Microbiota-Associated Therapy for Non-Alcoholic Steatohepatitis-Induced Liver Cancer: A Review



Abstract
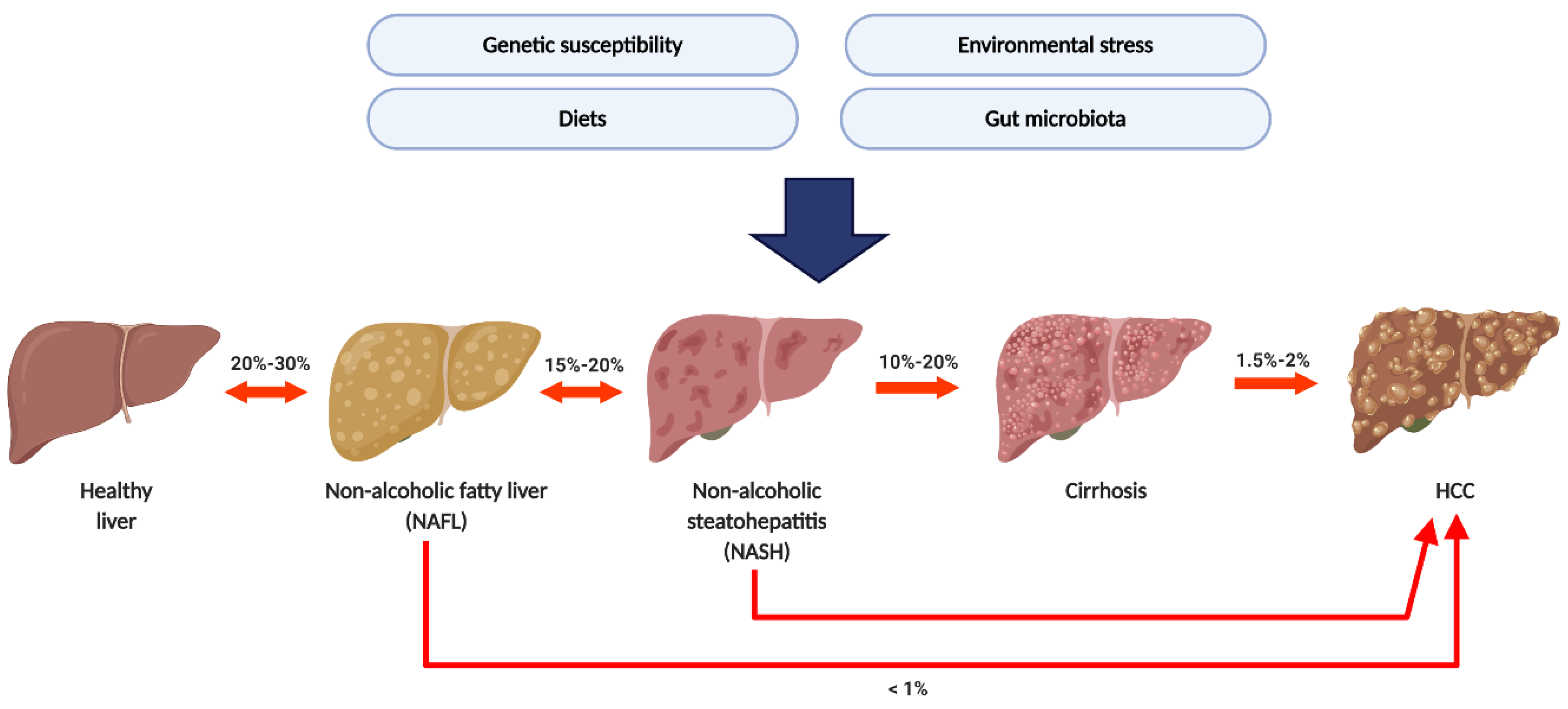
1. Introduction
2. From NAFL to NASH: The Gut Microbiota-Associated Mechanisms
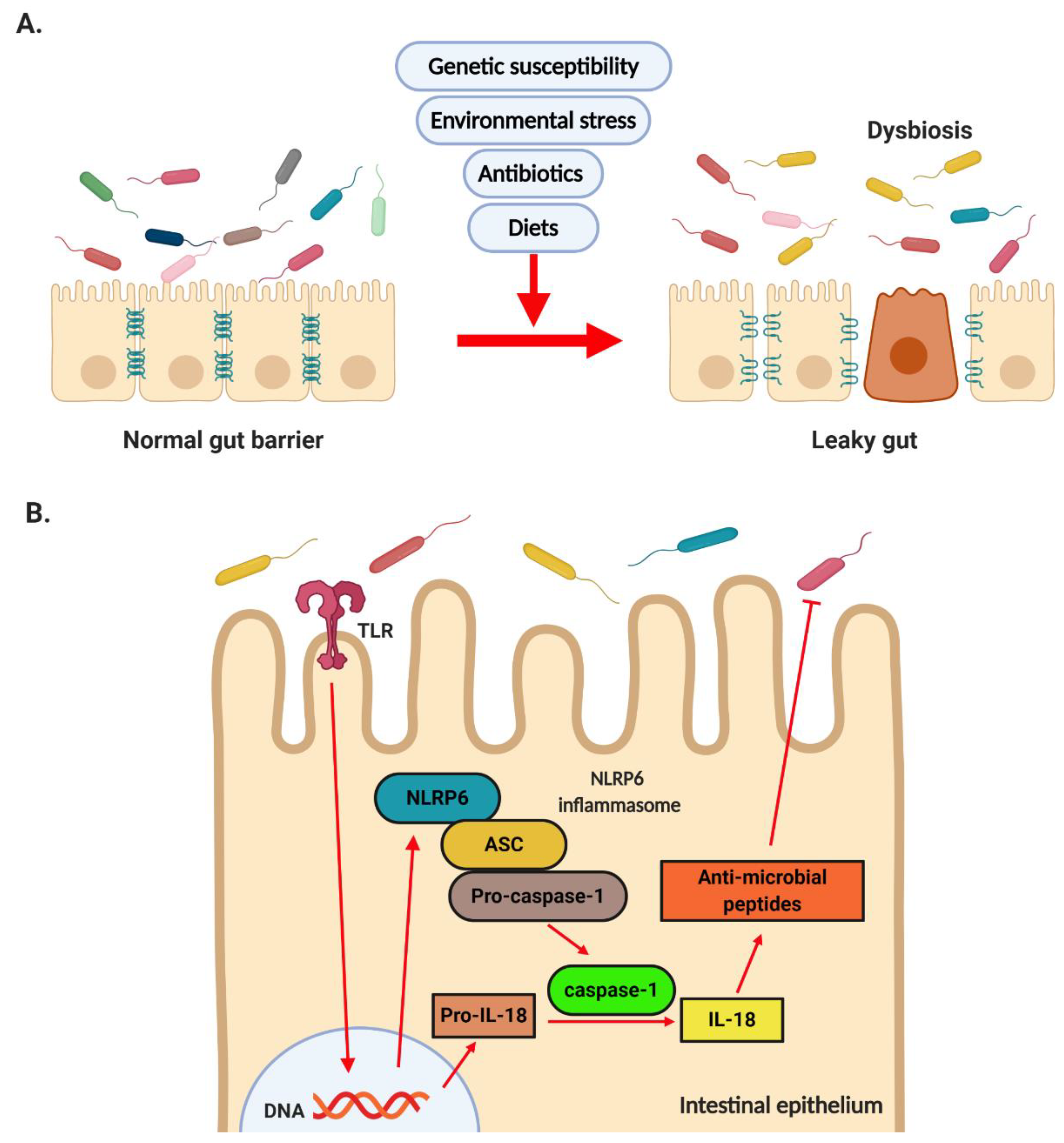
2.1. NASH and Dysbiosis
2.2. NASH and Leaky Gut
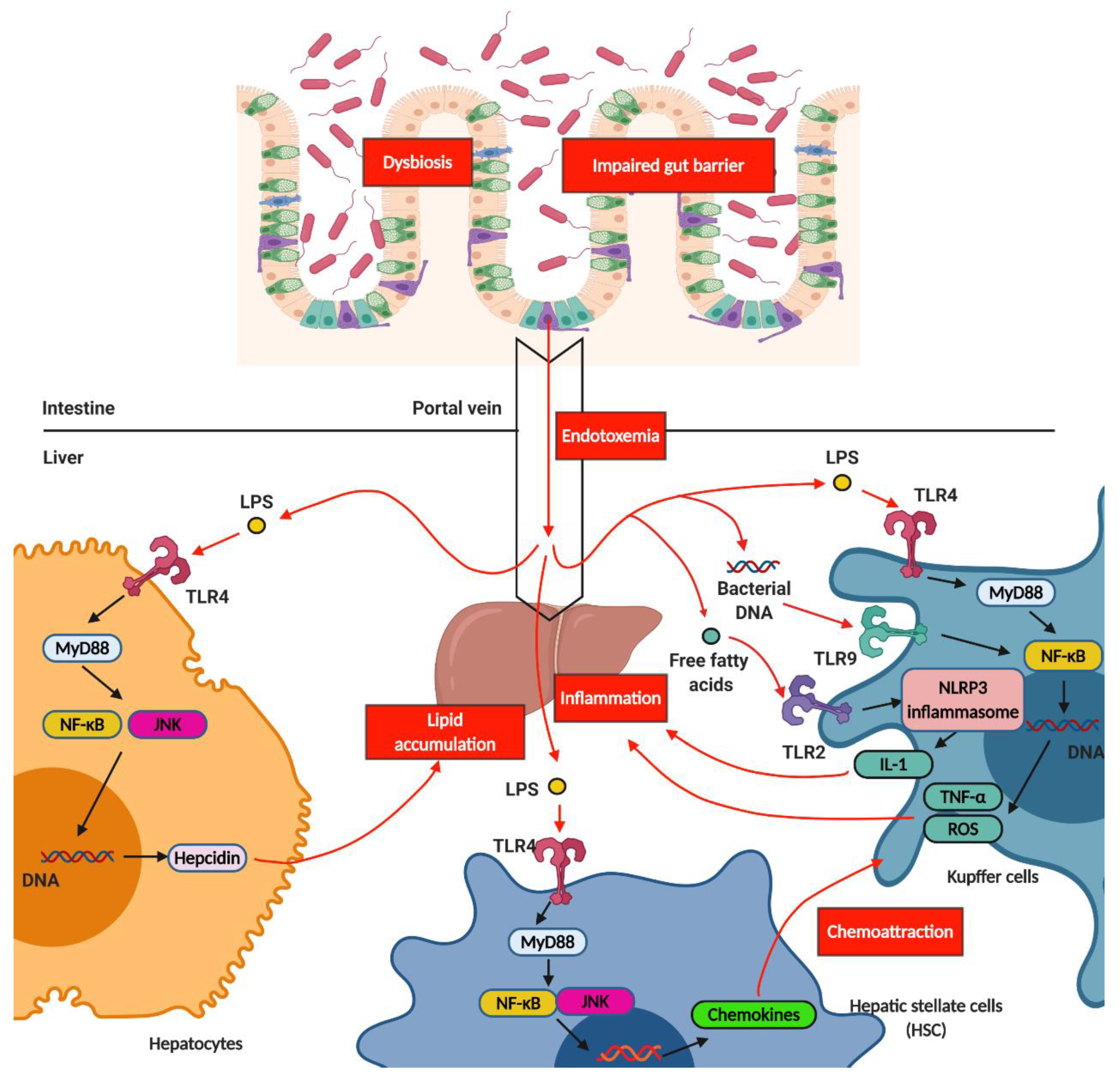
2.3. Gut Microbiota and Hepatic Inflammation
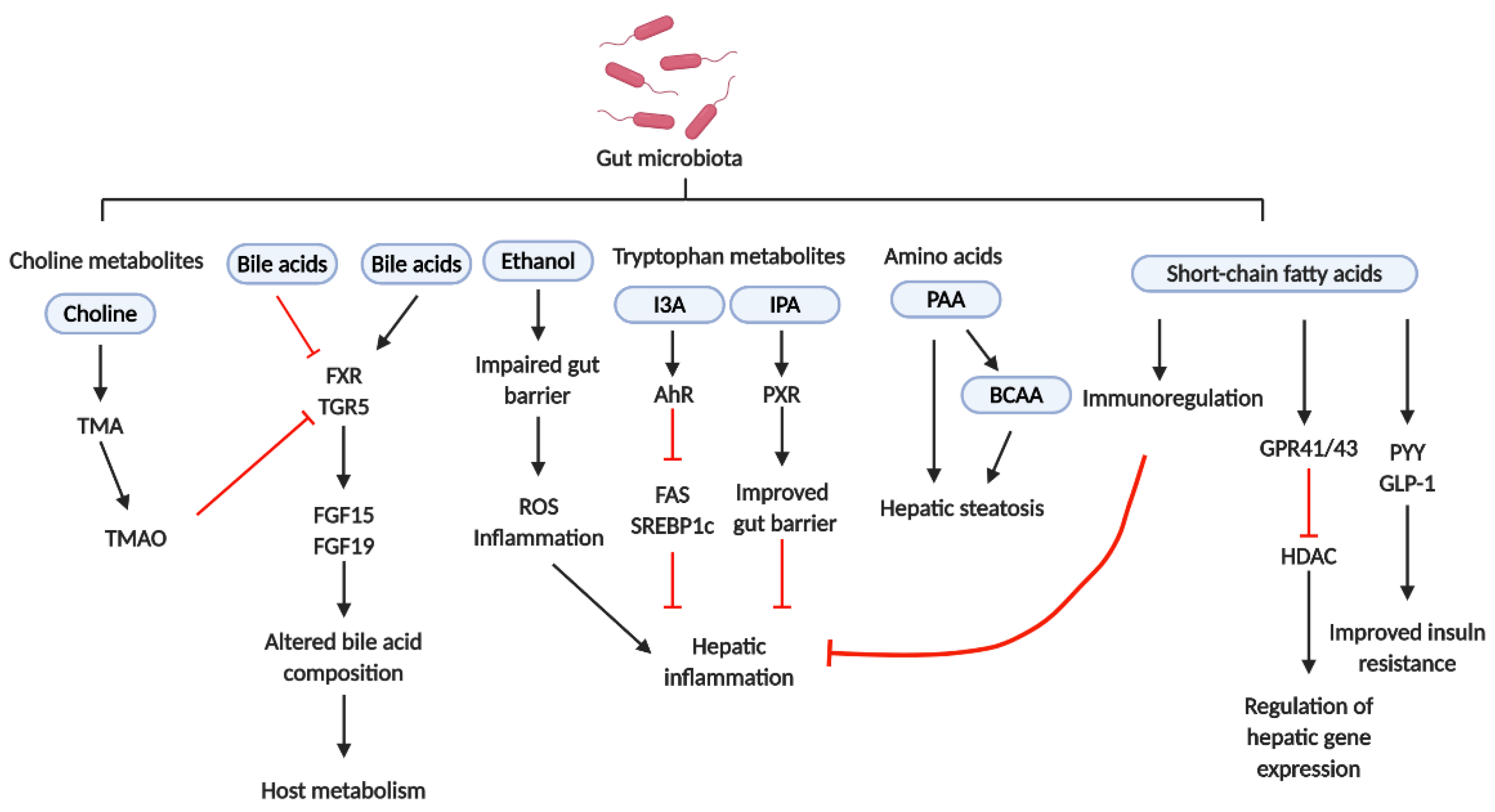
2.4. NASH and Gut Microbiota-Derived Metabolites
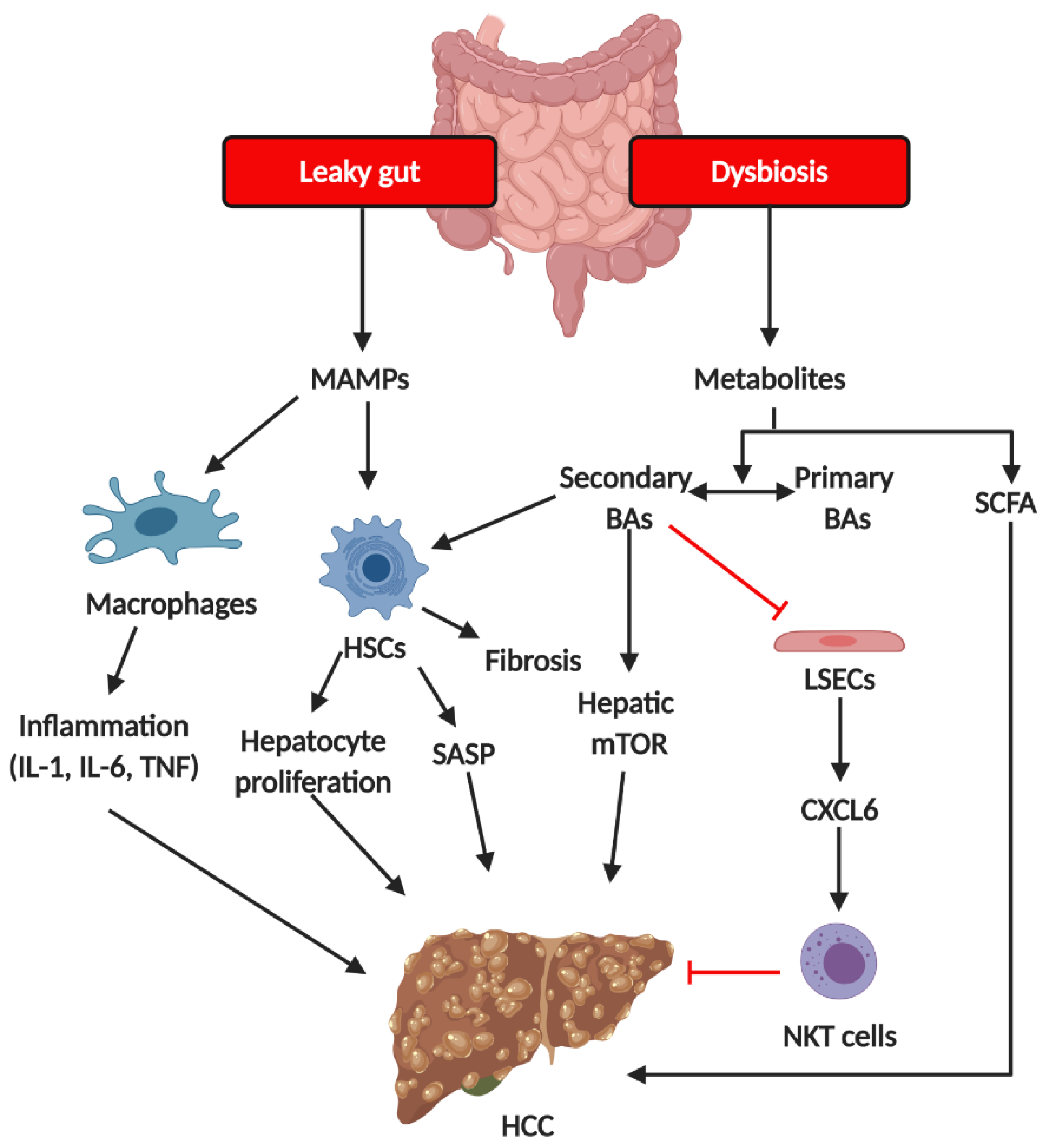
3. From NASH to HCC: The Gut Microbiota-Associated Mechanisms
4. Potential Therapeutic Strategies and Non-Invasive Diagnosis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HCC | hepatocellular carcinoma |
| NAFLD | non-alcoholic fatty liver disease |
| NASH | non-alcoholic steatohepatitis |
| HBV | hepatitis B viruses |
| HCV | hepatitis C viruses |
| SCFA | short chain fatty acid |
| ZO-1 | zonula occludens-1 |
| DSS | dextran sodium sulfate |
| IL10 | interleukin-10 |
| IL6 | interleukin-6 |
| PPAR | peroxisome proliferator-activated receptor |
| FMT | fecal microbial transplantation |
| HFDs | high-fat diets |
| NLRP6 | nucleotide-binding oligomerization domain -like receptor family pyrin domain containing 6 |
| ASC | apoptosis-associated speck-like protein containing a caspase recruitment domain |
| TLR4 | toll-like receptor 4 |
| HSCs | hepatic stellate cells |
| LSECs | liver sinusoidal endothelial cells |
| MyD88 | myeloid differentiation primary response 88 |
| TNF-α | tumor necrosis factor-α |
| ROS | reactive oxygen species |
| JNK | c-Jun N-terminal kinase |
| PAMPs | pathogen-associated molecular patterns |
| NEFAs | non-esterified fatty acids |
| S1P | sphingosine 1-phosphate |
| TRAIL | TNF-related apoptosis-inducing ligand |
| DAMP | damage-associated molecular pattern |
| GPR | G-protein coupled receptor |
| HDAC | histone deacetylase |
| CREB | cyclic adenosine monophosphate (cAMP) response element binding protein |
| BCAAs | branched-chain amino acids |
| AAAs | aromatic amino acids |
| ACADSB | acyl-CoA dehydrogenase short/branched chain |
| IA | indoleacrylic acid |
| IAA | indole-3-acetic acid |
| I3A | indole-3-aldehyde |
| IPA | indole-3-propionic acid |
| SREBP1c | sterol regulatory element-binding protein-1c |
| AhR | aryl-hydrocarbon receptor |
| PXR | pregnane X receptor |
| CA | cholic acid |
| CDCA | chenodeoxycholic acid |
| DCA | deoxycholic acid |
| LCA | lithocholic acid |
| FXR | farnesoid X receptor |
| TGR5 | takeda G-protein-coupled bile acid receptor 5 |
| OCA | obeticholic acid |
| FGF | fibroblast growth factor |
| VLDL | very low-density lipoprotein |
| TMA | trimethylamine |
| TMAO | trimethylamine-N-oxide |
| CXCL6 | chemokine ligand 6 |
References
- Sia, D.; Villanueva, A.; Friedman, S.L.; Llovet, J.M. Liver cancer cell of origin, molecular class, and effects on patient prognosis. Gastroenterology 2017, 152, 745–761. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.L. Global elimination of chronic hepatitis. N. Engl. J. Med. 2019, 380, 2041–2050. [Google Scholar] [CrossRef] [PubMed]
- Ghouri, Y.A.; Mian, I.; Rowe, J.H. Review of hepatocellular carcinoma: Epidemiology, etiology, and carcinogenesis. J. Carcinog. 2017, 16, 1. [Google Scholar]
- Sanyal, A.J. Past, present and future perspectives in nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.-G.; Kim, S.-U.; Wong, V.W.-S. New trends on obesity and NAFLD in Asia. J. Hepatol. 2017, 67, 862–873. [Google Scholar] [CrossRef]
- Estes, C.; Anstee, Q.M.; Arias-Loste, M.T.; Bantel, H.; Bellentani, S.; Caballeria, J.; Colombo, M.; Craxi, A.; Crespo, J.; Day, C.P.; et al. Modeling nafld disease burden in china, france, germany, italy, japan, spain, united kingdom, and united states for the period 2016–2030. J. Hepatol. 2018, 69, 896–904. [Google Scholar] [CrossRef]
- Ferolla, S.M.; Armiliato, G.N.; Couto, C.A.; Ferrari, T.C. The role of intestinal bacteria overgrowth in obesity-related nonalcoholic fatty liver disease. Nutrients 2014, 6, 5583–5599. [Google Scholar] [CrossRef]
- Rinella, M.; Charlton, M. The globalization of nonalcoholic fatty liver disease: Prevalence and impact on world health. Hepatology 2016, 64, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 2018, 67, 123–133. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, S.P.; Karlsen, T.H.; LaRusso, N.F. Cholangiocytes and the environment in primary sclerosing cholangitis: Where is the link? Gut 2017, 66, 1873–1877. [Google Scholar] [CrossRef] [PubMed]
- Brandl, K.; Kumar, V.; Eckmann, L. Gut-liver axis at the frontier of host-microbial interactions. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G413–G419. [Google Scholar] [CrossRef]
