Ionizing Radiation-Induced Epigenetic Modifications and Their Relevance to Radiation Protection



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Role of Radiation Biology in Radiation Protection
2.1. The Current Paradigm of Radiation Biology
- (i)
- The DNA damage in directly exposed cells is the main event for biological effects;
- (ii)
- the DNA damage occurs during, or very shortly after, irradiation of the nuclei in targeted cells;
- (iii)
- the potential for biological consequences can be expressed within one or two cell generations;
- (iv)
- at low doses, the biological effect is in direct proportion to the energy deposited in nuclear DNA.
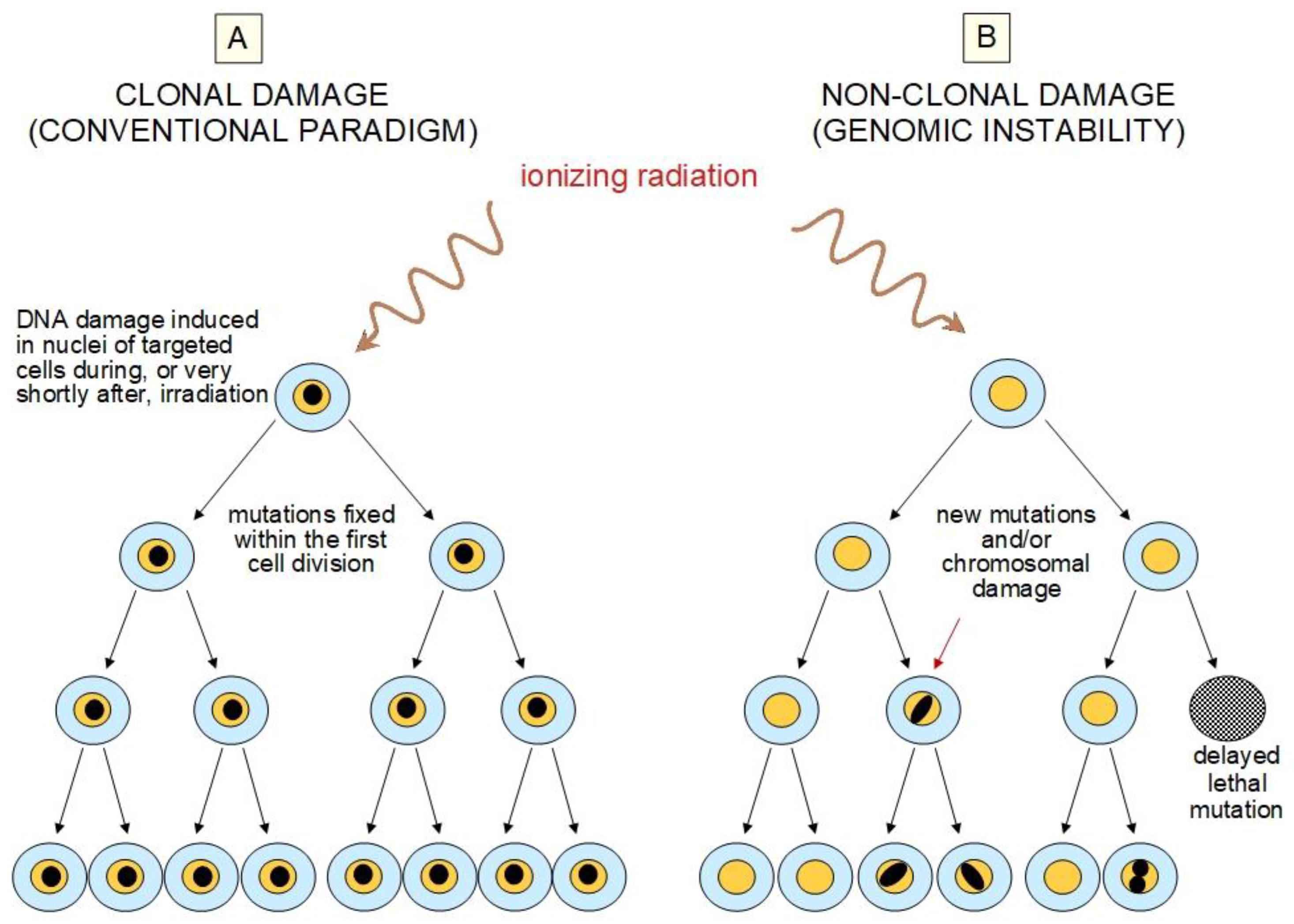
2.2. Challenges to the Current Paradigm
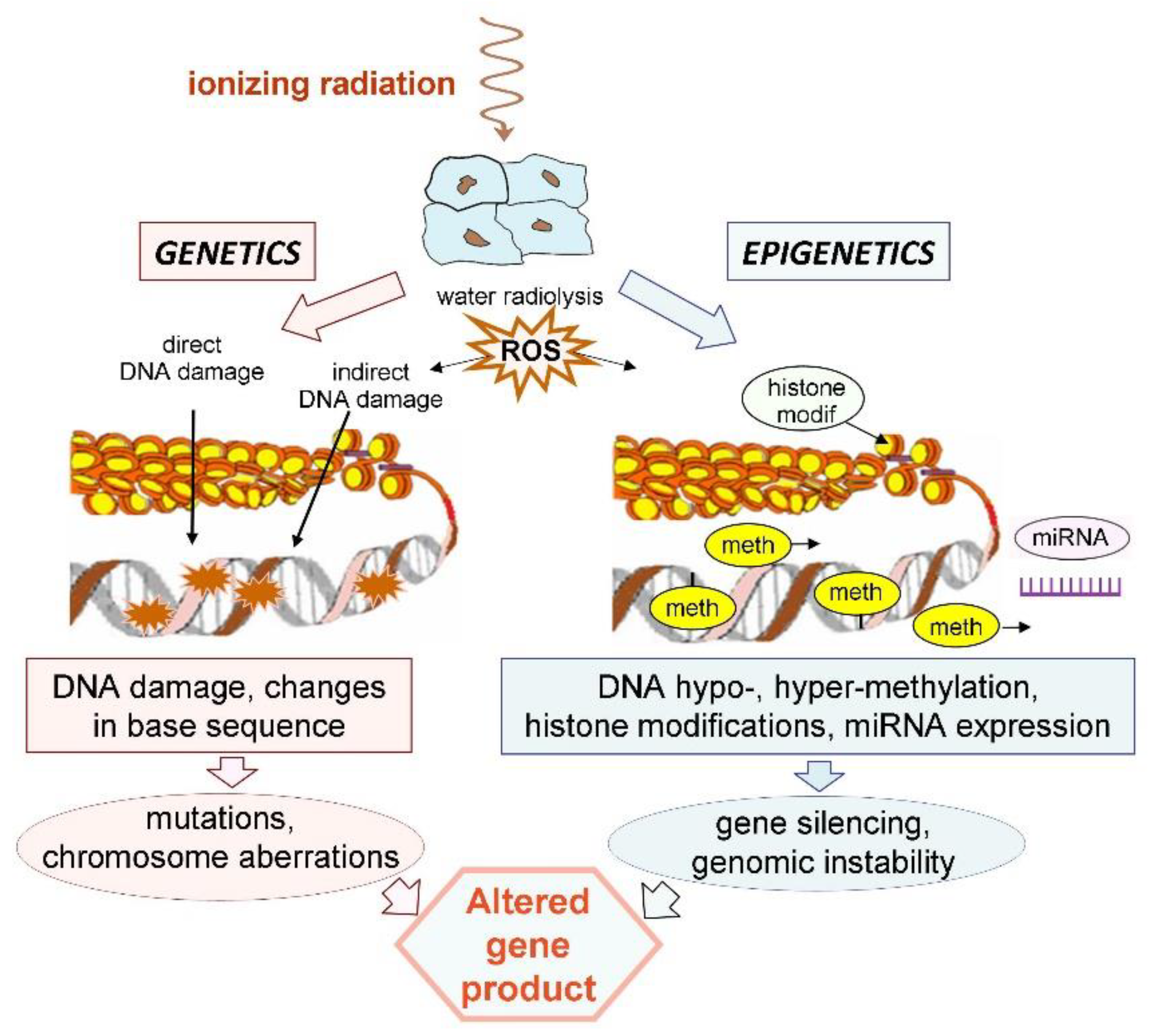
3. Ionizing Radiation Induces Epigenetic Changes
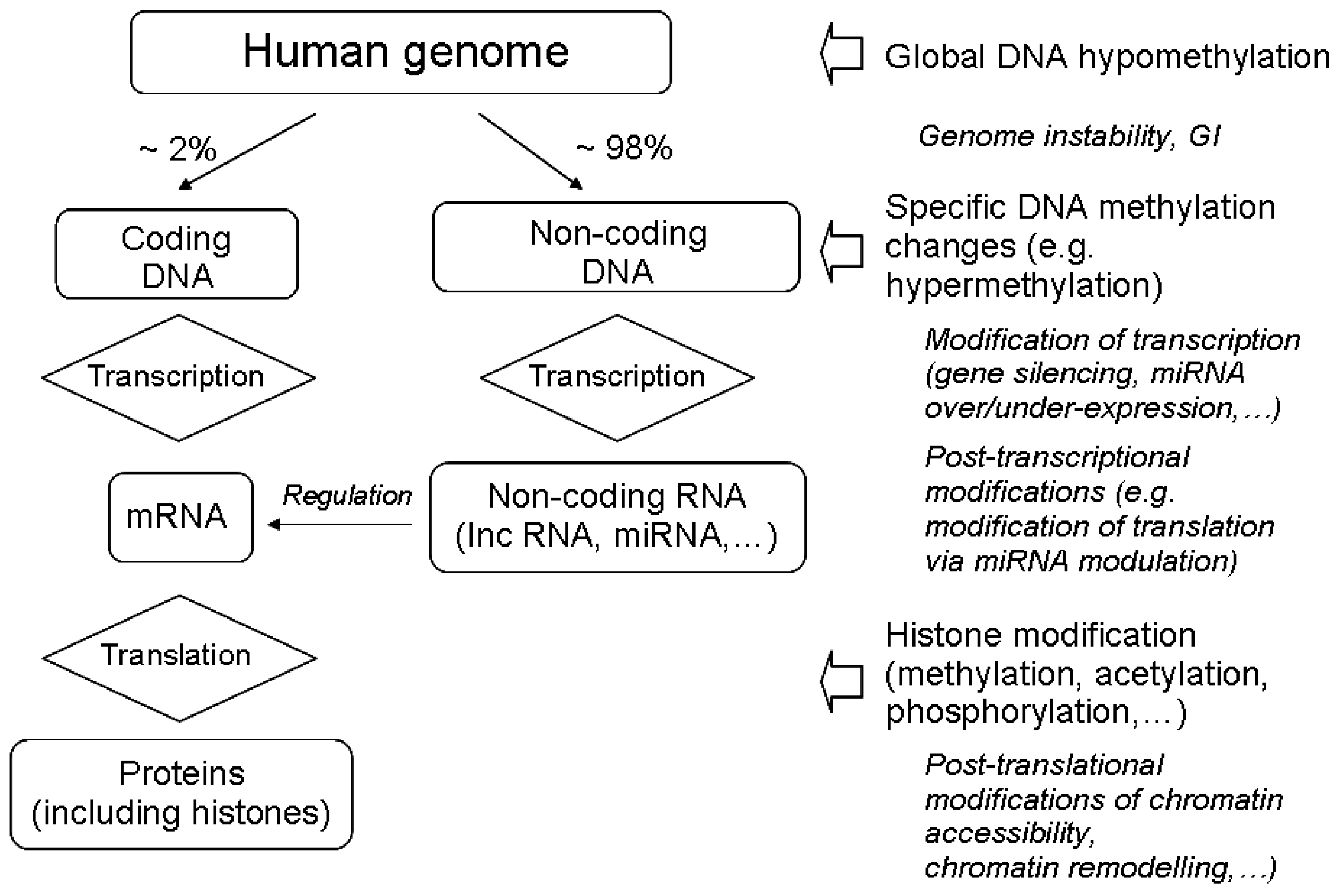
3.1. The Main Epigenetic Modifications
3.2. Radiation-Induced Changes in DNA Methylation
3.3. Radiation-Induced Histone Modifications
3.4. Radiation-Induced Modulation of Non-Coding RNA Expression
3.5. Radiation Quality May Affect Epigenetic Changes
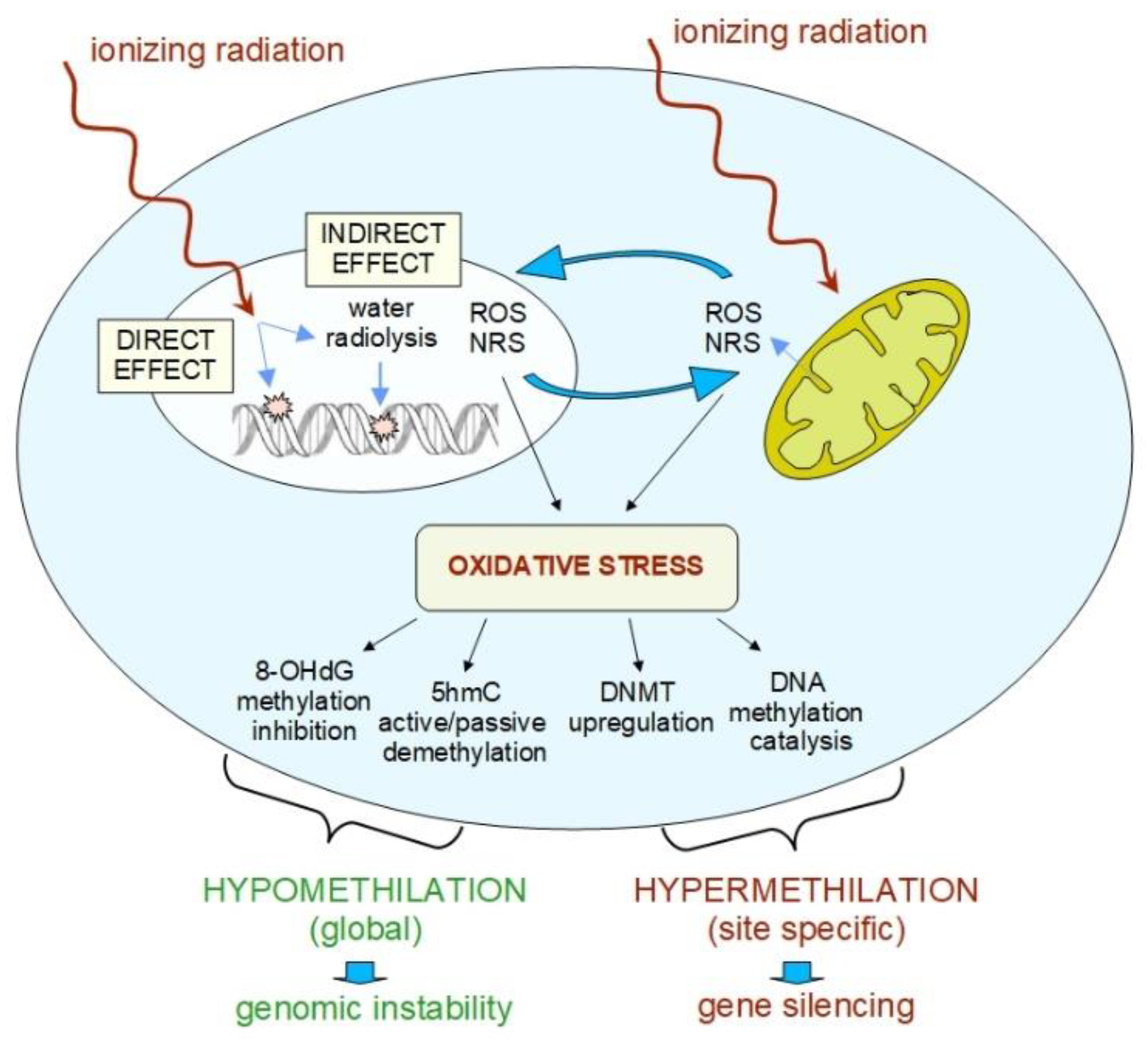
4. Basic Mechanisms of Radiation-Induced Epigenetic Changes
5. Epigenetic Changes Have a Role in Radiation-Induced NTE and AR
6. Epigenetics in Radiation Risk Assessment
6.1. Radiation-Induced Cancer
6.2. Transgenerational Effects
6.3. Non Cancer Effects
6.3.1. Possible Epigenetic Role in Radiation-Induced Cognitive Effects
6.3.2. Possible Epigenetic Role in Radiation-Induced Cardiovascular Effects
6.3.3. Possible Epigenetic Role in Radiation-Induced Cataract
6.4. Epigenetics and the Low Dose/Dose Rate Issue
7. Concluding Remarks and Perspectives
7.1. Epigenetics Is Needed in Radiobiology Paradigms
7.2. Implications in Radiation-Induced Cancer
7.3. Implications in Radiation-Induced Hereditary Effects
7.4. Implications in Radiation-Induced Non-Cancer Effects
7.5. Low-Level Exposures: Detrimental or Beneficial?
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| UNSCEAR | United Nations Scientific Committee on the Effects of Atomic Radiation |
| ICRP | International Commission on Radiological Protection |
| UNEP | United Nations Environment Programme |
| DSB | Double strand break |
| LET | Linear energy transfer |
| LNT | Linear No-Threshold |
| NTE | Non-targeted effects |
| AR | Adaptive response |
| BE | Bystander effect |
| GI | Genomic instability |
| C, G | Cytosine, Guanine |
| CpG | 5′—C—phosphate—G—3′ |
| DNMT | DNA methyltransferase |
| ncRNA | Non-coding RNA |
| lncRNA | Long non-coding RNA |
| miRNA | Micro RNA |
| ROS | Reactive oxygen species |
| RNS | Reactive nitrogen species |
| TE | Transposable element |
| LINE-1 | Long interspersed nucleotide element 1 |
