Ecto-Nucleotide Triphosphate Diphosphohydrolase-2 (NTPDase2) Deletion Increases Acetaminophen-Induced Hepatotoxicity

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
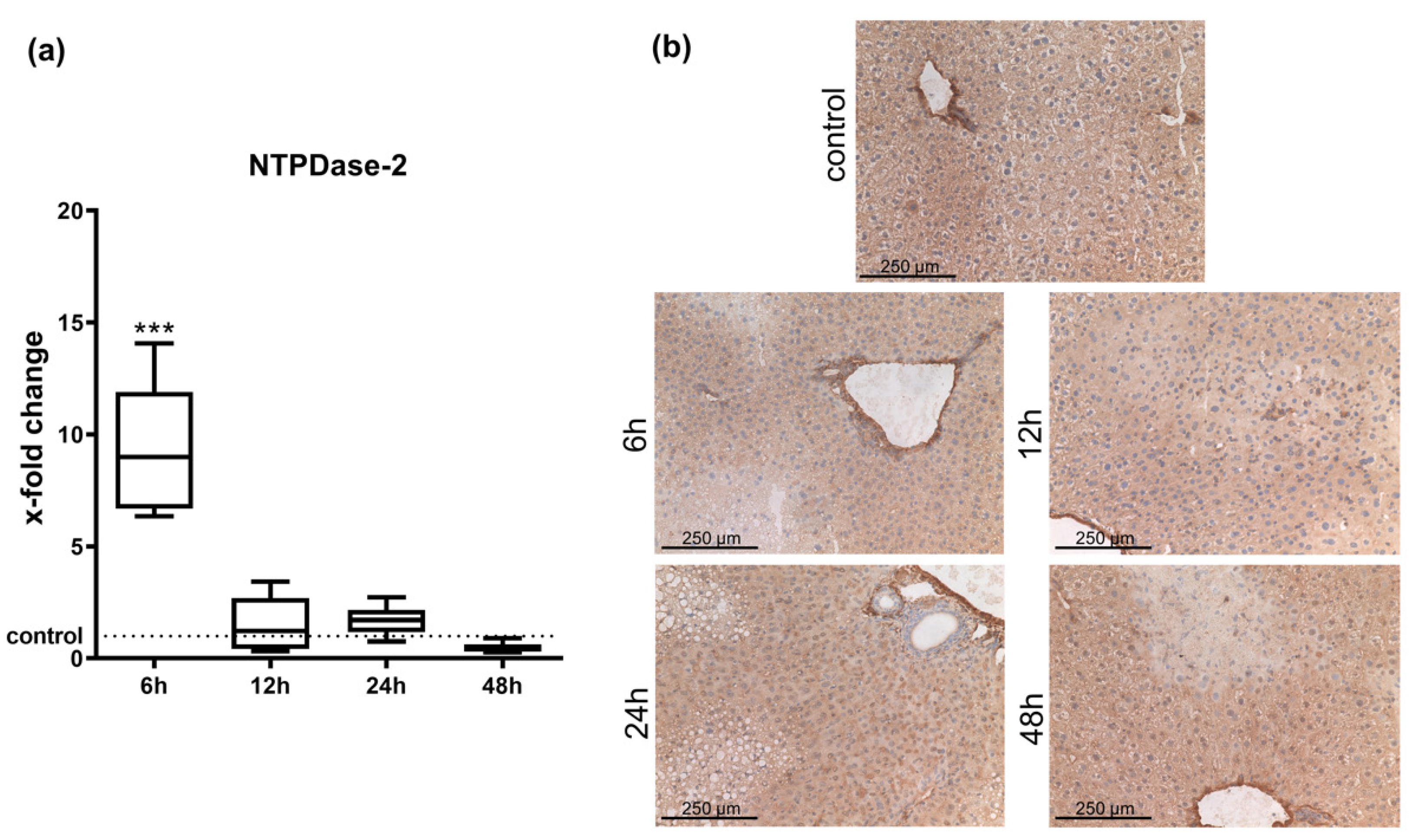
2.1. Hepatic Entpd2 Is Upregulated in Wild-Type (WT) Mice after APAP-Induced Acute Liver Injury
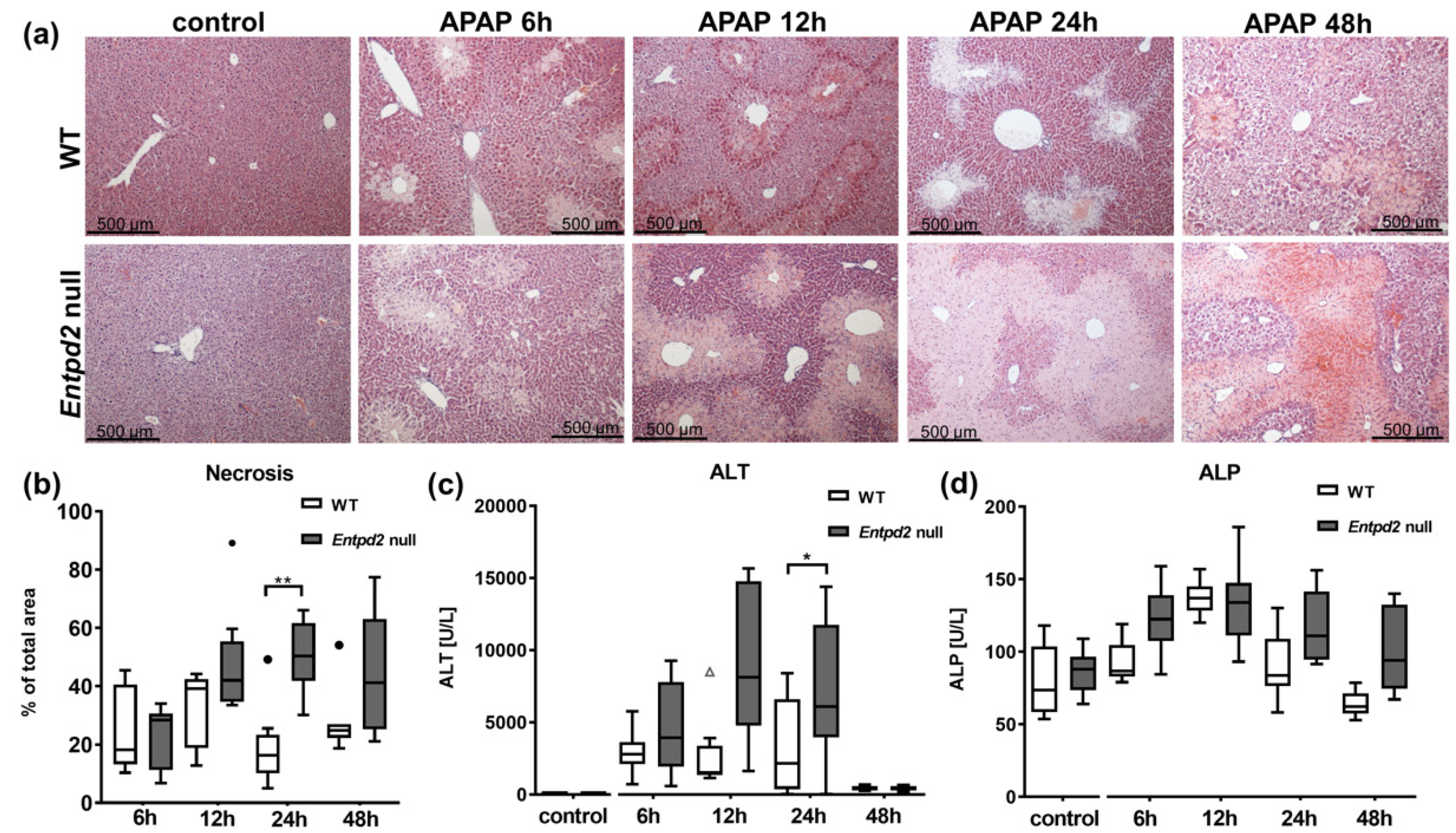
2.2. Liver Necrosis after APAP Treatment Is Exacerbated in Entpd2 Null Mice
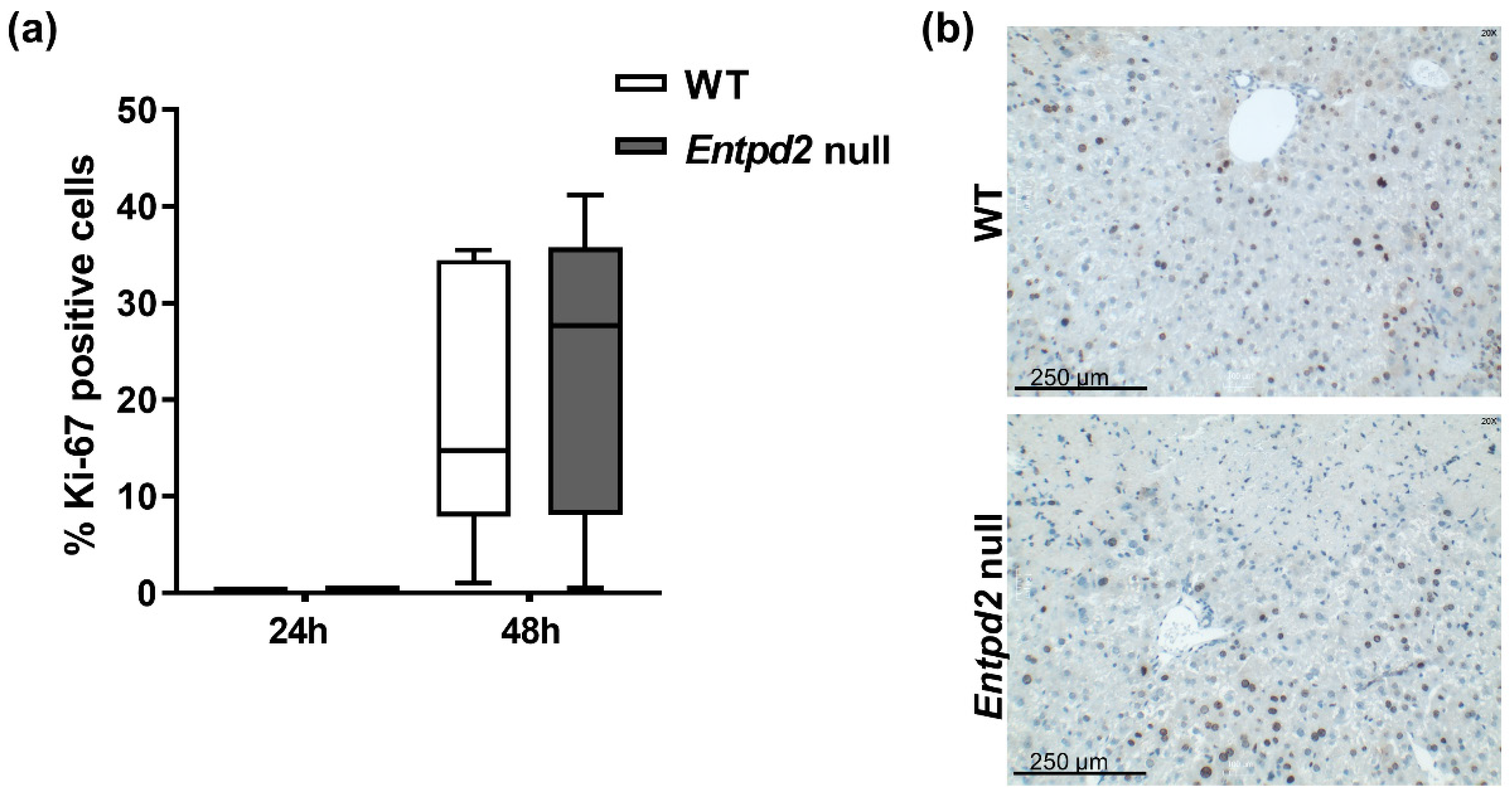
2.3. Liver Regeneration and Proliferation of Liver Cells Are Not Enhanced in Entpd2 Null Mice
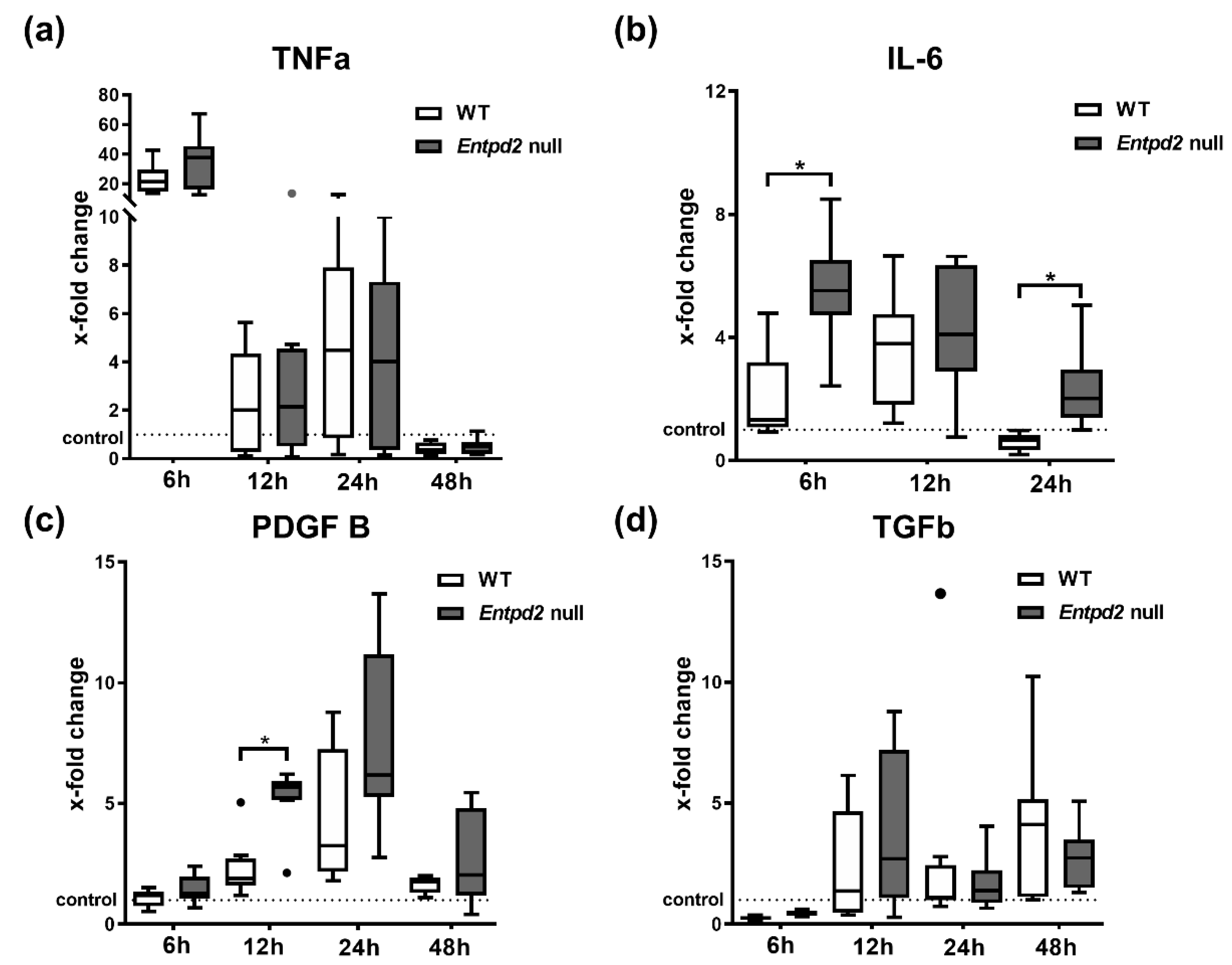
2.4. Hepatic Expression of IL-6 and PDGF-B Is Upregulated in Entpd2 Null Mice after APAP Intoxication
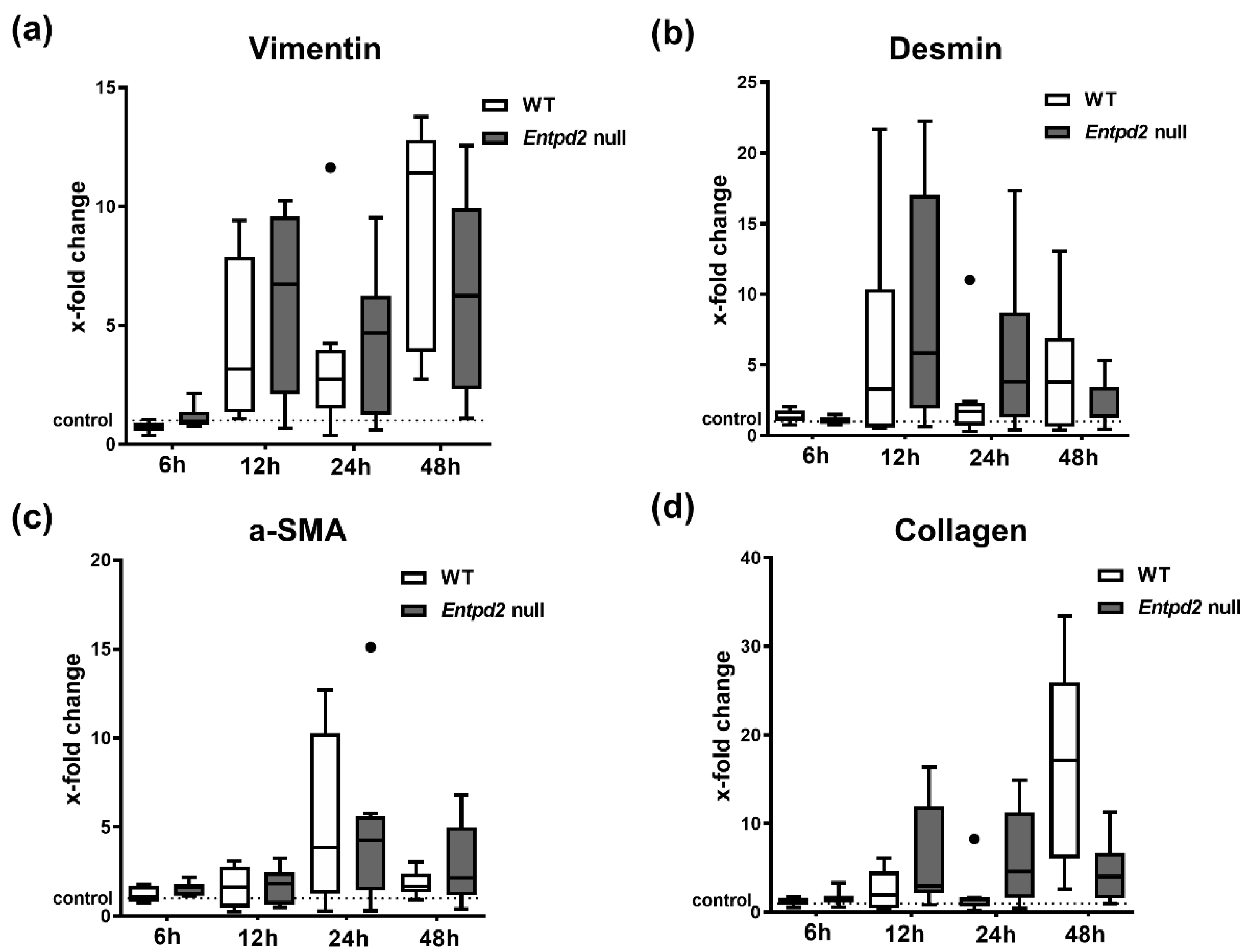
2.5. Hepatic Expression of Fibrosis-Associated Markers Is Triggered by APAP-Induced Liver Injury, Irrespective of Entpd2 Deletion
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Induction of Acute Liver Injury by APAP
4.3. Measurement of Serum ALT and ALP
4.4. RNA Isolation and Quantitative Real Time Polymerase Chain Reaction (qPCR)
4.5. Immunohistochemistry
4.6. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bernal, W.; Lee, W.M.; Wendon, J.; Larsen, F.S.; Williams, R. Acute liver failure: A curable disease by 2024? J. Hepatol. 2015, 62 (Suppl. 1), S112–S120. [Google Scholar] [CrossRef]
- Aminoshariae, A.; Khan, A. Acetaminophen: Old Drug, New Issues. J. Endod. 2015, 41, 588–593. [Google Scholar] [CrossRef]
