Role of P2Y Receptors in Platelet Extracellular Vesicle Release


 ,
,  , and
, and
Abstract
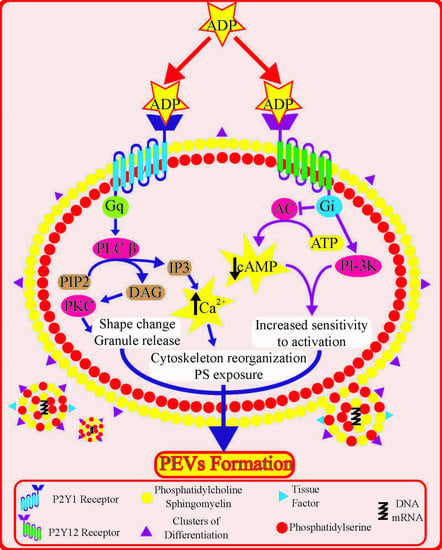
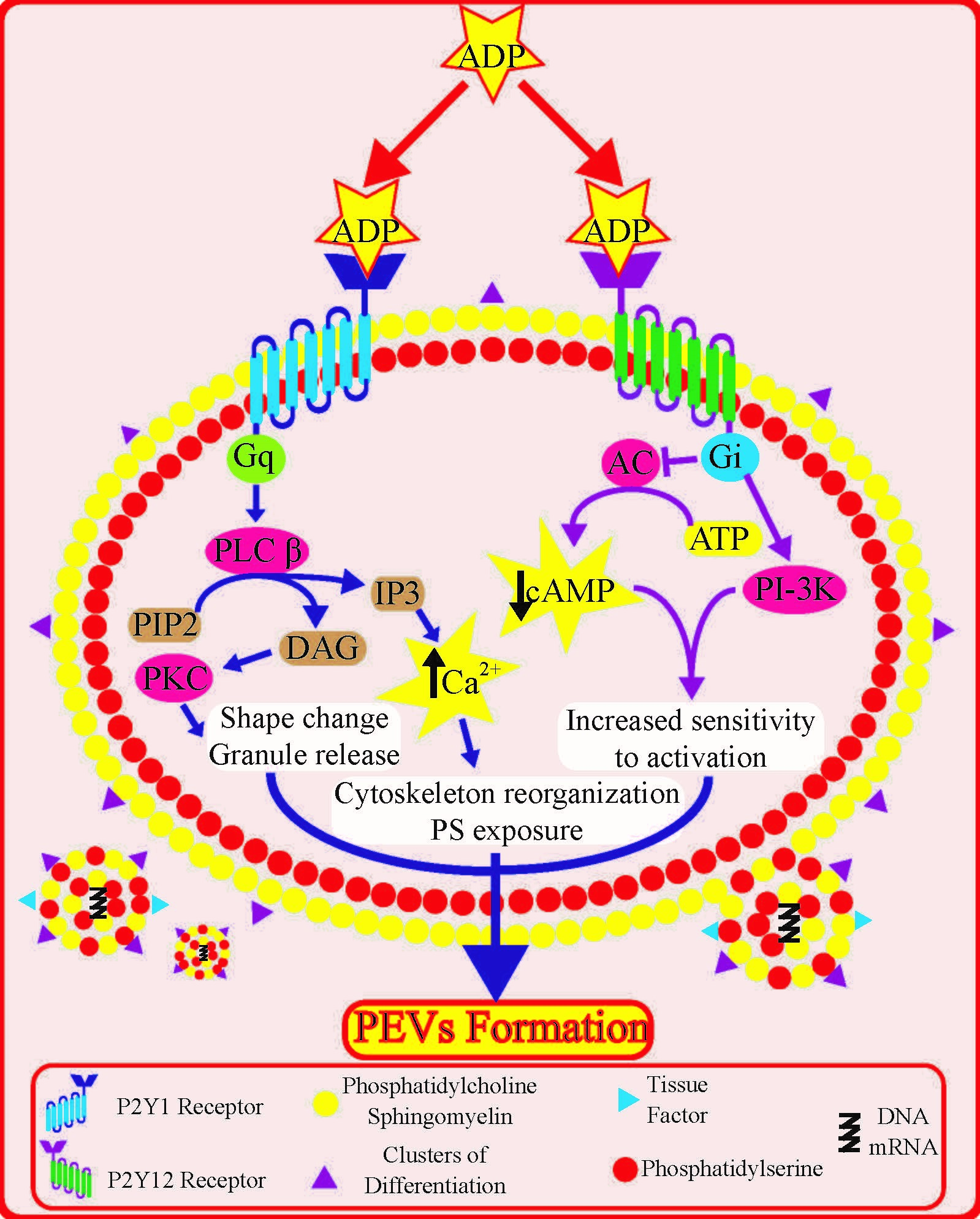
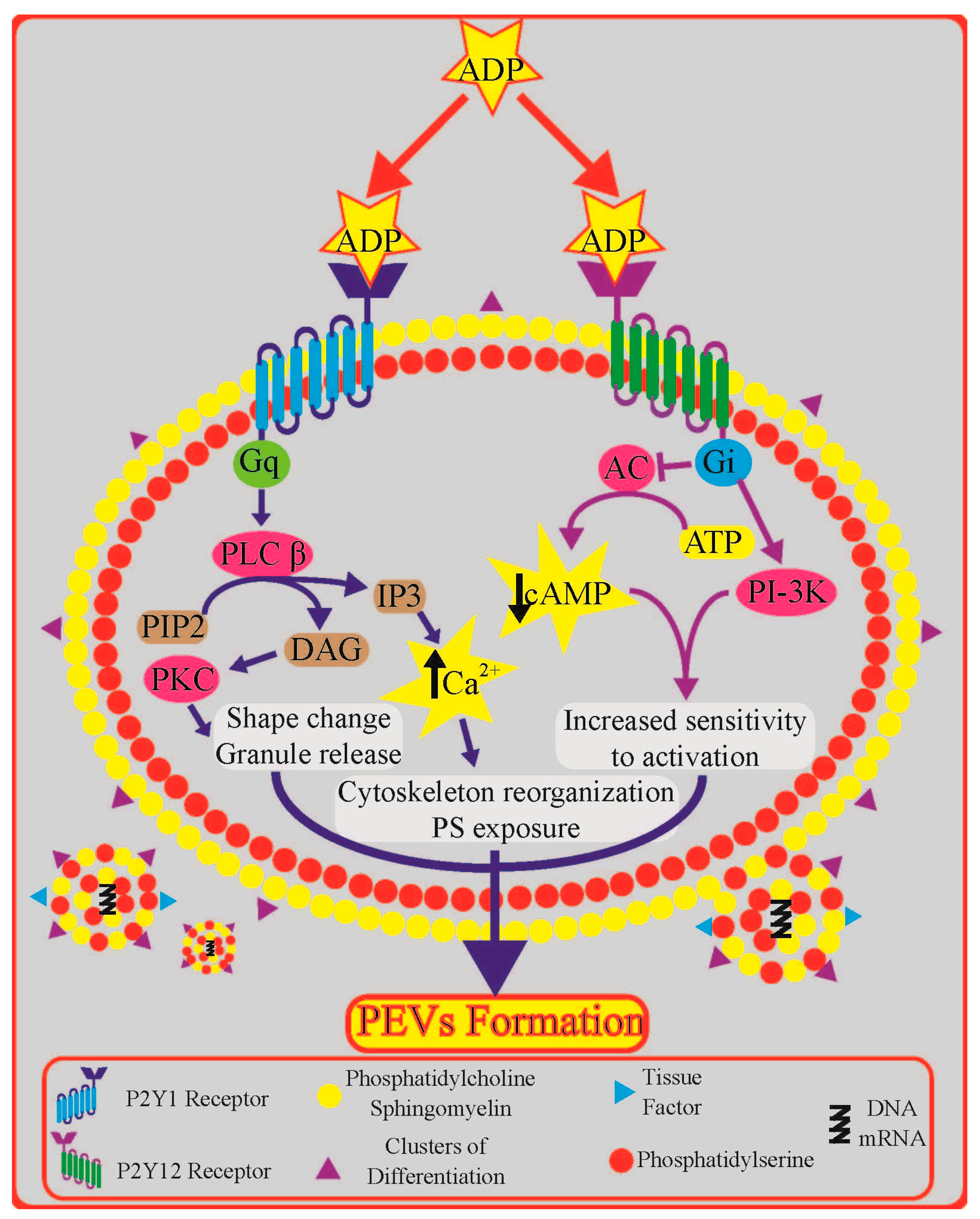
1. Introduction
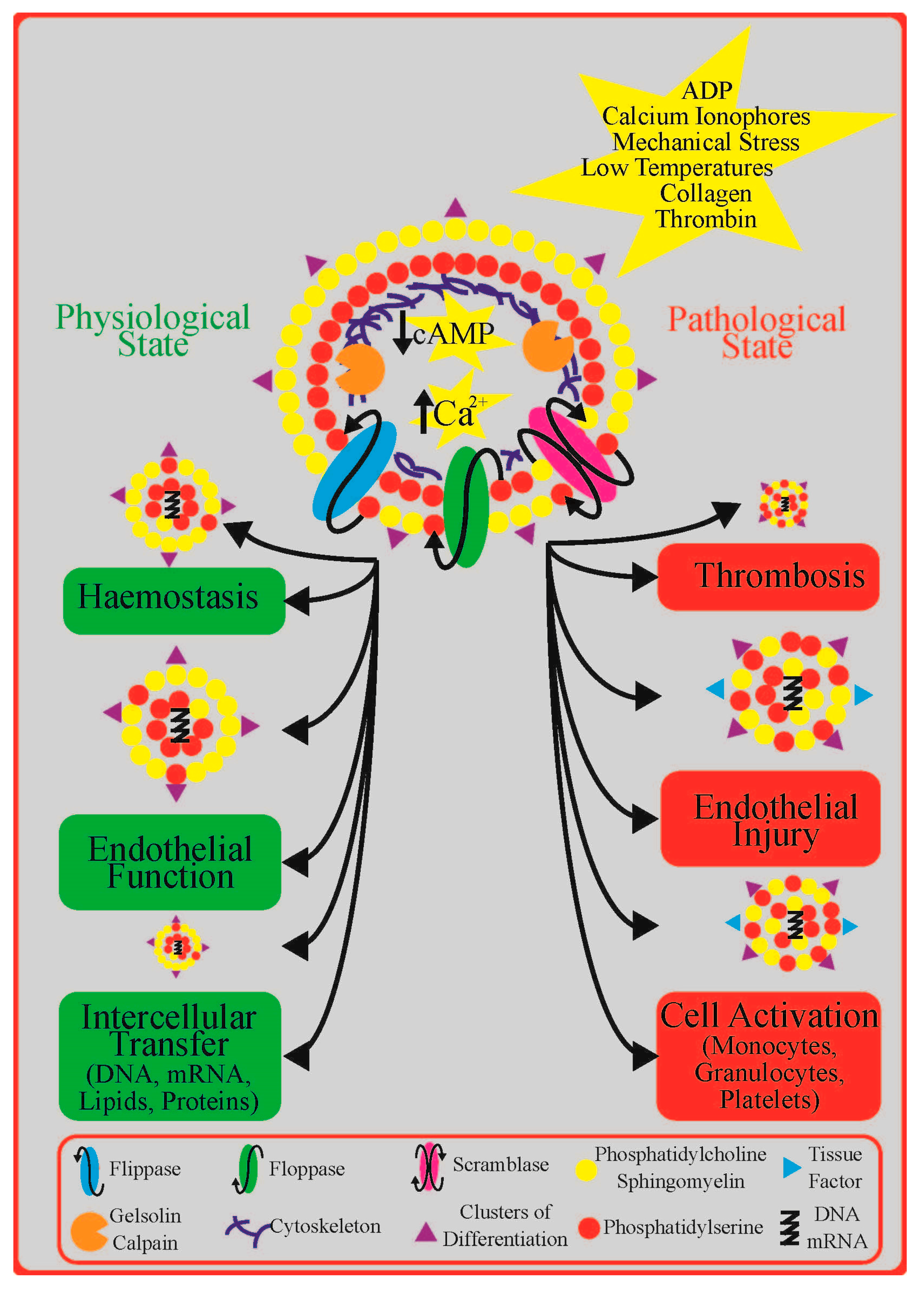
2. Platelet Extracellular Vesicles in Cardiovascular Disease: Diagnosis, Prognosis, and Treatment
3. Platelet P2Y Receptors: Role and Downstream Signalling
4. Role of Platelet P2Y Receptors in Platelet Extracellular Vesicle Formation
4.1. Insights from Signal Transduction Pathways
4.2. Insights from Experimental Studies
4.3. Insights from Antiplatelet Therapy
5. Other P2Y Receptors: P2Y13 and P2Y14
6. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AC | Adenylyl cyclase |
| ACS | Acute coronary syndromes |
| ADP | Adenosine diphosphate |
| AMI | Acute myocardial infarction |
| ATP | Adenosine triphosphate |
| cAMP | Cyclic adenosine monophosphate |
| Ca2+ | Calcium |
| CD | Cluster of differentiation |
| DAG | Diacylglycerol |
| Gi | GTP-binding protein i |
| Gq | GTP-binding protein q |
| IP3 | Inositol trisphosphate |
| PCI | Percutaneous coronary intervention |
| PIP2 | Phosphatidylinositol 4,5-bisphosphate |
| PI3-K | Phosphoinositide 3-kinase |
| PEVs | Platelet extracellular vesicles |
| PKC | Protein kinase C |
| PLC β | Phospholipase C β |
| PS | Phosphatidylserine |
| SCAD | Stable coronary artery disease |
| TF | Tissue factor |
| TRAP | Thrombin receptor-activating peptide |
References
- Jurk, K.; Kehrel, B.E. Platelet: Physiology and Biochemistry. Semin. Thromb. Hemost. 2005, 1, 381–392. [Google Scholar] [CrossRef]
- Willems, C.; van Aken, W.G. Production of prostacyclin by vascular endothelial cells. Pathophysiol. Haemost. Thromb. 1979, 8, 266–273. [Google Scholar] [CrossRef]
- Kim, S.; Kunapuli, S.P. P2Y12 receptor in platelet activation. Platelets 2011, 22, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Linden, M.D.; Jackson, D.E. Platelets: Pleiotropic roles in atherogenesis and atherothrombosis. Int. J. Biochem. Cell Biol. 2010, 42, 1762–1766. [Google Scholar] [CrossRef]
- Hechler, B.; Gachet, C. Purinergic Receptors in Thrombosis and Inflammation. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2307–2315. [Google Scholar] [CrossRef]