References
- ICRP. ICRP Publication 103: The 2007 Recommendations of the International Commission on Radiological Protection. Ann. ICRP 2007, 37, 1–332. [Google Scholar]
- United Nations Scientific Committee on the Effects of Atomic Radiation (UNSCEAR). Biological Mechanisms of Radiation Actions at Low Doses; United Nations: New York, NY, USA, 2012. [Google Scholar]
- ICRP. ICRP Publication 99: Low-dose Extrapolation of Radiation-related Cancer Risk. Ann. ICRP 2005, 35, 1–140. [Google Scholar]
- National Research Council. Health Risks from Exposure to Low Levels of Ionizing Radiation: BEIR VII Phase 2; The National Academies Press: Washington, DC, USA, 2006. [CrossRef]
- United Nations Environment Programme (UNEP). Radiation Effects and Sources; UNSCEAR: Vienna, Austria, 2016; Available online: http://hdl.handle.net/20.500.11822/7790 (accessed on 18 August 2020).
- Feil, R.; Fraga, M.R. Epigenetics and the environment: Emerging patterns and implications. Nat. Rev. Genet. 2012, 13, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Pacchierotti, F.; Spanò, M. Environmental Impact on DNA Methylation in the Germline: State of the Art and Gaps of Knowledge. BioMed. Res. Int. 2015, 1–23. [Google Scholar] [CrossRef]
- Goodhead, D.T. New radiobiological, radiation risk and radiation protection paradigms. Mutat. Res. 2010, 687, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Averbeck, D. Non-targeted effects as a paradigm breaking evidence. Mutat. Res. 2010, 687, 7–12. [Google Scholar] [CrossRef]
- Mothersill, C.; Seymour, C. Targets, pools, shoulders, and communication–a reflection on the evolution of low-dose radiobiology. Int. J. Radiat. Biol. 2019, 95, 851–860. [Google Scholar] [CrossRef]
- Schofield, P.N.; Kondratowicz, M. Evolving paradigms for the biological response to low dose ionizing radiation: The role of epigenetics. Int. J. Radiat. Biol. 2018, 94, 769–781. [Google Scholar] [CrossRef]
- Miousse, I.R.; Kutanzi, K.R.; Koturbash, I. Effects of Ionizing Radiation on DNA Methylation: From Experimental Biology to Clinical Applications. Int. J. Radiat. Biol. 2017, 93, 457–469. [Google Scholar] [CrossRef]
- O’Neill, P.; Wardman, P. Radiation chemistry comes before radiation biology. Int. J. Radiat. Biol. 2009, 85, 9–25. [Google Scholar] [CrossRef]
- Ward, J.F. Biochemistry of DNA Lesions. Radiat. Res. 1985, 104, S103–S111. [Google Scholar] [CrossRef]