- Gupta, H.; Youn, G.S.; Shin, M.J.; Suk, K.T. Role of gut microbiota in hepatocarcinogenesis. Microorganisms 2019, 7, 121. [Google Scholar] [CrossRef]
- Giau, V.V.; Wu, S.Y.; Jamerlan, A.; An, S.S.A.; Kim, S.; Hulme, J. Gut microbiota and their neuroinflammatory implications in Alzheimer’s disease. Nutrients 2018, 10, 1765. [Google Scholar] [CrossRef]
- Nishida, A.; Inoue, R.; Inatomi, O.; Bamba, S.; Naito, Y.; Andoh, A. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin. J. Gastroenterol. 2018, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ponziani, F.R.; Zocco, M.A.; Cerrito, L.; Gasbarrini, A.; Pompili, M. Bacterial translocation in patients with liver cirrhosis: Physiology, clinical consequences, and practical implications. Expert Rev. Gastroenterol. Hepatol. 2018, 12, 641–656. [Google Scholar] [CrossRef]
- Carotti, S.; Guarino, M.P.L.; Vespasiani-Gentilucci, U.; Morini, S. Starring role of toll-like receptor-4 activation in the gut-liver axis. World J. Gastrointest. Pathophysiol. 2015, 6, 99. [Google Scholar] [CrossRef] [PubMed]
- Del Chierico, F.; Nobili, V.; Vernocchi, P.; Russo, A.; De Stefanis, C.; Gnani, D.; Furlanello, C.; Zandonà, A.; Paci, P.; Capuani, G.; et al. Gut microbiota profiling of pediatric nonalcoholic fatty liver disease and obese patients unveiled by an integrated meta-omics-based approach. Hepatology 2017, 65, 451–464. [Google Scholar] [CrossRef]
- Mouzaki, M.; Wang, A.Y.; Bandsma, R.; Comelli, E.M.; Arendt, B.M.; Zhang, L.; Fung, S.; Fischer, S.E.; McGilvray, I.G.; Allard, J.P. Bile acids and dysbiosis in non-alcoholic fatty liver disease. PLoS ONE 2016, 11, e0151829. [Google Scholar] [CrossRef] [PubMed]
- Boursier, J.; Mueller, O.; Barret, M.; Machado, M.; Fizanne, L.; Araujo-Perez, F.; Guy, C.D.; Seed, P.C.; Rawls, J.F.; David, L.A. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016, 63, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Shen, F.; Zheng, R.-D.; Sun, X.-Q.; Ding, W.-J.; Wang, X.-Y.; Fan, J.-G. Gut microbiota dysbiosis in patients with non-alcoholic fatty liver disease. Hepatobiliary Pancreat. Dis. Int. 2017, 16, 375–381. [Google Scholar] [CrossRef]
- Loomba, R.; Seguritan, V.; Li, W.; Long, T.; Klitgord, N.; Bhatt, A.; Dulai, P.S.; Caussy, C.; Bettencourt, R.; Highlander, S.K.; et al. Gut microbiome-based metagenomic signature for non-invasive detection of advanced fibrosis in human nonalcoholic fatty liver disease. Cell Metab. 2017, 25, 1054–1062.e5. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, H.E.; Teterina, A.; Comelli, E.M.; Taibi, A.; Arendt, B.M.; Fischer, S.E.; Lou, W.; Allard, J.P. Nonalcoholic fatty liver disease is associated with dysbiosis independent of body mass index and insulin resistance. Sci. Rep. 2018, 8, 1–12. [Google Scholar]
- Duarte, S.M.B.; Stefano, J.T.; Miele, L.; Ponziani, F.; Souza-Basqueira, M.; Okada, L.; de Costa, F.B.; Toda, K.; Mazo, D.; Sabino, E.; et al. Gut microbiome composition in lean patients with NASH is associated with liver damage independent of caloric intake: A prospective pilot study. Nutr. Metab. Cardiovasc. Dis. 2018, 28, 369–384. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Putignani, L.; Mosca, A.; Del Chierico, F.; Vernocchi, P.; Alisi, A.; Stronati, L.; Cucchiara, S.; Toscano, M.; Drago, L. Bifidobacteria and lactobacilli in the gut microbiome of children with non-alcoholic fatty liver disease: Which strains act as health players? Arch. Med Sci. AMS 2018, 14, 81. [Google Scholar] [CrossRef]
- Million, M.; Maraninchi, M.; Henry, M.; Armougom, F.; Richet, H.; Carrieri, P.; Valero, R.; Raccah, D.; Vialettes, B.; Raoult, D. Obesity-associated gut microbiota is enriched in Lactobacillus reuteri and depleted in Bifidobacterium animalis and Methanobrevibacter smithii. Int. J. Obes. 2012, 36, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Jumpertz, R.; Le, D.S.; Turnbaugh, P.J.; Trinidad, C.; Bogardus, C.; Gordon, J.I.; Krakoff, J. Energy-balance studies reveal associations between gut microbes, caloric load, and nutrient absorption in humans. Am. J. Clin. Nutr. 2011, 94, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Luther, J.; Garber, J.J.; Khalili, H.; Dave, M.; Bale, S.S.; Jindal, R.; Motola, D.L.; Luther, S.; Bohr, S.; Jeoung, S.W.; et al. Hepatic injury in nonalcoholic steatohepatitis contributes to altered intestinal permeability. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 222–232.e2. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, V.; Miele, L.; Principessa, L.; Ferretti, F.; Villa, M.P.; Negro, V.; Grieco, A.; Alisi, A.; Nobili, V. Intestinal permeability is increased in children with non-alcoholic fatty liver disease, and correlates with liver disease severity. Dig. Liver Dis. 2014, 46, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Miele, L.; Valenza, V.; La Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Masciana, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1877–1887. [Google Scholar] [CrossRef]