- Pérez-Sen, R.; Gómez-Villafuertes, R.; Ortega, F.; Gualix, J.; Delicado, E.G.; Miras-Portugal, M.T. An update on P2Y 13 receptor signalling and function. Adv. Exp. Med. Biol. 2017, 1051, 139–168. [Google Scholar] [PubMed]
- Koupenova-Zamor, M.; Ravid, K. Biology of Platelet Purinergic Receptors and Implications for Platelet Heterogeneity. Front. Pharmacol. 2018, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Dovlatova, N.; Wijeyeratne, Y.D.; Fox, S.C.; Manolopoulos, P.; Johnson, A.J.; White, A.E.; Latif, M.L.; Ralevic, V.; Heptinstall, S. Detection of P2Y (14) protein in platelets and investigation of the role of P2Y (14) in platelet function in comparison with the EP (3) receptor. Thromb Haemost. 2008, 100, 261–270. [Google Scholar]
- Abbas, Z.S.B.; Latif, M.L.; Dovlatova, N.; Fox, S.C.; Heptinstall, S.; Dunn, W.R.; Ralevic, V. UDP-sugars activate P2Y14 receptors to mediate vasoconstriction of the porcine coronary artery. Vascul. Pharmacol. 2018, 103, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Gachet, C. P2 receptors, platelet function and pharmacological implications. Thromb. Haemost. 2008, 99, 466–472. [Google Scholar] [CrossRef]
- Gremmel, T.; Yanachkov, I.B.; Yanachkova, M.I.; Wright, G.E.; Wider, J.; Undyala, V.V.R.; Michelson, A.D.; Frelinger III, A.L.; Przyklenk, K. Synergistic inhibition of both P2Y1 and P2Y12 adenosine diphosphate receptors as novel approach to rapidly attenuate platelet-mediated thrombosis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Valgimigli, M.; Bueno, H.; Byrne, R.A.; Collet, J.-P.; Costa, F.; Jeppsson, A.; Jüni, P.; Kastrati, A.; Kolh, P.; Mauri, L.; et al. 2017 ESC focused update on dual antiplatelet therapy in coronary artery disease developed in collaboration with EACTS: The Task Force for dual antiplatelet therapy in coronary artery disease of the European Society of Cardiology (ESC) and of the European Association for Cardio-Thoracic Surgery (EACTS). Eur. Heart J. 2017, 39, 213–260. [Google Scholar] [CrossRef]
- Golebiewska, E.M.; Poole, A.W. Platelet secretion: From haemostasis to wound healing and beyond. Blood Rev. 2015, 29, 153–162. [Google Scholar] [CrossRef] [PubMed]
- van der Pol, E.; Boing, A.N.; Harrison, P.; Sturk, A.; Nieuwland, R. Classification, Functions, and Clinical Relevance of Extracellular Vesicles. Pharmacol. Rev. 2012, 64, 676–705. [Google Scholar] [CrossRef]