- Ward, J.F. The complexity of DNA damage: Relevance to biological consequences. Int. J. Radiat. Biol. 1994, 66, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Goodhead, D.T.; Thacker, J.; Cox, R. Weiss Lecture. Effects of radiations of different qualities on cells: Molecular mechanisms of damage and repair. Int. J. Radiat. Biol. 1993, 63, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Goodhead, D.T. Initial events in the cellular effects of ionising radiation: Clustered damage in DNA. Int. J. Radiat. Biol. 1994, 65, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Nikjoo, H.; O’Neill, P.; Wilson, W.E.; Goodhead, D.T. Computational Approach for Determining the Spectrum of DNA Damage Induced by Ionizing Radiation. Radiat. Res. 2001, 156, 577–583. [Google Scholar] [CrossRef]
- Hill, M.A. Radiation Track Structure: How the Spatial Distribution of Energy Deposition Drives Biological Response. Clin. Oncolol. 2001, 32, 75–83. [Google Scholar] [CrossRef]
- Prise, K.M.; Pinto, M.; Newman, H.C.; Michael, B.D. A review of studies of ionizing radiation-induced double-strand break clustering. Radiat. Res. 2001, 156, 572–576. [Google Scholar] [CrossRef]
- Prise, K.M.; Folkard, M.; Newman, H.C.; Michael, B.D. Effect of Radiation Quality on Lesion Complexity in Cellular DNA. Int. J. Radat. Biol. 1994, 66, 537–542. [Google Scholar] [CrossRef]
- Georgakilas, A.G.; O’Neill, P.; Stewart, R.D. Induction and repair of clustered DNA lesions: What do we know so far? Radiat. Res. 2013, 180, 100–109. [Google Scholar] [CrossRef]
- Mavragani, I.V.; Nikitaki, Z.; Kalospyros, S.A.; Georgakilas, A.G. Ionizing Radiation and Complex DNA Damage: From Prediction to Detection Challenges and Biological Significance. Cancers 2019, 11, 1789. [Google Scholar] [CrossRef]
- Anderson, R.M.; Stevens, D.L.; Goodhead, D.T. M-FISH analysis shows that complex chromosome aberrations induced by α-particle tracks are cumulative products of localized rearrangements. Proc. Natl. Acad. Sci. USA 2002, 99, 12167–12172. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.M.; Marsden, S.J.; Paice, S.J.; Bristow, A.E.; Kadhim, M.A.; Griffin, C.S.; Goodhead, D.T. Transmissible and Non transmissible Complex Chromosome Aberrations Characterized by Three-Color and mFISH Define a Biomarker of Exposure to High-LET α Particles. Radiat. Res. 2003, 159, 40–48. [Google Scholar] [CrossRef]
- Savage, J.R.; Simpson, P.J. FISH “painting” patterns resulting from complex exchanges. Mutat. Res. 1994, 312, 51–60. [Google Scholar] [CrossRef]
- Ritter, S.; Durante, M. Heavy-ion induced chromosomal aberrations: A review. Mutat. Res. Genet. Toxicol. Environ. Mut. 2010, 701, 38–46. [Google Scholar] [CrossRef]
- Loucas, B.D.; Durante, M.; Bailey, S.; Cornforth, M.N. Chromosome damage in human cells by γ-rays, α-particles and heavy ions: Track interactions in basic dose–response relationships. Radiat. Res. 2013, 179, 9–20. [Google Scholar] [CrossRef]
- Muller, H.J. Nobel Lecture. NobelPrize.org. Nobel Media AB. 2020. Available online: https://www.nobelprize.org/prizes/medicine/1946/muller/lecture/ (accessed on 18 August 2020).