- Bernal, W.; Wendon, J. Acute Liver Failure. N. Engl. J. Med. 2013, 369, 2525–2534. [Google Scholar] [CrossRef]
- Yoon, E.; Babar, A.; Choudhary, M.; Kutner, M.; Pyrsopoulos, N. Acetaminophen-Induced Hepatotoxicity: A Comprehensive Update. J. Clin. Transl. Hepatol. 2016, 4, 131–142. [Google Scholar] [CrossRef]
- Maes, M.; Vinken, M.; Jaeschke, H. Experimental models of hepatotoxicity related to acute liver failure. Toxicol. Appl. Pharmacol. 2016, 290, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Sitkovsky, M.V.; Robson, S.C. Purinergic Signaling during Inflammation. N. Engl. J. Med. 2012, 367, 2322–2333. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Purinergic signalling: Its unpopular beginning, its acceptance and its exciting future. BioEssays 2012, 34, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Beldi, G.; Wu, Y.; Sun, X.; Imai, M.; Enjyoji, K.; Csizmadia, E.; Candinas, D.; Erb, L.; Robson, S.C. Regulated Catalysis of Extracellular Nucleotides by Vascular CD39/ENTPD1 Is Required for Liver Regeneration. Gastroenterology 2008, 135, 1751–1760. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yoshida, O.; Kimura, S.; Jackson, E.K.; Robson, S.C.; Geller, D.A.; Murase, N.; Thomson, A.W. CD39 expression by hepatic myeloid dendritic cells attenuates inflammation in liver transplant ischemia-reperfusion injury in mice. Hepatology 2013, 58, 2163–2175. [Google Scholar] [CrossRef] [PubMed]