- Heijnen, B.H.F.G.; Schiel, A.E.; Fijnheer, R.; Geuze, H.J.; Sixma, J.J. Activated Platelets Release Two Types of Membrane Vesicles: Microvesicles by Surface Shedding and Exozomes Derived from Exocytosis of multivascular bodies and alpha-granules. Blood 1999, 94, 3791–3800. [Google Scholar] [CrossRef]
- Gasecka, A.; Böing, A.N.; Filipiak, K.J.; Nieuwland, R. Platelet extracellular vesicles as biomarkers for arterial thrombosis. Platelets 2017, 28, 228–234. [Google Scholar] [CrossRef]
- Tomaniak, M.; Gąsecka, A.; Filipiak, K.J. Cell-derived microvesicles in cardiovascular diseases and antiplatelet therapy monitoring — A lesson for future trials? Current evidence, recent progresses and perspectives of clinical application. Int. J. Cardiol. 2017, 226, 93–102. [Google Scholar] [CrossRef]
- Gąsecka, A.; Van Der Pol, E.; Nieuwland, R.; Stępień, E. Extracellular vesicles in post-infarct ventricular remodelling. Kardiol. Pol. 2018, 76, 69–76. [Google Scholar] [CrossRef]
- Gasecka, A.; Nieuwland, R.; Siljander, P.R.-M. Platelet-derived extracellular vesicles. In Platelets, 4th ed.; Michelson, A., Cattaneo, M., Frelinger, A., Newman, P., Eds.; Academic Press, Elsevier: Cambridge, MA, USA, 2019; pp. 401–416. [Google Scholar]
- Sun, C.; Zhao, W.-B.; Chen, Y.; Hu, H.-Y. Higher plasma concentrations of platelet microparticles in patients with acute coronary syndrome: A systematic review and meta-analysis. Can. J. Cardiol. 2016, 32. [Google Scholar] [CrossRef]
- Gasecka, A.; Nieuwland, R.; Budnik, M.; Dignat-George, F.; Eyileten, C.; Harrison, P.; Lacroix, R.; Leroyer, A.; Opolski, G.; Pluta, K.; et al. Ticagrelor attenuates the increase of extracellular vesicle concentrations in plasma after acute myocardial infarction compared to clopidogrel. J. Thromb. Haemost. 2020, 18, 609–623. [Google Scholar] [CrossRef]
- Stepien, E.; Surdacki, A. Platelet Reactivity And Circulating Platelet-Derived Microvesicles Are Differently Affected By P2Y 12 Receptor Antagonists. Int. J. Med. Sci. 2019, 16, 264–275. [Google Scholar] [CrossRef]
- Rank, A.; Nieuwland, R.; Crispin, A.; Grützner, S.; Iberer, M.; Toth, B.; Pihusch, R. Clearance of platelet microparticles in vivo. Platelets 2011, 22, 111–116. [Google Scholar] [CrossRef]