- Rohr, M.W.; Narasimhulu, C.A.; Rudeski-Rohr, T.A.; Parthasarathy, S. Negative effects of a high-fat diet on intestinal permeability: A review. Adv. Nutr. 2020, 11, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Lv, L.; Shi, D.; Ye, J.; Fang, D.; Guo, F.; Li, Y.; He, X.; Li, L. Protective effect of Akkermansia muciniphila against immune-mediated liver injury in a mouse model. Front. Microbiol. 2017, 8, 1804. [Google Scholar] [CrossRef] [PubMed]
- Lam, Y.Y.; Ha, C.W.; Campbell, C.R.; Mitchell, A.J.; Dinudom, A.; Oscarsson, J.; Cook, D.I.; Hunt, N.H.; Caterson, I.D.; Holmes, A.J.; et al. Increased gut permeability and microbiota change associate with mesenteric fat inflammation and metabolic dysfunction in diet-induced obese mice. PLoS ONE 2012, 7, e34233. [Google Scholar] [CrossRef] [PubMed]
- Ling, X.; Linglong, P.; Du Weixia, W.H. Protective effects of bifidobacterium on intestinal barrier function in LPS-induced enterocyte barrier injury of Caco-2 monolayers and in a rat NEC model. PLoS ONE 2016, 11, e0161635. [Google Scholar] [CrossRef] [PubMed]
- Lam, Y.Y.; Ha, C.W.; Hoffmann, J.M.; Oscarsson, J.; Dinudom, A.; Mather, T.J.; Cook, D.I.; Hunt, N.H.; Caterson, I.D.; Holmes, A.J.; et al. Effects of dietary fat profile on gut permeability and microbiota and their relationships with metabolic changes in mice. Obesity 2015, 23, 1429–1439. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; Zhang, B.; Li, S.; Fang, B.; Duan, W.; Zhang, J.; Song, J.; Wang, M. Vinegar extract ameliorates alcohol-induced liver damage associated with the modulation of gut microbiota in mice. Food Funct. 2020, 11, 2898–2909. [Google Scholar] [CrossRef] [PubMed]
- Din, A.U.; Hassan, A.; Zhu, Y.; Zhang, K.; Wang, Y.; Li, T.; Wang, Y.; Wang, G. Inhibitory effect of Bifidobacterium bifidum ATCC 29521 on colitis and its mechanism. J. Nutr. Biochem. 2020, 79, 108353. [Google Scholar] [CrossRef]
- Mouries, J.; Brescia, P.; Silvestri, A.; Spadoni, I.; Sorribas, M.; Wiest, R.; Mileti, E.; Galbiati, M.; Invernizzi, P.; Adorini, L.; et al. Microbiota-driven gut vascular barrier disruption is a prerequisite for non-alcoholic steatohepatitis development. J. Hepatol. 2019, 71, 1216–1228. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Wlodarska, M.; Thaiss, C.A.; Nowarski, R.; Henao-Mejia, J.; Zhang, J.-P.; Brown, E.M.; Frankel, G.; Levy, M.; Katz, M.N.; Philbrick, W.M.; et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell 2014, 156, 1045–1059. [Google Scholar] [CrossRef]
- Levy, M.; Thaiss, C.A.; Zeevi, D.; Dohnalova, L.; Zilberman-Schapira, G.; Mahdi, J.A.; David, E.; Savidor, A.; Korem, T.; Herzig, Y.; et al. Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell 2015, 163, 1428–1443. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-M.; Yu, R.; Zhang, L.-P.; Wen, S.-Y.; Wang, S.-J.; Zhang, X.-Y.; Xu, Q.; Kong, L.-D. Dietary fructose-induced gut dysbiosis promotes mouse hippocampal neuroinflammation: A benefit of short-chain fatty acids. Microbiome 2019, 7, 98. [Google Scholar] [CrossRef] [PubMed]
- Elinav, E.; Strowig, T.; Kau, A.L.; Henao-Mejia, J.; Thaiss, C.A.; Booth, C.J.; Peaper, D.R.; Bertin, J.; Eisenbarth, S.C.; Gordon, J.I.; et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 2011, 145, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Gäbele, E.; Dostert, K.; Hofmann, C.; Wiest, R.; Schölmerich, J.; Hellerbrand, C.; Obermeier, F. DSS induced colitis increases portal LPS levels and enhances hepatic inflammation and fibrogenesis in experimental NASH. J. Hepatol. 2011, 55, 1391–1399. [Google Scholar] [CrossRef] [PubMed]
- Rahman, K.; Desai, C.; Iyer, S.S.; Thorn, N.E.; Kumar, P.; Liu, Y.; Smith, T.; Neish, A.S.; Li, H.; Tan, S.; et al. Loss of junctional adhesion molecule a promotes severe steatohepatitis in mice on a diet high in saturated fat, fructose, and cholesterol. Gastroenterology 2016, 151, 733–746.e12. [Google Scholar] [CrossRef]
- Kolodziejczyk, A.A.; Zheng, D.; Shibolet, O.; Elinav, E. The role of the microbiome in NAFLD and NASH. EMBO Mol. Med. 2019, 11, e9302. [Google Scholar] [CrossRef]
- Verdam, F.J.; Rensen, S.S.; Driessen, A.; Greve, J.W.; Buurman, W.A. Novel evidence for chronic exposure to endotoxin in human nonalcoholic steatohepatitis. J. Clin. Gastroenterol. 2011, 45, 149–152. [Google Scholar] [CrossRef]
- Miura, K.; Kodama, Y.; Inokuchi, S.; Schnabl, B.; Aoyama, T.; Ohnishi, H.; Olefsky, J.M.; Brenner, D.A.; Seki, E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1β in mice. Gastroenterology 2010, 139, 323–334. [Google Scholar] [CrossRef]