- Rothweiler, S.; Feldbrügge, L.; Jiang, Z.G.; Csizmadia, E.; Longhi, M.S.; Vaid, K.; Enjyoji, K.; Popov, Y.V.; Robson, S.C. Selective deletion of ENTPD1/CD39 in macrophages exacerbates biliary fibrosis in a mouse model of sclerosing cholangitis. Purinergic Signal. 2019, 15, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Hoque, R.; Sohail, M.A.; Salhanick, S.; Malik, A.F.; Ghani, A.; Robson, S.C.; Mehal, W.Z. P2X7 receptor-mediated purinergic signaling promotes liver injury in acetaminophen hepatotoxicity in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G1171–G1179. [Google Scholar] [CrossRef] [PubMed]
- Schmelzle, M.; Splith, K.; Andersen, L.W.; Kornek, M.; Schuppan, D.; Jones-Bamman, C.; Nowak, M.; Toxavidis, V.; Salhanick, S.D.; Han, L.; et al. Increased plasma levels of microparticles expressing CD39 and CD133 in acute liver injury. Transplantation 2013, 95, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Jhandier, M.N.; Kruglov, E.A.; Lavoie, E.G.; Sévigny, J.; Dranoff, J.A. Portal fibroblasts regulate the proliferation of bile duct epithelia via expression of NTPDase2. J. Biol. Chem. 2005, 280, 22986–22992. [Google Scholar] [CrossRef] [PubMed]
- Feldbrügge, L.; Jiang, Z.G.; Csizmadia, E.; Mitsuhashi, S.; Tran, S.; Yee, E.U.; Rothweiler, S.; Vaid, K.A.; Sévigny, J.; Schmelzle, M.; et al. Distinct roles of ecto-nucleoside triphosphate diphosphohydrolase-2 (NTPDase2) in liver regeneration and fibrosis. Purinergic Signal. 