- van Es, N.; Bleker, S.; Sturk, A.; Nieuwland, R. Clinical Significance of Tissue Factor-Exposing Microparticles in Arterial and Venous Thrombosis. Semin. Thromb. Hemost. 2015, 41, 718–727. [Google Scholar] [CrossRef] [PubMed]
- Léon, C.; Freund, M.; Ravanat, C.; Baurand, A.; Cazenave, J.-P.; Gachet, C. Key Role of the P2Y1 Receptor in Tissue Factor--Induced Thrombin-Dependent Acute Thromboembolism: Studies in P2Y1-Knockout Mice and Mice Treated With a P2Y1 Antagonist. Circulation 2001, 103, 718–723. [Google Scholar]
- Hechler, B.; Zhang, Y.; Eckly, A.; Cazenave, J.-P.; Gachet, C.; Ravid, K. Lineage-specific overexpression of the P2Y1 receptor induces platelet hyper-reactivity in transgenic mice. J. Thromb. Haemost. 2003, 1, 155–163. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, X.; Liu, L.; Zaske, A.-M.; Zhou, Z.; Fu, Y.; Yang, X.; Conyers, J.L.; Li, M.; Dong, J.; et al. Contact-and agonist-regulated microvesiculation of human platelets. Thromb. Haemost. 2013, 110, 331–399. [Google Scholar]
- Yan, R.; Wang, Z.; Yuan, Y.; Cheng, H.; Dai, K. Role of cAMP-dependent protein kinase in the regulation of platelet procoagulant activity. Arch. Biochem. Biophys. 2009, 485, 41–48. [Google Scholar] [CrossRef]
- Morel, O.; Jesel, L.; Freyssinet, J.-M.; Toti, F. Cellular mechanisms underlying the formation of circulating microparticles. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 15–26. [Google Scholar] [CrossRef]
- Kahner, B.N.; Dorsam, R.T.; Kunapuli, S.P. Role of P2Y receptor subtypes in platelet-derived microparticle generation. Front Biosci. 2008, 13, 433–439. [Google Scholar] [CrossRef]
- Gasecka, A.; Nieuwland, R.; van der Pol, E.; Hajji, N.; Ćwiek, A.; Pluta, K.; Konwerski Michałand Filipiak, K.J. P2Y12 antagonist ticagrelor inhibits the release of procoagulant extracellular vesicles from activated platelets: Preliminary results. Cardiol. J. 2013. [Google Scholar] [CrossRef]
- Léon, C.; Ravanat, C.; Freund, M.; Cazenave, J.P.; Gachet, C. Differential involvement of the P2Y1and P2Y12receptors in platelet procoagulant activity. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1941–1947. [Google Scholar] [CrossRef] [PubMed]