- Szabo, G.; Petrasek, J. Inflammasome activation and function in liver disease. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 387. [Google Scholar] [CrossRef] [PubMed]
- Wree, A.; McGeough, M.D.; Peña, C.A.; Schlattjan, M.; Li, H.; Inzaugarat, M.E.; Messer, K.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation is required for fibrosis development in NAFLD. J. Mol. Med. 2014, 92, 1069–1082. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Cassader, M.; Paschetta, E.; Gambino, R. Bioactive lipid species and metabolic pathways in progression and resolution of nonalcoholic steatohepatitis. Gastroenterology 2018, 155, 282–302.e8. [Google Scholar] [CrossRef] [PubMed]
- Yue, F.; Xia, K.; Wei, L.; Xing, L.; Wu, S.; Shi, Y.; Lam, S.M.; Shui, G.; Xiang, X.; Russell, R.; et al. Effects of constant light exposure on sphingolipidomics and progression of NASH in high-fat-fed rats. J. Gastroenterol. Hepatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Cassader, M.; Gambino, R. Non-alcoholic steatohepatitis: Emerging molecular targets and therapeutic strategies. Nat. Rev. Drug Discov. 2016, 15, 249. [Google Scholar] [CrossRef]
- Kaufman, R.J. Orchestrating the unfolded protein response in health and disease. J. Clin. Investig. 2002, 110, 1389–1398. [Google Scholar] [CrossRef]
- Hirsova, P.; Ibrabim, S.H.; Gores, G.J.; Malhi, H. Lipotoxic lethal and sublethal stress signaling in hepatocytes: Relevance to NASH pathogenesis. J. Lipid Res. 2016, 57, 1758–1770. [Google Scholar] [CrossRef]
- Ibrahim, S.H.; Hirsova, P.; Gores, G.J. Non-alcoholic steatohepatitis pathogenesis: Sublethal hepatocyte injury as a driver of liver inflammation. Gut 2018, 67, 963–972. [Google Scholar] [CrossRef]
- Canfora, E.E.; Meex, R.C.; Venema, K.; Blaak, E.E. Gut microbial metabolites in obesity, NAFLD and T2DM. Nat. Rev. Endocrinol. 2019, 15, 261–273. [Google Scholar] [CrossRef]
- Chambers, E.S.; Viardot, A.; Psichas, A.; Morrison, D.J.; Murphy, K.G.; Zac-Varghese, S.E.; MacDougall, K.; Preston, T.; Tedford, C.; Finlayson, G.S.; et al. Effects of targeted delivery of propionate to the human colon on appetite regulation, body weight maintenance and adiposity in overweight adults. Gut 2015, 64, 1744–1754. [Google Scholar] [CrossRef]
- Zhou, D.; Pan, Q.; Liu, X.L.; Yang, R.X.; Chen, Y.W.; Liu, C.; Fan, J.G. Clostridium butyricum B1 alleviates high-fat diet-induced steatohepatitis in mice via enterohepatic immunoregulation. J. Gastroenterol. Hepatol. 2017, 32, 1640–1648. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.J.; Goldsworthy, S.M.; Barnes, A.A.; Eilert, M.M.; Tcheang, L.; Daniels, D.; Muir, A.I.; Wigglesworth, M.J.; Kinghorn, I.; Fraser, N.J. The Orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J. Biol. Chem. 2003, 278, 11312–11319. [Google Scholar] [CrossRef]
- Le Poul, E.; Loison, C.; Struyf, S.; Springael, J.-Y.; Lannoy, V.; Decobecq, M.-E.; Brezillon, S.; Dupriez, V.; Vassart, G.; Van Damme, J.; et al. Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J. Biol. Chem. 2003, 278, 25481–25489. [Google Scholar] [CrossRef] [PubMed]
- Li, B.-Y.; Xu, X.-Y.; Gan, R.-Y.; Sun, Q.-C.; Meng, J.-M.; Shang, A.; Mao, Q.-Q.; Li, H.-B. Targeting Gut Microbiota for the Prevention and Management of Diabetes Mellitus by Dietary Natural Products. Foods 2019, 8, 440. [Google Scholar] [CrossRef] [PubMed]
- Schilderink, R.; Verseijden, C.; de Jonge, W.J. Dietary inhibitors of histone deacetylases in intestinal immunity and homeostasis. Front. Immunol. 2013, 4, 226. [Google Scholar] [CrossRef]
- Iida, A.; Kuranuki, S.; Yamamoto, R.; Uchida, M.; Ohta, M.; Ichimura, M.; Tsuneyama, K.; Masaki, T.; Seike, M.; Nakamura, T. Analysis of amino acid profiles of blood over time and biomarkers associated with non-alcoholic steatohepatitis in STAM mice. Exp. Anim. 2019, 68, 417–428. [Google Scholar] [CrossRef]
- Hoyles, L.; Fernandez-Real, J.-M.; Federici, M.; Serino, M.; Abbott, J.; Charpentier, J.; Heymes, C.; Luque, J.L.; Anthony, E.; Barton, R.H.; et al. Molecular phenomics and metagenomics of hepatic steatosis in non-diabetic obese women. Nat. Med. 2018, 24, 1070–1080. [Google Scholar] [CrossRef]
- Wang, T.J.; Larson, M.G.; Vasan, R.S.; Cheng, S.; Rhee, E.P.; McCabe, E.; Lewis, G.D.; Fox, C.S.; Jacques, P.F.; Fernandez, C. Metabolite profiles and the risk of developing diabetes. Nat. Med. 2011, 17, 448. [Google Scholar] [CrossRef]
- Menni, C.; Fauman, E.; Erte, I.; Perry, J.R.; Kastenmüller, G.; Shin, S.-Y.; Petersen, A.-K.; Hyde, C.; Psatha, M.; Ward, K.J.; et al. Biomarkers for type 2 diabetes and impaired fasting glucose using a nontargeted metabolomics approach. Diabetes 2013, 62, 4270–4276. [Google Scholar] [CrossRef]