- United Nations Scientific Committee on the Effects of Atomic Radiation (UNSCEAR). Sources, Effects and Risks of Ionizing Radiation; United Nations: New York, NY, USA, 1988. [Google Scholar]
- Ledford, H. Language: Disputed definitions. Nature 2008, 455, 1023–1028. [Google Scholar] [CrossRef]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Develop. 2009, 23, 781–783. [Google Scholar] [CrossRef]
- Christensen, B.C.; Marsit, C.J. Epigenomics in environmental health. Front. Genet. 2011, 2, 1–10. [Google Scholar] [CrossRef]
- Head, J.A.; Dolinoy, D.C.; Basu, N. Epigenetics for Ecotoxicologists. Environ. Toxicol. Chem. 2012, 31, 221–227. [Google Scholar] [CrossRef]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef]
- Saxonov, S.; Berg, P.; Brutlag, D.L. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. USA 2006, 103, 1412–1417. [Google Scholar] [CrossRef] [PubMed]
- Bird, A.P. DNA methylation versus gene expression. J. Embryol. Exp. Morph. 1984, 83, 31–40. Available online: https://dev.biologists.org/content/develop/83/Supplement/31.full.pdf (accessed on 18 August 2020). [PubMed]
- Greenberg, M.V.C.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell. Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef] [PubMed]
- Madakashira, B.P.; Sadler, K.C. DNA Methylation, Nuclear Organization, and Cancer. Front. Genet. 2017. [Google Scholar] [CrossRef]
- Kim, M.; Costello, J. DNA methylation: An epigenetic mark of cellular memory. Exper. Mol. Med. 2017, 49, e322. [Google Scholar] [CrossRef]
- Margueron, R.; Trojer, P.; Reinberg, D. The key to development: Interpreting the histone code? Curr. Opin. Genet. Dev. 2005, 15, 163–176. [Google Scholar] [CrossRef]
- Allfrey, V.G.; Faulkner, R.; Mirsky, A.E. Acetylation and Methylation of Histones and Their Possible Role in the Regulation of RNA Synthesis. Proc. Natl. Acad. Sci. USA 1964, 51, 786–794. [Google Scholar] [CrossRef]
- Tharmalingam, S.; Sreetharan, S.; Kulesza, A.V.; Boreham, D.R.; Tai, T.C. Low-Dose Ionizing Radiation Exposure, Oxidative Stress and Epigenetic Programing of Health and Disease. Radiat. Res. 2017, 188, 525–538. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Morozova, N.; Zinovyev, A.; Nonne, N.; Pritchard, L.-L.; Gorban, A.N.; Harel-Bellan, A. Kinetic signatures of microRNA modes of action. RNA 2012, 18, 1635–1655. [Google Scholar] [CrossRef]
- Mao, A.; Liu, Y.; Zhang, H.; Di, C.; Sun, C. MicroRNA expression and biogenesis in cellular response to ionizing radiation. DNA Cell Biol. 2014, 33, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Carthew, R.W.; Sontheime, E.J. Origins and Mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef] [PubMed]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef] [PubMed]
- Morris, K.V. Long antisense non-coding RNAs function to direct epigenetic complexes that regulate transcription in human cells. Epigenetics 2009, 4, 296–301. [Google Scholar] [CrossRef]
- Koturbash, I.; Zemp, F.J.; Pogribny, I.; Kovalchuk, O. Small molecules with big effects: The role of the microRNAome in cancer and carcinogenesis. Mutat. Res. Genet. Toxicol. Environ. Mut. 2011, 722, 94–105. [Google Scholar] [CrossRef]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef]
- Friedman, R.C.; Farh, K.K.H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef]
- Shkumatava, A.; Stark, A.; Sive, H.; Bartel, D.P. Coherent but overlapping expression of microRNAs and their targets during vertebrate development. Genes Develop. 2009, 23, 466–481. [Google Scholar] [CrossRef]
- Fatica, A.; Bozzoni, I. Long non-coding RNAs: New players in cell differentiation and development. Nat. Rev. Genet. 2014, 15, 7–21. [Google Scholar] [CrossRef]
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Ann. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef]
- Anastasiadou, E.; Jacob, L.S.; Slack, F.J. Non-coding RNA networks in cancer. Nat. Rev. Cancer 2018, 18, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Kalinich, J.F.; Catravas, G.N.; Snyder, S.L. The effect of gamma radiation on DNA methylation. Radiat. Res. 1989, 117, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Goetz, W.; Morgan, M.N.M.; Baulch, J.E. The effect of radiation quality on genomic DNA methylation profiles in irradiated human cell lines. Radiat. Res. 2011, 175, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.H.; Park, A.K.; Dong, S.M.; Ahn, J.H.; Park, W.Y. Global analysis of CpG methylation reveals epigenetic control of the radiosensitivity in lung cancer cell lines. Oncogene 2010, 29, 4725–4731. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, M.A.; Omaruddin, R.A. Differential DNA Methylation Alterations in Radiation-Sensitive and Resistant Cells. DNA Cell Biol. 2012, 31, 908–916. [Google Scholar] [CrossRef] [PubMed]
- Tawa, R.; Kimura, Y.; Komura, J.; Miyamura, Y.; Kurishita, A.; Sasaki, M.S.; Sakurai, H.; Ono, T. Effects of X-ray irradiation on genomic DNA methylation levels in mouse tissues. J. Radiat. Res. 1998, 39, 271–278. [Google Scholar] [CrossRef]
- Pogribny, I.; Raiche, J.; Slovack, M.; Kovalchuk, O. Dose-dependence, sex- and tissue-specificity, and persistence of radiation-induced genomic DNA methylation changes. Biochem. Biophys. Res. Commun. 2004, 320, 1253–1261. [Google Scholar] [CrossRef]
- Raiche, J.; Rodriguez-Juarez, R.; Pogribny, I.; Kovalchuk, O. Sex- and tissue-specific expression of maintenance and de novo DNA methyltransferases upon low dose X-irradiation in mice. Biochem. Biophys. Res. Commun. 2004, 325, 39–47. [Google Scholar] [CrossRef]
- Koturbash, I.; Pogribny, I.; Kovalchuk, O. Stable loss of global DNA methylation in the radiation-target tissue-A possible mechanism contributing to radiation carcinogenesis? Biochem. Biophys. Res. Commun. 2005, 337, 526–533. [Google Scholar] [CrossRef]
- Giotopoulos, G.; McCormick, C.; Cole, C.; Zanker, A.; Jawad, M.; Brown, R. DNA methylation during mouse hemopoietic differentiation and radiation-induced leukemia. Exp. Hematol. 2006, 34, 1462–1470. [Google Scholar] [CrossRef]