2018, 14, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.W.; Rothweiler, S.; Wei, G.; Ikenaga, N.; Liu, S.B.; Sverdlov, D.Y.; Vaid, K.A.; Longhi, M.S.; Kuang, M.; Robson, S.C.; et al. The ectonucleotidase ENTPD1/CD39 limits biliary injury and fibrosis in mouse models of sclerosing cholangitis. Hepatol. Commun. 2017, 1, 957–972. [Google Scholar] [CrossRef] [PubMed]
- Deaglio, S.; Dwyer, K.M.; Gao, W.; Friedman, D.; Usheva, A.; Erat, A.; Chen, J.F.; Enjyoji, K.; Linden, J.; Oukka, M.; et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 2007, 204, 1257–1265. [Google Scholar] [CrossRef]
- Allard, B.; Longhi, M.S.; Robson, S.C.; Stagg, J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol. Rev. 2017, 276, 121–144. [Google Scholar] [CrossRef]
- Dranoff, J.A.; Kruglov, E.A.; Robson, S.C.; Braun, N.; Zimmermann, H.; Sévigny, J. The ecto-nucleoside triphosphate diphosphohydrolase NTPDase2/CD39L1 is expressed in a novel functional compartment within the liver. Hepatology 2002, 36, 1135–1144. [Google Scholar] [CrossRef]
- Feldbrügge, L.; Moss, A.C.; Yee, E.U.; Csizmadia, E.; Mitsuhashi, S.; Longhi, M.S.; Sandhu, B.; Stephan, H.; Wu, Y.; Cheifetz, A.S.; et al. Expression of Ecto-nucleoside Triphosphate Diphosphohydrolases-2 and -3 in the Enteric Nervous System Affects Inflammation in Experimental Colitis and Crohn’s Disease. J. Crohns Colitis 2017, 11, 1113–1123. [Google Scholar] [CrossRef]