- Pedersen, H.K.; Gudmundsdottir, V.; Nielsen, H.B.; Hyotylainen, T.; Nielsen, T.; Jensen, B.A.; Forslund, K.; Hildebrand, F.; Prifti, E.; Falony, G.; et al. Human gut microbes impact host serum metabolome and insulin sensitivity. Nature 2016, 535, 376–381. [Google Scholar] [CrossRef]
- Tanaka, H.; Fukahori, S.; Baba, S.; Ueno, T.; Sivakumar, R.; Yagi, M.; Asagiri, K.; Ishii, S.; Tanaka, Y. Branched-Chain Amino Acid–Rich Supplements Containing Microelements Have Antioxidant Effects on Nonalcoholic Steatohepatitis in Mice. J. Parenter. Enter. Nutr. 2016, 40, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Honda, T.; Ishigami, M.; Luo, F.; Lingyun, M.; Ishizu, Y.; Kuzuya, T.; Hayashi, K.; Nakano, I.; Ishikawa, T.; Feng, G.-G.; et al. Branched-chain amino acids alleviate hepatic steatosis and liver injury in choline-deficient high-fat diet induced NASH mice. Metabolism 2017, 69, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zhao, S.; Yan, W.; Xia, Y.; Chen, X.; Wang, W.; Zhang, J.; Gao, C.; Peng, C.; Yan, F.; et al. Branched chain amino acids cause liver injury in obese/diabetic mice by promoting adipocyte lipolysis and inhibiting hepatic autophagy. EBioMedicine 2016, 13, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.; Ding, Y.; Saedi, N.; Choi, M.; Sridharan, G.V.; Sherr, D.H.; Yarmush, M.L.; Alaniz, R.C.; Jayaraman, A.; Lee, K. Gut microbiota-derived tryptophan metabolites modulate inflammatory response in hepatocytes and macrophages. Cell Rep. 2018, 23, 1099–1111. [Google Scholar] [CrossRef] [PubMed]
- Konopelski, P.; Konop, M.; Gawrys-Kopczynska, M.; Podsadni, P.; Szczepanska, A.; Ufnal, M. Indole-3-propionic acid, a tryptophan-derived bacterial metabolite, reduces weight gain in rats. Nutrients 2019, 11, 591. [Google Scholar] [CrossRef]
- Jennis, M.; Cavanaugh, C.; Leo, G.; Mabus, J.; Lenhard, J.; Hornby, P. Microbiota-derived tryptophan indoles increase after gastric bypass surgery and reduce intestinal permeability in vitro and in vivo. Neurogastroenterol. Motil. 2018, 30, e13178. [Google Scholar] [CrossRef]
- Venkatesh, M.; Mukherjee, S.; Wang, H.; Li, H.; Sun, K.; Benechet, A.P.; Qiu, Z.; Maher, L.; Redinbo, M.R.; Phillips, R.S.; et al. Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and Toll-like receptor 4. Immunity 2014, 41, 296–310. [Google Scholar] [CrossRef]
- Cope, K.; Risby, T.; Diehl, A.M. Increased gastrointestinal ethanol production in obese mice: Implications for fatty liver disease pathogenesis. Gastroenterology 2000, 119, 1340–1347. [Google Scholar] [CrossRef]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef]
- Fuster, D.; Samet, J.H. Alcohol use in patients with chronic liver disease. N. Engl. J. Med. 2018, 379, 1251–1261. [Google Scholar] [CrossRef]
- Yuan, J.; Chen, C.; Cui, J.; Lu, J.; Yan, C.; Wei, X.; Zhao, X.; Li, N.; Li, S.; Xue, G. Fatty liver disease caused by high-alcohol-producing Klebsiella pneumoniae. Cell Metab. 2019, 30, 675–688. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.S.; Baker, R.D.; Liu, W.; Nowak, N.J.; Zhu, L. Role of alcohol metabolism in non-alcoholic steatohepatitis. PLoS ONE 2010, 5, e9570. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, U.C.; Goel, A.; Quigley, E.M. Gut microbiota abnormalities, small intestinal bacterial overgrowth, and non-alcoholic fatty liver disease: An emerging paradigm. Indian J. Gastroenterol. 2020, 39, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Sipe, L.M.; Chaib, M.; Pingili, A.K.; Pierre, J.F.; Makowski, L. Microbiome, bile acids, and obesity: How microbially modified metabolites shape anti-tumor immunity. Immunol. Rev. 2020, 295, 220–239. [Google Scholar] [CrossRef]
- Gérard, P. Metabolism of cholesterol and bile acids by the gut microbiota. Pathogens 2014, 3, 14–24. [Google Scholar] [CrossRef]
- Sayin, S.I.; Wahlström, A.; Felin, J.; Jäntti, S.; Marschall, H.-U.; Bamberg, K.; Angelin, B.; Hyötyläinen, T.; Orešič, M.; Bäckhed, F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013, 17, 225–235. [Google Scholar] [CrossRef]
- Adams, L.A.; Wang, Z.; Liddle, C.; Melton, P.E.; Ariff, A.; Chandraratna, H.; Tan, J.; Ching, H.; Coulter, S.; de Boer, B.; et al. Bile acids associate with specific gut microbiota, low-level alcohol consumption and liver fibrosis in patients with non-alcoholic fatty liver disease. Liver Int. 2020, 40, 1356–1365. [Google Scholar] [CrossRef]
- Jiang, C.; Xie, C.; Li, F.; Zhang, L.; Nichols, R.G.; Krausz, K.W.; Cai, J.; Qi, Y.; Fang, Z.-Z.; Takahashi, S.; et al. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J. Clin. Investig. 2015, 125, 386–402. [Google Scholar] [CrossRef]
- Jahn, D.; Rau, M.; Hermanns, H.M.; Geier, A. Mechanisms of enterohepatic fibroblast growth factor 15/19 signaling in health and disease. Cytokine Growth Factor Rev. 2015, 26, 625–635. [Google Scholar] [CrossRef]