- Klose, R.J.; Bird, A.P. Genomic DNA methylation: The mark and its mediators. Trends Biochem. Sci. 2006, 31, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Schübeler, D. Genomic patterns of DNA methylation: Targets and function of an epigenetic mark. Curr. Opin. Cell Biol. 2007, 19, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. Epigenetic determinants of cancer. Cold Spring Harb. Perspect. Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Collins, L.J.; Penny, D. The RNA infrastructure: Dark matter of the eukaryotic cell? Trends Genet. 2009, 25, 120–128. [Google Scholar] [CrossRef]
- Miousse, I.R.; Koturbash, I. The Fine LINE: Methylation Drawing the Cancer Landscape. Biomed. Res. Int. 2015, 131547. [Google Scholar] [CrossRef]
- Tanaka, A.; Nakatani, Y.; Hamada, N.; Jinno-Oue, A.; Shimizu, N.; Wada, S. Ionising irradiation alters the dynamics of human long interspersed nuclear elements 1 (LINE1) retrotransposon. Mutagenesis 2012, 27, 599–607. [Google Scholar] [CrossRef]
- Miousse, I.R.; Chalbot, M.C.; Lumen, A.; Ferguson, A.; Kavouras, I.G.; Koturbash, I. Response of transposable elements to environmental stressors. Mutat. Res. Rev. Mutat. Res. 2015, 765, 19–39. [Google Scholar] [CrossRef]
- Kovalchuk, O.; Baulch, J.E. Epigenetic changes and nontargeted radiation effects-Is there a link? Environ. Mol. Mutagen. 2008, 49, 16–25. [Google Scholar] [CrossRef]
- Goetz, W.; Morgan, M.N.M.; Belliveau, B.J.; Baulch, J.E. Effects of high and low LET radiation exposure on DNA methylation. Environ. Mol. Mutagen. 2009, 50, 575. [Google Scholar]
- Koturbash, I. Michael Fry Award Lecture: When DNA is Actually Not a Target: Radiation Epigenetics as a Tool to Understand and Control Cellular Response to Ionizing Radiation. Radiat Res. 2018, 190, 5–11. [Google Scholar] [CrossRef]
- Mendonca, M.S.; Antoniono, R.J.; Redpath, J.L. Delayed Heritable Damage and Epigenetics in Radiation-Induced Neoplastic Transformation of Human Hybrid Cells. Radiat. Res. 1993, 134, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.J.; Schulz, W.A. Causes and consequences of DNA hypomethylation in human cancer. Biochem. Cell Biol. 2005, 83, 296–321. [Google Scholar] [CrossRef] [PubMed]
- Toyota, M.; Issa, J.P. The role of DNA hypermethylation in human neoplasia. Electrophoresis 2000, 21, 329–333. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome-biological and translational implications. Nat. Rev. Cancer 2012, 11, 726–734. [Google Scholar] [CrossRef]
- Su, S.; Jin, Y.; Zhang, W.; Yang, L.; Shen, Y.; Cao, Y.; Tong, J. Aberrant promoter methylation of p16 (INK4a) and O(6)-methylguanine-DNA methyltransferase genes in workers at a Chinese uranium mine. J. Occup. Health 2006, 48, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Lyon, C.M.; Klinge, D.M.; Liechty, K.C.; Gentry, F.D.; March, T.H.; Kang, T.; Gilliland, F.D.; Adamova, G.; Rusinova, G.; Telnov, V.; et al. Radiation-induced lung adenocarcinoma is associated with increased frequency of genes, inactivated by promoter hypermethylation. Radiat. Res. 2007, 168, 409–414. [Google Scholar] [CrossRef]
- Antwih, K.M.; Gabbara, W.D.; Lancaster, D.M.; Ruden, S.P.; Zielske, S.P. Radiation-induced epigenetic DNA methylation modification of radiation-response pathways. Epigenetics 2013, 8, 839–848. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef]
- Pilch, D.R.; Sedelnikova, O.A.; Redon, C.; Celeste, A.; Nussenzweig, A.; Bonner, W.M. Characteristics of gamma-H2AX foci at DNA double-strand breaks sites. Biochem. Cell Biol. 2003, 81, 123–129. [Google Scholar] [CrossRef]
- Pogribny, I.; Koturbash, I.; Tryndyak, V.; Hudson, D.; Stevenson, S.M.; Sedelnikova, O.; Bonner, W.; Kovalchuk, O. Fractionated low-dose radiation exposure leads to accumulation of DNA damage and profound alterations in DNA and histone methylation in the murine thymus. Mol. Cancer Res. 2005, 3, 553–561. [Google Scholar] [CrossRef]
- Tryndyak, V.P.; Kovalchuk, O.; Pogribny, I.P. Loss of DNA methylation and histone H4 lysine 20 trimethylation in human breast cancer cells is associated with aberrant expression of DNA methyltransferase 1, Suv4-20h2 histone methyltransferase and methyl-binding proteins. Cancer Biol. Ther. 2006, 5, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Acuna, L.; Di Tomaso, M.V.; Palitti, F.; Martinez-Lopez, W. Histone posttranslational modifications in DNA damage response. Cytogenet. Genome Res. 2010, 128, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Averbeck, N.B.; Durante, M. Protein acetylation within the cellular response to radiation. J. Cell. Physiol. 2011, 226, 962–967. [Google Scholar] [CrossRef] [PubMed]
- Metheetrairut, C.; Slack, F.J. MicroRNAs in the Ionizing Radiation Response and in Radiotherapy. Curr. Opin. Genet. Dev. 2013, 23, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Aypar, U.; Morgan, W.F.; Baulch, J.E. Radiation-induced epigenetic alterations after low and high LET irradiations. Mutat. Res. 2011, 707, 24–33. [Google Scholar] [CrossRef]
- Wagner-Ecker, M.; Schwager, C.; Wirkner, U.; Abdollahi, A.; Huber, P.E. MicroRNA expression after ionizing radiation in human endothelial cells. Radiat. Oncol. 2010, 5, 25. [Google Scholar] [CrossRef]
- Chaudhry, M.A.; Kreger, B.; Omaruddin, R.A. Transcriptional modulation of micro-RNA in human cells differing in radiosensitivity. Int. J. Radiat. Biol. 2010, 86, 569–583. [Google Scholar] [CrossRef]
- Halimi, M.; Asghari, S.M.; Sariri, R.; Moslemi, D.; Parsian, H. Cellular Response to Ionizing Radiation: A MicroRNA Story. Int. J. Mol. Cell Med. 2012, 1, 178–184. [Google Scholar]
- Gong, P.; Zhang, T.; He, D.; Hsieh, J.T. MicroRNA-145 Modulates Tumor Sensitivity to Radiation in Prostate Cancer. Radiat. Res. 2015, 184, 630–638. [Google Scholar] [CrossRef]
- El Bezawy, R.; Tinelli, S.; Tortoreto, M.; Doldi, V.; Zuco, V.; Folini, M.; Stucchi, C.; Rancati, T.; Valdagni, R.; Gandellini, P.; et al. MiR-205 enhances radiation sensitivity of prostate cancer cells by impairing DNA damage repair through PKCε and ZEB1 inhibition. J. Exp. Clin. Cancer Res. 2019, 38, 51. [Google Scholar] [CrossRef]
- Chaudhry, M.A. Radiation-induced microRNA: Discovery, functional analysis, and cancer radiotherapy. J. Cell. Biochem. 2014, 115, 436–449. [Google Scholar] [CrossRef] [PubMed]
- Cellini, F.; Morganti, A.G.; Genovesi, D.; Silvestris, N.; Valentini, V. Role of microRNA in response to ionizing radiations: Evidences and potential impact on clinical practice for radiotherapy. Molecules 2014, 19, 5379–5401. [Google Scholar] [CrossRef] [PubMed]