- Takano, K.; Asazuma, N.; Satoh, K.; Yatomi, Y.; Ozaki, Y. Collagen-induced generation of platelet-derived microparticles in whole blood is dependent on ADP released from red blood cells and calcium ions. Platelets 2004, 15, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Milanowski, L.; Rasul, F.; Gajda, S.N.; Eyileten, C.; Siller-Matula, J.; Postula, M. Genetic Variability of SRC Family Kinases and Its Association with Platelet Hyperreactivity and Clinical Outcomes: A Systematic Review. Curr. Pharm. Des. 2018, 24, 628–640. [Google Scholar] [CrossRef] [PubMed]
- Biesinger, B.S.; Gasecka, A.; Perkmann, T.; Wojta, J.; Lesiak, M.; Grygier, M.; Eyileten, C.; Postuła, M.; Filipiak, K.J.; Toma, A.; et al. Inflammatory state does not affect the antiplatelet efficacy of potent P2Y12 inhibitors in ACS. Platelets 2020, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Eyileten, C.; Soplinska, A.; Pordzik, J.; Siller-Matula, J.M.; Postuła, M. Effectiveness of antiplatelet drugs under therapeutic hypothermia: A comprehensive review. Clin. Pharmacol. Ther. 2019, 106, 993–1005. [Google Scholar] [CrossRef] [PubMed]
- Kubisa, M.J.; Jezewski, M.P.; Gasecka, A.; Siller-Matula, J.M.; Postuła, M. Ticagrelor – toward more efficient platelet inhibition and beyond. Ther. Clin. Risk Manag. 2018, 14, 129–140. [Google Scholar] [CrossRef]
- Tomaniak, M.; Kołtowski, L.; Kochman, J.; Huczek, Z.; Rdzanek, A.; Pietrasik, A.; Gasecka, A.; Gajda, S.; Opolski, G.; Filipiak, K.J. Can prasugrel decrease the extent of periprocedural myocardial injury during elective percutaneous coronary intervention? Pol. Arch. Intern. Med. 2017, 127, 730–740. [Google Scholar] [CrossRef][Green Version]
- Behan, M.W.H.; Fox, S.C.; Heptinstall, S.; Storey, R.F. Inhibitory effects of P2Y12 receptor antagonists on TRAP-induced platelet aggregation, procoagulant activity, microparticle formation and intracellular calcium responses in patients with acute coronary syndromes. Platelets 2005, 16, 73–80. [Google Scholar] [CrossRef]
- Judge, H.M.; Buckland, R.J.; Sugidachi, A.; Jakubowski, J.A.; Storey, R.F. The active metabolite of prasugrel effectively blocks the platelet P2Y12 receptor and inhibits procoagulant and pro-inflammatory platelet responses. Platelets 2008, 19, 125–133. [Google Scholar] [CrossRef]
- Hechler, B.; Eckly, A.; Ohlmann, P.; Cazenave, J.P.; Gachet, C. The P2Y1receptor, necessary but not sufficient to support full ADP- induced platelet aggregation, is not the target of the drug clopidogrel. Br. J. Haematol. 1998, 103, 858–866. [Google Scholar] [CrossRef]