- De Wit, N.; Derrien, M.; Bosch-Vermeulen, H.; Oosterink, E.; Keshtkar, S.; Duval, C.; Bosch, J.; Kleerebezem, M.; Müller, M.; van der Meer, R. Saturated fat stimulates obesity and hepatic steatosis and affects gut microbiota composition by an enhanced overflow of dietary fat to the distal intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G589–G599. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Mason, A.; Luketic, V.; Lindor, K.; Gordon, S.C.; Mayo, M.; Kowdley, K.V.; Vincent, C.; Bodhenheimer, H.C., Jr.; Parés, A.; et al. Efficacy of obeticholic acid in patients with primary biliary cirrhosis and inadequate response to ursodeoxycholic acid. Gastroenterology 2015, 148, 751–761.e8. [Google Scholar] [CrossRef] [PubMed]
- Verbeke, L.; Mannaerts, I.; Schierwagen, R.; Govaere, O.; Klein, S.; Elst, I.; Windmolders, P.; Farre, R.; Wenes, M.; Mazzone, M.; et al. FXR agonist obeticholic acid reduces hepatic inflammation and fibrosis in a rat model of toxic cirrhosis. Sci. Rep. 2016, 6, 1–12. [Google Scholar]
- Devkota, S.; Wang, Y.; Musch, M.W.; Leone, V.; Fehlner-Peach, H.; Nadimpalli, A.; Antonopoulos, D.A.; Jabri, B.; Chang, E.B. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/− mice. Nature 2012, 487, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Islam, K.S.; Fukiya, S.; Hagio, M.; Fujii, N.; Ishizuka, S.; Ooka, T.; Ogura, Y.; Hayashi, T.; Yokota, A. Bile acid is a host factor that regulates the composition of the cecal microbiota in rats. Gastroenterology 2011, 141, 1773–1781. [Google Scholar] [CrossRef] [PubMed]
- Katsuma, S.; Hirasawa, A.; Tsujimoto, G. Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem. Biophys. Res. Commun. 2005, 329, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Alarcon, C.; Wicksteed, B.; Rhodes, C. Exendin 4 controls insulin production in rat islet beta cells predominantly by potentiation of glucose-stimulated proinsulin biosynthesis at the translational level. Diabetologia 2006, 49, 2920–2929. [Google Scholar] [CrossRef]
- Smallwood, T.; Allayee, H.; Bennett, B.J. Choline metabolites: Gene by diet interactions. Curr. Opin. Lipidol. 2016, 27, 33. [Google Scholar] [CrossRef]
- Romano, K.A.; Vivas, E.I.; Amador-Noguez, D.; Rey, F.E. Intestinal microbiota composition modulates choline bioavailability from diet and accumulation of the proatherogenic metabolite trimethylamine-N-oxide. MBio 2015, 6. [Google Scholar] [CrossRef]
- Wu, W.-K.; Chen, C.-C.; Liu, P.-Y.; Panyod, S.; Liao, B.-Y.; Chen, P.-C.; Kao, H.-L.; Kuo, H.-C.; Kuo, C.-H.; Chiu, T.H.; et al. Identification of TMAO-producer phenotype and host–diet–gut dysbiosis by carnitine challenge test in human and germ-free mice. Gut 2019, 68, 1439–1449. [Google Scholar] [CrossRef]
- Chen, Y.-M.; Liu, Y.; Zhou, R.-F.; Chen, X.-L.; Wang, C.; Tan, X.-Y.; Wang, L.-J.; Zheng, R.-D.; Zhang, H.-W.; Ling, W.-H.; et al. Associations of gut-flora-dependent metabolite trimethylamine-N-oxide, betaine and choline with non-alcoholic fatty liver disease in adults. Sci. Rep. 2016, 6, 1–9. [Google Scholar]
- Tan, X.; Liu, Y.; Long, J.; Chen, S.; Liao, G.; Wu, S.; Li, C.; Wang, L.; Ling, W.; Zhu, H. Trimethylamine N-Oxide Aggravates Liver Steatosis through Modulation of Bile Acid Metabolism and Inhibition of Farnesoid X Receptor Signaling in Nonalcoholic Fatty Liver Disease. Mol. Nutr. Food Res. 2019, 63, 1900257. [Google Scholar]
- Grąt, M.; Wronka, K.; Krasnodębski, M.; Lewandowski, Z.; Kosińska, I.; Grąt, K.; Stypułkowski, J.; Rejowski, S.; Wasilewicz, M.; Gałęcka, M. Profile of gut microbiota associated with the presence of hepatocellular cancer in patients with liver cirrhosis. In Transplantation Proceedings; Elsevier: Amsterdam, The Netherlands, 2016; pp. 1687–1691. [Google Scholar]
- Ni, J.; Huang, R.; Zhou, H.; Xu, X.; Li, Y.; Cao, P.; Zhong, K.; Ge, M.; Chen, X.; Hou, B.; et al. Analysis of the relationship between the degree of dysbiosis in gut microbiota and prognosis at different stages of primary hepatocellular carcinoma. Front. Microbiol. 2019, 10, 1458. [Google Scholar] [CrossRef]
- Ponziani, F.R.; Bhoori, S.; Castelli, C.; Putignani, L.; Rivoltini, L.; Del Chierico, F.; Sanguinetti, M.; Morelli, D.; Sterbini, F.P.; Petito, V.; et al. Hepatocellular carcinoma is associated with gut microbiota profile and inflammation in nonalcoholic fatty liver disease. Hepatology 2019, 69, 107–120. [Google Scholar] [PubMed]
- Dapito, D.H.; Mencin, A.; Gwak, G.-Y.; Pradere, J.-P.; Jang, M.-K.; Mederacke, I.; Caviglia, J.M.; Khiabanian, H.; Adeyemi, A.; Bataller, R.; et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell 2012, 21, 504–516. [Google Scholar] [PubMed]
- Schwabe, R.F.; Greten, T.F. Gut microbiome in HCC–Mechanisms, diagnosis and therapy. J. Hepatol. 2020, 72, 230–238. [Google Scholar] [CrossRef]