- Grubišić, V.; Perez-Medina, A.L.; Fried, D.E.; Sévigny, J.; Robson, S.C.; Galligan, J.J.; Gulbransen, B.D. NTPDase1 and -2 are expressed by distinct cellular compartments in the mouse colon and differentially impact colonic physiology and function after DSS colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 317, G314–G332. [Google Scholar] [CrossRef]
- Liñán-Rico, A.; Turco, F.; Ochoa-Cortes, F.; Harzman, A.; Needleman, B.J.; Arsenescu, R.; Abdel-Rasoul, M.; Fadda, P.; Grants, I.; Whitaker, E.; et al. Molecular Signaling and Dysfunction of the Human Reactive Enteric Glial Cell Phenotype: Implications for GI infection, IBD, POI, Neurological, Motility and GI Disorders. Inflamm. Bowel Dis. 2016, 22, 1812–1834. [Google Scholar] [CrossRef] [PubMed]
- James, L.P.; Kurten, R.C.; Lamps, L.W.; McCullough, S.; Hinson, J.A. Tumour Necrosis Factor Receptor 1 and Hepatocyte Regeneration in Acetaminophen Toxicity: A Kinetic Study of Proliferating Cell Nuclear Antigen and Cytokine Expression. Basic Clin. Pharmacol. Toxicol. 2005, 97, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Bhushan, B.; Walesky, C.; Manley, M.; Gallagher, T.; Borude, P.; Edwards, G.; Monga, S.P.S.; Apte, U. Pro-Regenerative Signaling after Acetaminophen-Induced Acute Liver Injury in Mice Identified Using a Novel Incremental Dose Model. Am. J. Pathol. 2014, 184, 3013–3025. [Google Scholar] [CrossRef] [PubMed]