- Marta, G.N.; Garicochea, B.; Carvalho, A.L.; Real, J.M.; Kowalski, L.P. MicroRNAs, cancer and ionizing radiation: Where are we? Rev. Assoc. Med. Bras. 2015, 61, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Lima, F.; Ding, D.; Goetz, W.; Yang, A.J.; Baulch, J. High LET 56Fe ion irradiation induces tissue-specific changes in DNA methylation in the mouse. Environ. Mol. Mutagen. 2014, 55, 266–277. [Google Scholar] [CrossRef]
- Nzabarushimana, E.; Miousse, I.R.; Shao, L.; Chang, J.; Allen, A.R.; Turner, J.; Stewart, B.; Raber, J.; Koturbash, I. Long-term epigenetic effects of exposure to low doses of 56Fe in the mouse lung. J. Radiat. Res. 2014, 55, 823–828. [Google Scholar] [CrossRef]
- Miousse, I.R.; Shao, L.J.; Chang, J.H.; Feng, W.; Wang, Y.Y.; Allen, A.R. Exposure to low-dose Fe-56-ion radiation induces long-term epigenetic alterations in mouse bone marrow hematopoietic progenitor and stem cells. Radiat. Res. 2014, 182, 92–101. [Google Scholar] [CrossRef]
- Kennedy, E.M.; Conneely, K.N.; Vertino, P.M. Epigenetic Memory of Space Radiation Exposure. Available online: https://three-jsc.nasa.gov/articles/Vertino.pdf (accessed on 30 July 2014).
- Durante, M.; Cucinotta, F.A. Heavy ion carcinogenesis and human space exploration. Nat. Rev. Cancer 2008, 8, 465–472. [Google Scholar] [CrossRef]
- Cucinotta, F.A. Space Radiation Risks for Astronauts on Multiple International Space Station Missions. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Morano, A.; Angrisano, T.; Russo, G.; Landi, R.; Pezone, A.; Bartollino, S.; Zuchegna, C.; Babbio, F.; Bonapace, I.M.; Allen, B.; et al. Targeted DNA methylation by homology-directed repair in mammalian cells. Transcription reshapes methylation on the repaired gene. Nucleic Acids Res. 2014, 42, 804–821. [Google Scholar] [CrossRef]
- Prior, S.; Miousse, I.R.; Nzabarushimana, E.; Pathak, R.; Skinner, C.; Kutanzi, K.R.; Allen, A.R.; Raber, J.; Tackett, A.J.; Hauer-Jensen, M.; et al. Densely ionizing radiation affects DNA methylation of selective LINE-1 elements. Environ. Res. 2016, 150, 470–481. [Google Scholar] [CrossRef]
- Belinsky, S.A.; Klinge, D.M.; Liechty, K.C.; March, T.H.; Kang, T.; Gilliland, F.D.; Sotnic, N.; Adamova, G.; Rusinova, G.; Telnov, V. Plutonium targets the p16 gene for inactivation by promoter hypermethylation in human lung adenocarcinoma. Carcinogenesis 2004, 25, 1063–1067. [Google Scholar] [CrossRef]
- Templin, T.; Amundson, S.A.; Brenner, D.J.; Smilenov, L.B. Whole mouse blood microRNA as biomarkers for exposure to γ-rays and 56Fe ions. Int. J. Radiat. Biol. 2011, 87, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Templin, T.; Young, E.F.; Smilenov, L.B. Proton radiation-induced miRNA signatures in mouse blood: Characterization and comparison with 56Fe-ion and gamma radiation. Int. J. Radiat. Biol. 2012, 88, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.F. DNA damage produced by ionizing radiation in mammalian cells: Identities, mechanisms of formation, and reparability. Prog. Nucleic Acid Res. Mol. Biol. 1988, 35, 95–125. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, P.; Fielden, E.M. Primary Free Radical Processes in DNA. Adv. Radiat. Biol. 1993, 17, 53–120. [Google Scholar] [CrossRef]
- De Lara, C.M.; Jenner, T.J.; Townsend, K.M.S.; Marsden, S.J.; O’Neill, P. The Effect of Dimethyl Sulfoxide on the Induction of DNA Double-Strand Breaks in V79-4 Mammalian Cells by Alpha Particles. Radiat. Res. 1995, 144, 43–49. [Google Scholar] [CrossRef]
- Azzam, E.I.; Jay-Gerin, J.P.; Pain, D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012, 327, 48–60. [Google Scholar] [CrossRef]
- Shrishrimal, S.; Kosmacek, E.A.; Oberley-Deegan, R.E. Reactive Oxygen Species Drive Epigenetic Changes in Radiation-Induced Fibrosis. Oxid. Med. Cell. Longev. 2019, 2019. [Google Scholar] [CrossRef]
- Cerda, S.; Weitzman, S.A. Influence of oxygen radical injury on DNA methylation. Mutat. Res. 1997, 386, 141–152. [Google Scholar] [CrossRef]
- Franco, R.; Schoneveld, O.; Georgakilas, A.G.; Panayiotidis, M.I. Oxidative stress, DNA methylation and carcinogenesis. Cancer Lett. 2008, 266, 6–11. [Google Scholar] [CrossRef]
- Ziech, D.; Franco, R.; Pappa, A.; Panayiotidis, M.I. Reactive Oxygen Species (ROS)-Induced genetic and epigenetic alterations in human carcinogenesis. Mutat. Res. 2011, 711, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Branco, M.R.; Ficz, G.; Reik, W. Uncovering the role of 5- hydroxymethylcytosine in the epigenome. Nat. Rev. Genet. 2012, 13, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Efimova, O.A.; Koltsova, A.S.; Krapivin, M.I.; Tikhonov, A.V.; Pendina, A.A. Environmental Epigenetics and Genome Flexibility: Focus on 5-Hydroxymethylcytosine. Int. J. Mol. Sci. 2020, 21, 3223. [Google Scholar] [CrossRef] [PubMed]
- Maltseva, D.V.; Baykov, A.A.; Jeltsch, A.; Gromova, E.S. Impact of 7,8-dihydro-8-oxoguanine on methylation of the CpG site by Dnmt3a. Biochemistry 2009, 48, 1361–1368. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, M.; Dellino, G.I.; Gambino, V.; Roda, N.; Pelicci, P.G. On the epigenetic role of guanosine oxidation. Redox Biol. 2020, 29, 101398. [Google Scholar] [CrossRef] [PubMed]
- Shaughnessy, D.T.; McAllister, K.; Worth, L.; Haugen, A.C.; Meyer, J.N.; Domann, F.E.; Houten, B.V.; Mostoslavsky, R.; Bultman, S.J.; Baccarelli, A.A.; et al. Mitochondria, energetics, epigenetics, and cellular responses to stress. Environ. Health Perspect. 2014, 122, 1271–1278. [Google Scholar] [CrossRef]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef]
- Szumiel, I. Ionizing radiation-induced oxidative stress, epigenetic changes and genomic instability: The pivotal role of mitochondria. Int. J. Radiat. Biol. 2015, 91, 1–12. [Google Scholar] [CrossRef]
- Baulch, J.E. Radiation-induced genomic instability, epigenetic mechanisms and the mitochondria: A dysfunctional ménage a trois? Int. J. Radiat. Biol. 2019, 95, 516–525. [Google Scholar] [CrossRef]
- Wu, Q.; Ni, X. ROS-Mediated DNA Methylation Pattern Alterations in Carcinogenesis. Curr. Drug Targets 2015, 16, 13–19. [Google Scholar] [CrossRef]