- Ma, C.; Han, M.; Heinrich, B.; Fu, Q.; Zhang, Q.; Sandhu, M.; Agdashian, D.; Terabe, M.; Berzofsky, J.A.; Fako, V. Gut microbiome–mediated bile acid metabolism regulates liver cancer via NKT cells. Science 2018, 360, eaan5931. [Google Scholar] [CrossRef]
- Yamada, S.; Takashina, Y.; Watanabe, M.; Nagamine, R.; Saito, Y.; Kamada, N.; Saito, H. Bile acid metabolism regulated by the gut microbiota promotes non-alcoholic steatohepatitis-associated hepatocellular carcinoma in mice. Oncotarget 2018, 9, 9925. [Google Scholar]
- Francescone, R.; Hou, V.; Grivennikov, S.I. Microbiome, inflammation and cancer. Cancer J. 2014, 20, 181. [Google Scholar] [CrossRef]
- Gerber, L.; Otgonsuren, M.; Mishra, A.; Escheik, C.; Birerdinc, A.; Stepanova, M.; Younossi, Z. Non-alcoholic fatty liver disease (NAFLD) is associated with low level of physical activity: A population-based study. Aliment. Pharmacol. Ther. 2012, 36, 772–781. [Google Scholar] [CrossRef]
- Promrat, K.; Kleiner, D.E.; Niemeier, H.M.; Jackvony, E.; Kearns, M.; Wands, J.R.; Fava, J.L.; Wing, R.R. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology 2010, 51, 121–129. [Google Scholar]
- Vilar-Gomez, E.; Martinez-Perez, Y.; Calzadilla-Bertot, L.; Torres-Gonzalez, A.; Gra-Oramas, B.; Gonzalez-Fabian, L.; Friedman, S.L.; Diago, M.; Romero-Gomez, M. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology 2015, 149, 367–378.e5. [Google Scholar] [CrossRef] [PubMed]
- Janssen, A.W.; Houben, T.; Katiraei, S.; Dijk, W.; Boutens, L.; Van Der Bolt, N.; Wang, Z.; Brown, J.M.; Hazen, S.L.; Mandard, S.; et al. Modulation of the gut microbiota impacts nonalcoholic fatty liver disease: A potential role for bile acids. J. Lipid Res. 2017, 58, 1399–1416. [Google Scholar] [CrossRef] [PubMed]
- Safari, Z.; Gérard, P. The links between the gut microbiome and non-alcoholic fatty liver disease (NAFLD). Cell. Mol. Life Sci. 2019, 76, 1541–1558. [Google Scholar] [PubMed]
- Mahana, D.; Trent, C.M.; Kurtz, Z.D.; Bokulich, N.A.; Battaglia, T.; Chung, J.; Müller, C.L.; Li, H.; Bonneau, R.A.; Blaser, M.J. Antibiotic perturbation of the murine gut microbiome enhances the adiposity, insulin resistance, and liver disease associated with high-fat diet. Genome Med. 2016, 8, 1–20. [Google Scholar]
- McBurney, M.I.; Davis, C.; Fraser, C.M.; Schneeman, B.O.; Huttenhower, C.; Verbeke, K.; Walter, J.; Latulippe, M.E. Establishing what constitutes a healthy human gut microbiome: State of the science, regulatory considerations, and future directions. J. Nutr. 2019, 149, 1882–1895. [Google Scholar]
- Pachikian, B.D.; Essaghir, A.; Demoulin, J.B.; Catry, E.; Neyrinck, A.M.; Dewulf, E.M.; Sohet, F.M.; Portois, L.; Clerbaux, L.A.; Carpentier, Y.A.; et al. Prebiotic approach alleviates hepatic steatosis: Implication of fatty acid oxidative and cholesterol synthesis pathways. Mol. Nutr. Food Res. 2013, 57, 347–359. [Google Scholar]
- Daubioul, C.; Horsmans, Y.; Lambert, P.; Danse, E.; Delzenne, N.M. Effects of oligofructose on glucose and lipid metabolism in patients with nonalcoholic steatohepatitis: Results of a pilot study. Eur. J. Clin. Nutr. 2005, 59, 723–726. [Google Scholar]
- Kumar, M.; Verma, V.; Nagpal, R.; Kumar, A.; Gautam, S.K.; Behare, P.V.; Grover, C.R.; Aggarwal, P.K. Effect of probiotic fermented milk and chlorophyllin on gene expressions and genotoxicity during AFB1-induced hepatocellular carcinoma. Gene 2011, 490, 54–59. [Google Scholar] [CrossRef]
- Monem, S.M.A. Probiotic therapy in patients with nonalcoholic steatohepatitis in Zagazig University hospitals. Euroasian J. Hepatogastroenterol. 2017, 7, 101. [Google Scholar] [CrossRef]
- Meroni, M.; Longo, M.; Dongiovanni, P. The role of probiotics in nonalcoholic fatty liver disease: A new insight into therapeutic strategies. Nutrients 2019, 11, 2642. [Google Scholar]
- Gupta, A.; Khanna, S. Fecal microbiota transplantation. JAMA 2017, 318, 102. [Google Scholar] [CrossRef] [PubMed]
- Khoruts, A.; Dicksved, J.; Jansson, J.K.; Sadowsky, M.J. Changes in the composition of the human fecal microbiome after bacteriotherapy for recurrent Clostridium difficile-associated diarrhea. J. Clin. Gastroenterol. 2010, 44, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Van Nood, E.; Vrieze, A.; Nieuwdorp, M.; Fuentes, S.; Zoetendal, E.G.; de Vos, W.M.; Visser, C.E.; Kuijper, E.J.; Bartelsman, J.F.; Tijssen, J.G.; et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. N. Engl. J. Med. 2013, 368, 407–415. [Google Scholar] [PubMed]
- Surawicz, C.M.; Brandt, L.J.; Binion, D.G.; Ananthakrishnan, A.N.; Curry, S.R.; Gilligan, P.H.; McFarland, L.V.; Mellow, M.; Zuckerbraun, B.S. Guidelines for Diagnosis, Treatment, and Prevention of Clostridium difficileInfections. Am. J. Gastroenterol. 2013, 108, 478–498. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Pan, Q.; Shen, F.; Cao, H.-X.; Ding, W.-J.; Chen, Y.-W.; Fan, J.-G. Total fecal microbiota transplantation alleviates high-fat diet-induced steatohepatitis in mice via beneficial regulation of gut microbiota. Sci. Rep. 2017, 7, 1–11. [Google Scholar]