- Amaral, S.S.; Oliveira, A.G.; Marques, P.E.; Quintão, J.L.; Pires, D.A.; Resende, R.R.; Sousa, B.R.; Melgaço, J.G.; Pinto, M.A.; Russo, R.C.; et al. Altered responsiveness to extracellular ATP enhances acetaminophen hepatotoxicity. Cell Commun. Signal. 2013, 11, 10. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; Ramachandran, A. Mechanisms and pathophysiological significance of sterile inflammation during acetaminophen hepatotoxicity. Food Chem. Toxicol. 2020, 138, 111240. [Google Scholar] [CrossRef]
- Xie, Y.; Williams, C.D.; McGill, M.R.; Lebofsky, M.; Ramachandran, A.; Jaeschke, H. Purinergic receptor antagonist A438079 protects against acetaminophen-induced liver injury by inhibiting p450 isoenzymes, not by inflammasome activation. Toxicol. Sci. 2013, 131, 325–335. [Google Scholar] [CrossRef]
- Xu, H.; Li, H.; Cao, D.; Wu, Y.; Chen, Y. Tumor necrosis factor α (TNF-α) receptor-I is required for TNF-α-mediated fulminant virus hepatitis caused by murine hepatitis virus strain-3 infection. Immunol. Lett. 2014, 158, 25–32. [Google Scholar] [CrossRef]
- Chastre, A.; Bélanger, M.; Beauchesne, E.; Nguyen, B.N.; Desjardins, P.; Butterworth, R.F. Inflammatory cascades driven by tumor necrosis factor-alpha play a major role in the progression of acute liver failure and its neurological complications. PLoS ONE 2012, 7, e49670. [Google Scholar] [CrossRef]
- Gao, R.Y.; Wang, M.; Liu, Q.; Feng, D.; Wen, Y.; Xia, Y.; Colgan, S.P.; Eltzschig, H.K.; Ju, C. Hypoxia-Inducible Factor-2α Reprograms Liver Macrophages to Protect Against Acute Liver Injury Through the Production of Interleukin-6. Hepatology 2020, 71, 2105–2117. [Google Scholar] [CrossRef]