- Traby, L.; Kaider, A.; Kollars, M.; Eichinger, S.; Wolzt, M.; Kyrle, P.A. Effects of clopidogrel with or without aspirin on the generation of extracellular vesicles in the microcirculation and in venous blood: A randomized placebo controlled trial. Thromb. Res. 2018, 167, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Judge, H.M.; Buckland, R.J.; Holgate, C.E.; Storey, R.F. Glycoprotein IIb/IIIa and P2Y12 receptor antagonists yield additive inhibition of platelet aggregation, granule secretion, soluble CD40L release and procoagulant responses. Platelets 2005, 16, 398–407. [Google Scholar] [CrossRef] [PubMed]
- França, C.N.; Pinheiro, L.F.M.; Izar, M.C.O.; Brunialti, M.K.C.; Salomão, R.; Bianco, H.T.; Kasmas, S.H.; Barbosa, S.P.; de Nucci, G.; Fonseca, F.A.H. Endothelial Progenitor Cell Mobilization and Platelet Microparticle Release Are Influenced by Clopidogrel Plasma Levels in Stable Coronary Artery Disease. Circ. J. 2012, 76, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Camargo, L.M.; Franc, C.M.; Izar, M.C.; Bianco, H.T.; Lins, L.S.; Barbosa, S.P.; Pinheiro, L.F.; Fonseca, F.A.H. Effects of simvastatin/ezetimibe on microparticles, endothelial progenitor cells and platelet aggregation in subjects with coronary heart disease under antiplatelet therapy. Braz. J. Med. Biol. Res. 2014, 47, 432–437. [Google Scholar] [CrossRef]
- Haller, P.M.; Stojkovic, S.; Piackova, E.; Andric, T.; Wisgrill, L.; Spittler, A.; Wojta, J.; Huber, K.; Jäger, B. The association of P2Y12 inhibitors with pro-coagulatory extracellular vesicles and microRNAs in stable coronary artery disease. Platelets 2020, 31, 497–504. [Google Scholar] [CrossRef]
- Gemmell, C.H.; Sefton, M.V.; Yeo, E.L. Platelet-derived microparticle formation involves glycoprotein IIb-IIIa. Inhibition by RGDS and a Glanzmann’s thrombasthenia defect. J. Biol. Chem. 1993, 268, 14586–14589. [Google Scholar]
- Gasecka, A.; Nieuwland, R.; Budnik, M.; Dignat-George, F.; Eyileten, C.; Harrison, P.; Huczek, Z.; Kapłon-Cieślicka, A.; Lacroix, R.; Opolski, G.; et al. Randomized controlled trial protocol to investigate the antiplatelet therapy effect on extracellular vesicles (AFFECT EV) in acute myocardial infarction. Platelets 2018, 31, 26–32. [Google Scholar] [CrossRef]
