognostic and predictive immune markers in cancer. Curr. Opin. Immunol. 2013, 25, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef] [PubMed]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019, 565, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Shae, D.; Baljon, J.J.; Wehbe, M.; Becker, K.W.; Sheehy, T.L.; Wilson, J.T. At the bench: Engineering the next generation of cancer vaccines. J. Leukoc. Biol. 2019, 1–19. [Google Scholar] [CrossRef]
- Hilf, N.; Kuttruff-Coqui, S.; Frenzel, K.; Bukur, V.; Stevanović, S.; Gouttefangeas, C.; Platten, M.; Tabatabai, G.; Dutoit, V.; van der Burg, S.H.; et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 2019, 565, 240–245. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Tanyi, J.L.; Bobisse, S.; Ophir, E.; Tuyaerts, S.; Roberti, A.; Genolet, R.; Baumgartner, P.; Stevenson, B.J.; Iseli, C.; Dangaj, D.; et al. Personalized cancer vaccine effectively mobilizes antitumor T cell immunity in ovarian cancer. Sci. Transl. Med. 2018, 10, 1–14. [Google Scholar] [CrossRef]
- Ghorani, E.; Reading, J.L.; Henry, J.Y.; de Massy, M.R.; Rosenthal, R.; Turati, V.; Joshi, K.; Furness, A.J.S.; Ben Aissa, A.; Saini, S.K.; et al. The T cell differentiation landscape is shaped by tumour mutations in lung cancer. Nat. Cancer 2020, 1, 546–561. [Google Scholar] [CrossRef]
- Mogues, T.; Goodrich, M.E.; Ryan, L.; LaCourse, R.; North, R.J. The Relative Importance of T Cell Subsets in Immunity and Immunopathology of Airborne Mycobacterium tuberculosis Infection in Mice. J. Exp. Med. 2001, 193, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Huang, D.; Chen, C.Y.; Halliday, L.; Wang, R.C.; Chen, Z.W. CD4+ T Cells Contain Early Extrapulmonary Tuberculosis (TB) Dissemination and Rapid TB Progression and Sustain Multieffector Functions of CD8+ T and CD3− Lymphocytes: Mechanisms of CD4+ T Cell Immunity. J. Immunol. 2014, 192, 2120–2132. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.L.; Rutledge, T.; Green, A.M.; Bigbee, M.; Fuhrman, C.; Klein, E.; Flynn, J.L. CD4 T Cell Depletion Exacerbates Acute Mycobacterium tuberculosis While Reactivation of Latent Infection Is Dependent on Severity of Tissue Depletion in Cynomolgus Macaques. AIDS Res. Hum. Retrovir. 2012, 28, 1693–1702. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Huang, D.; Wang, R.C.; Shen, L.; Zeng, G.; Yao, S.; Shen, Y.; Halliday, L.; Fortman, J.; McAllister, M.; et al. A critical role for CD8 T cells in a nonhuman primate model of tuberculosis. PLoS Pathog. 2009, 5, 1–10. [Google Scholar] [CrossRef]
- Kagina, B.M.N.; Abel, B.; Scriba, T.J.; Hughes, E.J.; Keyser, A.; Soares, A.; Gamieldien, H.; Sidibana, M.; Hatherill, M.; Gelderbloem, S.; et al. Specific T cell frequency and cytokine expression profile do not correlate with protection against tuberculosis after bacillus Calmette-Guérin vaccination of newborns. Am. J. Respir. Crit. Care Med. 2010, 182, 1073–1079. [Google Scholar] [CrossRef]


{kind=link}
| Summary Table | ||
| T Cells | ||
| Tuberculosis | TME | |
| Role | Activation of M. tuberculosis infected macrophages to stimulate mycobacterial killing Possess an effector role that has potential to be modified through immunotherapy [2] | Elimination of Cancer Cells Immunomodulation of the TME with an effector role that has been modified through immunotherapy [20] |
| Cytokines | IFNγ, TNF-α, IL-6, IL-12, IL-1β | |
| Immunotherapies | Anti-PD-1/PD-L1 | Anti-PD-1/PD-L1, CAR T cells, TIGIT, OX40, 4-1BB, LAG3, TIM-3, Monalizumab, CTLA-4 |
| Macrophages | ||
| Tuberculosis | TME | |
| Role | The main reservoir for M. tuberculosis Work in conjunction with T cells to kill mycobacteria [2] | Immunomodulatory roles through cytokine production Effector role that can be modified through immunotherapy [21,22,23] |
| Cytokines | iNOS, IFNγ, TNF-α, IL-6, IL-1β, IL-10, IL-4, IL-2 | |
| Immunotherapies | CD40 agonists [23] Rifampicin [24] | CD40 agonists [23] β-Glucan [25] STAT3 [22] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bickett, T.E.; Karam, S.D. Tuberculosis–Cancer Parallels in Immune Response Regulation. Int. J. Mol. Sci. 2020, 21, 6136. https://doi.org/10.3390/ijms21176136
Bickett TE, Karam SD. Tuberculosis–Cancer Parallels in Immune Response Regulation. International Journal of Molecular Sciences. 2020; 21(17):6136. https://doi.org/10.3390/ijms21176136
Chicago/Turabian StyleBickett, Thomas E., and Sana D. Karam. 2020. "Tuberculosis–Cancer Parallels in Immune Response Regulation" International Journal of Molecular Sciences 21, no. 17: 6136. https://doi.org/10.3390/ijms21176136
APA StyleBickett, T. E., & Karam, S. D. (2020). Tuberculosis–Cancer Parallels in Immune Response Regulation. International Journal of Molecular Sciences, 21(17), 6136. https://doi.org/10.3390/ijms21176136