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojärvi, J.; Kootte, R.S.; Bartelsman, J.F.; Dallinga–Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012, 143, 913–916.e7. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Salzman, N.H.; Acharya, C.; Sterling, R.K.; White, M.B.; Gavis, E.A.; Fagan, A.; Hayward, M.; Holtz, M.L.; Matherly, S.; et al. Fecal Microbial Transplant Capsules Are Safe in Hepatic Encephalopathy: A Phase 1, Randomized, Placebo-Controlled Trial. Hepatology 2019, 70, 1690–1703. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Kakiyama, G.; Savidge, T.; Takei, H.; Kassam, Z.A.; Fagan, A.; Gavis, E.A.; Pandak, W.M.; Nittono, H.; Hylemon, P.B.; et al. Antibiotic-associated disruption of microbiota composition and function in cirrhosis is restored by fecal transplant. Hepatology 2018, 68, 1549–1558. [Google Scholar] [CrossRef]
- Leylabadlo, H.E.; Ghotaslou, R.; Kafil, H.S.; Feizabadi, M.M.; Moaddab, S.Y.; Farajnia, S.; Sheykhsaran, E.; Sanaie, S.; Shanehbandi, D.; Baghi, H.B. Non-alcoholic fatty liver diseases: From role of gut microbiota to microbial-based therapies. Eur. J. Clin. Microbiol. Infect. Dis. 2020, 39, 613–627. [Google Scholar] [CrossRef]
- Oh, T.G.; Kim, S.M.; Caussy, C.; Fu, T.; Guo, J.; Bassirian, S.; Singh, S.; Madamba, E.V.; Bettencourt, R.; Richards, L.; et al. A Universal Gut-Microbiome-Derived Signature Predicts Cirrhosis. Cell Metab. 2020. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolites | Effects | References |
| Acetate | HDAC inhibition. | [65] |
| Propionate | Induces PYY and GLP-1 release. HDAC inhibition. | [60,65] |
| Butyrate | Promotes Th2, Th22, or Treg cell differentiation, further preventing hepatic inflammation. HDAC inhibition. | [61,65] |
| PAA | Induces steatosis. | [67] |
| BCAA | Alleviates hepatic steatosis and liver injury by suppressing FAS gene expression and protein levels. Suppresses the progression of NASH by reducing oxidative stress. | [71,72] |
| Exacerbates hepatic oxidative stress, increases hepatic apoptosis. | [73] | |
| Tryptamine | Reduces production of pro-inflammatory cytokines and migration of macrophages. | [74] |
| I3A | Reduces production of pro-inflammatory cytokines and migration of macrophages. Reduces the expression of FAS and SREBP1c. | [74] |
| IPA | Reduces weight gain. Improves intestinal barrier function. | [75,76] |
| Ethanol | Transfer of high-alcohol-producing Klebsiella pneumoniae by oral gavage into mice induces NAFLD. | [81] |
| Obeticholic acid (OCA) | Decreases hepatic inflammation by inhibition of pro-inflammatory cytokines. Decreases fibrogenesis by inhibition of pro-fibrotic cytokines. Inhibits LSEC and Kupffer cell activation. | [92] |
| Cholic acid (CA) Deoxycholic acid(DCA) | Changes in the composition of gut microbiota. | [94] |
| TMAO | Increases hepatic TG accumulation and lipogenesis. Shifts hepatic BA composition toward FXR-antagonistic activity. | [101] |
| Human Studies | ||
| Metabolites | Effects | References |
| Propionate | Prevents weight gain and insulin resistance. | [60] |
| BCAA | Positive correlation with insulin resistance and steatosis. | [67,68,69] |
| IPA | Negative correlation with obesity. | [75,76] |
| Ethanol | Positive correlation with NASH. | [79] |
| DCA | Associated with fibrosis in NAFLD. | [87] |
| OCA | Reduction in ALP, ALT and GGT. | [91] |
| TMAO | Positively correlated with NAFLD. Positively correlated with the serum levels of total BA and hepatic CYP7A1 mRNA. | [100,101] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-H.; Wu, W.-K.; Wu, M.-S. Microbiota-Associated Therapy for Non-Alcoholic Steatohepatitis-Induced Liver Cancer: A Review. Int. J. Mol. Sci. 2020, 21, 5999. https://doi.org/10.3390/ijms21175999
Chen Y-H, Wu W-K, Wu M-S. Microbiota-Associated Therapy for Non-Alcoholic Steatohepatitis-Induced Liver Cancer: A Review. International Journal of Molecular Sciences. 2020; 21(17):5999. https://doi.org/10.3390/ijms21175999
Chicago/Turabian StyleChen, Yi-Hsun, Wei-Kai Wu, and Ming-Shiang Wu. 2020. "Microbiota-Associated Therapy for Non-Alcoholic Steatohepatitis-Induced Liver Cancer: A Review" International Journal of Molecular Sciences 21, no. 17: 5999. https://doi.org/10.3390/ijms21175999
APA StyleChen, Y.-H., Wu, W.-K., & Wu, M.-S. (2020). Microbiota-Associated Therapy for Non-Alcoholic Steatohepatitis-Induced Liver Cancer: A Review. International Journal of Molecular Sciences, 21(17), 5999. https://doi.org/10.3390/ijms21175999