- Wallentin, L.; Becker, R.C.; Budaj, A.; Cannon, C.P.; Emanuelsson, H.; Held, C.; Horrow, J.; Husted, S.; James, S.; Katus, H.; et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N. Engl. J. Med. 2009, 361, 1045–1057. [Google Scholar] [CrossRef]
- Taus, F.; Meneguzzi, A.; Castelli, M.; Minuz, P. Platelet-derived extracellular vesicles as target of antiplatelet agents. What is the evidence? Front. Pharmacol. 2019, 10, 1256. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Paoletta, S.; Katritch, V.; Wu, B.; Gao, Z.-G.; Zhao, Q.; Stevens, R.C.; Kiselev, E. Nucleotides acting at P2Y receptors: Connecting structure and function. Mol. Pharmacol. 2015, 88, 220–230. [Google Scholar] [CrossRef]
- Coumans, F.A.W.; Brisson, A.R.; Buzas, E.I.; Dignat-George, F.; Drees, E.E.E.; El-Andaloussi, S.; Emanueli, C.; Gasecka, A.; Hendrix, A.; Hill, A.F.; et al. Methodological guidelines to study extracellular vesicles. Circ. Res. 2017, 120, 1632–1648. [Google Scholar] [CrossRef]
- van der Pol, E.; Böing, A.N.; Gool, E.L.; Nieuwland, R. Recent developments in the nomenclature, presence, isolation, detection and clinical impact of extracellular vesicles. J. Thromb. Haemost. 2016, 14, 48–56. [Google Scholar] [CrossRef] [PubMed]
- van der Pol, E.; Van Gemert, M.J.C.; Sturk, A.; Nieuwland, R.; Van Leeuwen, T.G.; Nieuwl, R.; Van Leeuwen, T.G. Single vs. swarm detection of microparticles and exosomes by flow cytometry. J. Thromb. Haemost. 2012, 10, 919–930. [Google Scholar] [CrossRef] [PubMed]
- van der Pol, E.; Sturk, A.; van Leeuwen, T.G.; Nieuwland, R.; Coumans, F.A.W. Standardization of extracellular vesicle measurements by flow cytometry through vesicle diameter approximation. J. Thromb. Haemost. 2018, 16, 1236–1245. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.; van der Pol, E.; Pálmai, M.; Garcia-Diez, R.; Gollwitzer, C.; Krumrey, M.; Fraikin, J.-L.; Gasecka, A.; Hajji, N.; van Leeuwen, T.G.; et al. Hollow organosilica beads as reference particles for optical detection of extracellular vesicles. J. Thromb. Haemost. 2018, 16. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.A.; van der Pol, E.; Arkesteijn, G.J.A.; Bremer, M.; Brisson, A.; Coumans, F.; Dignat-George, F.; Duggan, E.; Ghiran, I.; Giebel, B.; et al. MIFlowCyt-EV: A framework for standardized reporting of extracellular vesicle flow cytometry experiments. J. Extracell. Vesicles. 2020, 9, 1713526. [Google Scholar] [CrossRef]