- Li, S.Q.; Zhu, S.; Han, H.M.; Lu, H.J.; Meng, H.Y. IL-6 trans-signaling plays important protective roles in acute liver injury induced by acetaminophen in mice. J. Biochem. Mol. Toxicol. 2015, 29, 288–297. [Google Scholar] [CrossRef]
- Mekala, S.; Tulimilli, S.V.; Geesala, R.; Manupati, K.; Dhoke, N.R.; Das, A. Cellular crosstalk mediated by platelet-derived growth factor BB and transforming growth factor β during hepatic injury activates hepatic stellate cells. Can. J. Physiol. Pharmacol. 2018, 96, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Gampe, K.; Stefani, J.; Hammer, K.; Brendel, P.; Pötzsch, A.; Enikolopov, G.; Enjyoji, K.; Acker-Palmer, A.; Robson, S.C.; Zimmermann, H. NTPDase2 and Purinergic Signaling Control Progenitor Cell Proliferation in Neurogenic Niches of the Adult Mouse Brain. Stem Cells 2014. [Google Scholar] [CrossRef] [PubMed]
- Vandenbeuch, A.; Anderson, C.B.; Parnes, J.; Enjyoji, K.; Robson, S.C.; Finger, T.E.; Kinnamon, S.C. Role of the ectonucleotidase NTPDase2 in taste bud function. Proc. Natl. Acad. Sci. USA 2013, 110, 14789–14794. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.L.; Sullivan, S.L.; Lavoie, E.G.; Sevigny, J.; Finger, T.E. Nucleoside triphosphate diphosphohydrolase-2 is the ecto-ATPase of type I cells in taste buds. J. Comp. Neurol. 2006, 497, 1–12. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feldbrügge, L.; Splith, K.; Kämmerer, I.; Richter, S.; Riddermann, A.; Ortiz Galindo, S.A.; Krenzien, F.; Müller, T.; Csizmadia, E.; Pratschke, J.; et al. Ecto-Nucleotide Triphosphate Diphosphohydrolase-2 (NTPDase2) Deletion Increases Acetaminophen-Induced Hepatotoxicity. Int. J. Mol. Sci. 2020, 21, 5998. https://doi.org/10.3390/ijms21175998
Feldbrügge L, Splith K, Kämmerer I, Richter S, Riddermann A, Ortiz Galindo SA, Krenzien F, Müller T, Csizmadia E, Pratschke J, et al. Ecto-Nucleotide Triphosphate Diphosphohydrolase-2 (NTPDase2) Deletion Increases Acetaminophen-Induced Hepatotoxicity. International Journal of Molecular Sciences. 2020; 21(17):5998. https://doi.org/10.3390/ijms21175998
Chicago/Turabian StyleFeldbrügge, Linda, Katrin Splith, Ines Kämmerer, Sandra Richter, Anna Riddermann, Santiago Andres Ortiz Galindo, Felix Krenzien, Tobias Müller, Eva Csizmadia, Johann Pratschke, and et al. 2020. "Ecto-Nucleotide Triphosphate Diphosphohydrolase-2 (NTPDase2) Deletion Increases Acetaminophen-Induced Hepatotoxicity" International Journal of Molecular Sciences 21, no. 17: 5998. https://doi.org/10.3390/ijms21175998
APA StyleFeldbrügge, L., Splith, K., Kämmerer, I., Richter, S., Riddermann, A., Ortiz Galindo, S. A., Krenzien, F., Müller, T., Csizmadia, E., Pratschke, J., Robson, S. C., & Schmelzle, M. (2020). Ecto-Nucleotide Triphosphate Diphosphohydrolase-2 (NTPDase2) Deletion Increases Acetaminophen-Induced Hepatotoxicity. International Journal of Molecular Sciences, 21(17), 5998. https://doi.org/10.3390/ijms21175998