- Kietzmann, T.; Petry, A.; Shvetsova, A.; Gerhold, J.M.; Görlach, A. The epigenetic landscape related to reactive oxygen species formation in the cardiovascular system. Br. J. Pharmacol. 2017, 174, 1533–1554. [Google Scholar] [CrossRef]
- Qian, W.; Miki, D.; Zhang, H.; Liu, Y.; Zhang, X.; Tang, K.; Kan, Y.; La, H.; Li, X.; Li, S.; et al. A histone acetyltransferase regulates active DNA demethylation in Arabidopsis. Science 2012, 336, 1445–1448. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Yamaguchi, L.; Nakanishi, M. Regulation of maintenance DNA methylation via histone ubiquitylation. J. Biochem. 2016, 159, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Fuks, F. DNA methylation and histone modifications: Teaming up to silence genes. Curr. Opin. Genet. Dev. 2005, 15, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K. DNA methylation and chromatin–unraveling the tangled web. Oncogene 2002, 21, 5361–5379. [Google Scholar] [CrossRef] [PubMed]
- Cameron, E.; Bachman, K.; Myöhänen, S.; Herman, J.G.; Baylin, S.B. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat. Genet. 1999, 21, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Cedar, H.; Bergman, Y. Linking DNA methylation and histone modification: Patterns and paradigms. Nat. Rev. Genet. 2009, 10, 295–304. [Google Scholar] [CrossRef]
- Rose, N.R.; Klose, R.J. Understanding the relationship between DNA methylation and histone lysine methylation. Biochim. Biophys. Acta 2014, 1839, 1362–1372. [Google Scholar] [CrossRef]
- Du, J.; Johnson, L.M.; Jacobsen, S.E.; Patel, D.J. DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol. 2015, 16, 519–532. [Google Scholar] [CrossRef]
- Sun, X.; He, Y.; Huang, C.; Ma, T.-T.; Li, J. The epigenetic feedback loop between DNA methylation and microRNAs in fibrotic disease with an emphasis on DNA methyltransferases. Cell. Signal. 2013, 25, 1870–1876. [Google Scholar] [CrossRef]
- Wang, S.; Wu, W.; Claret, F.X. Mutual regulation of microRNAs and DNA methylation in human cancers. Epigenetics 2017, 12, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Huan, T.; Mendelson, M.; Joehanes, R.; Yao, C.; Liu, C.; Song, C.; Bhattacharya, A.; Rong, J.; Tanriverdi, K.; Keefe, J.; et al. Epigenome-wide association study of DNA methylation and microRNA expression highlights novel pathways for human complex traits. Epigenetics 2020, 15, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Jin, C. Histone variants in environmental-stress-induced DNA damage repair. Mutat. Res. 2019, 780, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Seymour, C.B.; Mothersill, C.E.; Alper, T. High yields of lethal mutations in somatic mammalian cells that survive ionizing radiation. Int. J. Radiat. Biol. 1986, 50, 167–179. [Google Scholar] [CrossRef]
- Nagasawa, H.; Little, J.B. Induction of sister chromatid exchanges by extremely low doses of α-particles. Cancer Res. 1992, 52, 6394–6396. [Google Scholar]
- Kadhim, M.A.; Macdonald, D.A.; Goodhead, D.T.; Lorimore, S.A.; Marsden, S.J.; Wright, E.G. Transmission of chromosomal instability after plutonium alpha-particle irradiation. Nature 1992, 355, 738–740. [Google Scholar] [CrossRef]
- Marder, B.A.; Morgan, W.F. Delayed chromosomal instability induced by DNA damage. Mol. Cell. Biol. 1993, 13, 6667–6677. [Google Scholar] [CrossRef]
- Mothersill, C.; Seymour, C. Radiation-induced bystander effects: Past history and future directions. Radiat. Res. 2001, 155, 759–767. [Google Scholar] [CrossRef]
- Morgan, W.F. Non-targeted and delayed effects of exposure to ionizing radiation: I. Radiation-induced genomic instability and bystander effects in vitro. Radiat. Res. 2003, 159, 567–580. [Google Scholar] [CrossRef]
- Morgan, W.F. Non-targeted and delayed effects of exposure to ionizing radiation: II. Radiation-induced genomic instability and bystander effects in vivo, clastogenic factors and transgenerational effects. Radiat. Res. 2003, 159, 581–596. [Google Scholar] [CrossRef]
- Olivieri, G.; Bodycote, J.; Wolff, S. Adaptive response of human lymphocytes to low concentrations of radioactive thymidine. Science 1984, 223, 594–597. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, H.; Little, J.B. Unexpected sensitivity to the induction of mutations by very low doses of alpha particle radiation: Evidence for a bystander effect. Radiat. Res. 1999, 152, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Blyth, B.J.; Sykes, P.J. Radiation-Induced Bystander Effects: What Are They, and How Relevant Are They to Human Radiation Exposures? Radiat. Res. 2011, 176, 139–157. [Google Scholar] [CrossRef] [PubMed]
- Koturbash, I.; Boyko, A.; Rodriguez-Juarez, R.; McDonald, R.J.; Tryndyak, V.P.; Kovalchuk, I.; Pogribny, I.P.; Kovalchuk, O. Role of epigenetic effectors in maintenance of the long-term persistent bystander effect in spleen in vivo. Carcinogenesis 2007, 28, 1831–1838. [Google Scholar] [CrossRef]
- Mancuso, M.; Pasquali, E.; Leonardi, S.; Tanori, M.; Rebessi, S.; Di Majo, V.; Pazzaglia, S.; Toni, M.P.; Pimpinella, M.; Covelli, V.; et al. Oncogenic bystander radiation effects in Patched heterozygous mouse cerebellum. Proc. Natl. Acad. Sci. USA 2008, 105, 12445–12450. [Google Scholar] [CrossRef]
- Kadim, M.; Salomaa, S.; Wright, E.; Hildebrandt, G.; Belyakov, O.V.; Prise, K.M.; Little, M.P. Non-targeted effects of ionising radiation-implications for low dose risk. Mutat. Res. 2013, 752, 84–98. [Google Scholar] [CrossRef]
- Campa, A.; Balduzzi, M.; Dini, V.; Esposito, G.; Tabocchini, M.A. The complex interactions between radiation induced non-targeted effects and cancer. Cancer Lett. 2015, 356, 126–136. [Google Scholar] [CrossRef]
- Lorimore, S.A.; Coates, P.J.; Wright, E.G. Radiation-induced genomic instability and bystander effects: Inter-related nontargeted effects of exposure to ionizing radiation. Oncogene 2003, 22, 7058–7069. [Google Scholar] [CrossRef]
- Huang, L.; Kim, P.M.; Nickoloff, J.A.; Morgan, W.F. Targeted and nontargeted effects of low-dose ionizing radiation on delayed genomic instability in human cells. Cancer Res. 2007, 67, 1099–1104. [Google Scholar] [CrossRef]