- Van Deun, J.; Mestdagh, P.; Agostinis, P.; Akay, Ö.; Anand, S.; Anckaert, J.; Martinez, Z.A.; Baetens, T.; Beghein, E.; Bertier, L.; et al. EV-TRACK: Transparent reporting and centralizing knowledge in extracellular vesicle research. Nat. Methods. 2017, 14, 228–232. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Condition | Study Group | Control Group | Sample Size | Effect on EVs | Ref. |
|---|---|---|---|---|---|
| Healthy volunteers | Clopidogrel | Aspirin | n = 44 | ~ Platele ~ TF ~ Endothelial | [42] |
| Prasugrel | No inhibitor | n = 18 | ↓ Platelet | [40] | |
| Cangrelor | No inhibitor | Data not provided | ↓ Platelet | [43] | |
| SCAD | Clopidogrel | Aspirin | n = 26 | ~ Platelet | [44] |
| Clopidogrel | Aspirin | n = 20 | ~ Platelet ~ Endothelial | [45] | |
| ACS | Clopidogrel | Aspirin | n = 12 | ↓ Platelet | [39] |
| Ticagrelor | Clopidogrel | n = 60 | ↓ Platelet ↓ Phosphatidylserine+ ↓ Fibrinogen+ ↓ Leukocyte ~ Endothelial ~ Erythrocyte | [21] | |
| Ticagrelor, Prasugrel, Clopidogrel | Aspirin | n = 62 | ~ Platelet ~ Endothelial ~ Monocyte ~ Erythrocyte | [46] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gąsecka, A.; Rogula, S.; Eyileten, C.; Postuła, M.; Jaguszewski, M.J.; Kochman, J.; Mazurek, T.; Nieuwland, R.; Filipiak, K.J. Role of P2Y Receptors in Platelet Extracellular Vesicle Release. Int. J. Mol. Sci. 2020, 21, 6065. https://doi.org/10.3390/ijms21176065
Gąsecka A, Rogula S, Eyileten C, Postuła M, Jaguszewski MJ, Kochman J, Mazurek T, Nieuwland R, Filipiak KJ. Role of P2Y Receptors in Platelet Extracellular Vesicle Release. International Journal of Molecular Sciences. 2020; 21(17):6065. https://doi.org/10.3390/ijms21176065
Chicago/Turabian StyleGąsecka, Aleksandra, Sylwester Rogula, Ceren Eyileten, Marek Postuła, Miłosz J. Jaguszewski, Janusz Kochman, Tomasz Mazurek, Rienk Nieuwland, and Krzysztof J. Filipiak. 2020. "Role of P2Y Receptors in Platelet Extracellular Vesicle Release" International Journal of Molecular Sciences 21, no. 17: 6065. https://doi.org/10.3390/ijms21176065
APA StyleGąsecka, A., Rogula, S., Eyileten, C., Postuła, M., Jaguszewski, M. J., Kochman, J., Mazurek, T., Nieuwland, R., & Filipiak, K. J. (2020). Role of P2Y Receptors in Platelet Extracellular Vesicle Release. International Journal of Molecular Sciences, 21(17), 6065. https://doi.org/10.3390/ijms21176065