- Lorimore, S.A.; Kadhim, M.A.; Pocock, D.A.; Papworth, D.; Stevens, D.L.; Goodhead, D.T.; Wright, E.G. Chromosomal instability in the descendants of unirradiated surviving cells after alpha-particle irradiation. Proc. Natl. Acad. Sci. USA 1998, 95, 5730–5733. [Google Scholar] [CrossRef]
- Schwartz, J.L. Variability: The common factor linking low dose-induced genomic instability, adaptation and bystander effects. Mutat. Res. 2007, 616, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Mothersill, C.; Seymour, C. Changing paradigms in radiobiology. Mutat. Res. Rev. Mutat. Res. 2012, 750, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Morgan, W.F.; Sowa, M.B. Non-targeted effects induced by ionizing radiation: Mechanisms and potential impact on radiation induced health effects. Cancer Lett. 2015, 356, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.J. The bystander effect. Health Phys. 2003, 85, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Sowa, M.B.; Goetz, W.; Baulch, J.E.; Pyles, D.N.; Dziegielewski, J.; Yovino, S.; Snyder, A.R.; De Toledo, S.M.; Azzam, E.I.; Morgan, W.F. Lack of evidence for low-LET radiation induced bystander response in normal human fibroblasts and colon carcinoma cells. Int. J. Radiat. Biol. 2010, 86, 102–113. [Google Scholar] [CrossRef]
- Sowa, M.B.; Goetz, W.; Baulch, J.E.; Lewis, A.J.; Morgan, W.F. No evidence for a low linear energy transfer adaptive response in irradiated RKO cells. Radiat. Prot. Dos. 2011, 143, 311–314. [Google Scholar] [CrossRef]
- Ilnytskyy, Y.; Kovalchuk, O. Non-targeted radiation effects-An epigenetic connection. Mutat. Res. 2011, 714, 113–125. [Google Scholar] [CrossRef]
- Manti, L.; Jamali, M.; Prise, K.M.; Michael, B.D.; Trott, K.R. Genomic Instability in Chinese Hamster Cells After Exposure to X Rays or Alpha Particles of Different Mean Linear Energy Transfer. Radiat. Res. 1997, 147, 22–28. [Google Scholar] [CrossRef]
- Morgan, W.F.; Corcoran, J.; Hartmann, A.; Kaplan, M.I.; Limoli, C.L.; Ponnaiya, B. DNA double-strand breaks, chromosomal rearrangements, and genomic instability. Mutat. Res. 1998, 404, 125–128. [Google Scholar] [CrossRef]
- Snyder, A.R.; Morgan, W.F. Differential induction and activation of NF-κB transcription complexes in radiation-induced chromosomally unstable cell lines. Environ. Mol. Mut. 2005, 45, 177–187. [Google Scholar] [CrossRef]
- Bright, S.; Kadhim, M. The future impacts of non-targeted effects. Int. J. Radiat. Biol. 2018, 94, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Merrifield, M.; Kovalchuk, O. Epigenetics in radiation biology: A new research frontier. Front. Genet. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Rugo, R.E.; Mutamba, J.T.; Mohan, K.; Yee, T.; Chaillet, J.R.; Greenberger, J.S.; Engelward, B.P. Methyltransferases mediate cell memory of a genotoxic insult. Oncogene 2011, 30, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Sisakht, M.; Darabian, M.; Mahmoodzadeh, A.; Bazi, A.; Shafiee, S.M.; Mokarram, P.; Khoshdel, Z. The role of radiation induced oxidative stress as a regulator of radio-adaptive responses. Int. J. Radiat. Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Filkowski, J.N.; Ilnytskyy, Y.; Tamminga, J.; Koturbash, I.; Golubov, A.; Bagnyukova, T.; Pogribny, I.P.; Kovalchuk, O. Hypomethylation and genome instability in the germline of exposed parents and their progeny is associated with altered miRNA expression. Carcinogenesis 2010, 1, 1110–1115. [Google Scholar] [CrossRef]
- Guéguen, Y.; Bontemps, A.; Ebrahimian, T.G. Adaptive responses to low doses of radiation or chemicals: Their cellular and molecular mechanisms. Cell. Mol. Life Sci. 2019, 76, 1255–1273. [Google Scholar] [CrossRef]
- Esposito, G.; Campa, A.; Pinto, M.; Simone, G.; Tabocchini, M.A.; Belli, M. Adaptive response: Modelling and experimental studies. Radiat. Prot. Dosim. 2011, 143, 320–324. [Google Scholar] [CrossRef]
- Timp, W.; Feinberg, A.P. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat. Rev. Cancer 2013, 13, 497–510. [Google Scholar] [CrossRef]
- Kim, J.G.; Park, M.T.; Heo, K.; Yang, K.M.; Yi, J.M. Epigenetics Meets Radiation Biology as a New Approach in Cancer Treatment. Int. J. Mol. Sci. 2013, 14, 15059–15073. [Google Scholar] [CrossRef]
- Altucci, L.; Clarke, N.; Nebbioso, A.; Scognamiglio, A.; Gronemeyer, H. Acute myeloid leukemia: Therapeutic impact of epigenetic drugs. Int. J. Biochem. Cell Biol. 2005, 37, 1752–1762. [Google Scholar] [CrossRef]
- Jones, P.A.; Gonzalgo, M.L. Altered DNA methylation and genome instability: A new pathway to cancer? Proc. Natl. Acad. Sci. USA 1997, 94, 2103–2105. [Google Scholar] [CrossRef] [PubMed]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instability in colorectal cancers. Nature 1997, 386, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Zingg, J.M.; Jones, P.A. Genetic and epigenetic aspects of DNA methylation on genome expression, evolution, mutation and carcinogenesis. Carcinogenesis 1997, 18, 869–882. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Epstein, R.J. Programmed genetic instability: A tumor-permissive mechanism for maintaining the evolvability of higher species through methylation-dependent mutation of DNA repair genes in the male germ line. Mol. Biol. Evol. 2008, 25, 1737–1749. [Google Scholar] [CrossRef][Green Version]
- Feinberg, A.P.; Tycko, B. The history of cancer epigenetics. Nat. Rev. Cancer 2004, 4, 143–153. [Google Scholar] [CrossRef]
- Rodriguez, J.; Frigola, J.; Vendrell, E.; Risques, R.A.; Fraga, M.F.; Morales, C.; Moreno, V.; Esteller, M.; Capellà, G.; Ribas, M.; et al. Chromosomal instability correlates with genome-wide DNA demethylation in human primary colorectal cancers. Cancer Res. 2006, 66, 8462–9468. [Google Scholar] [CrossRef]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability-an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef]
- Esteller, M.; Silva, J.M.; Dominguez, G.; Bonilla, F.; Matias-Guiu, X.; Lerma, E.; Bussaglia, E.; Prat, J.; Harkes, I.C.; Repasky, E.A.; et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J. Natl. Canc. Inst. 2000, 92, 564–569. [Google Scholar] [CrossRef]
- Kanwal, R.; Gupta, S. Epigenetics and cancer. J. Appl. Physiol. 2010, 109, 598–605. [Google Scholar] [CrossRef]
- Durso, D.F.; Bacalini, M.G.; Faria do Valle, I.; Pirazzini, C.; Bonafé, M.; Castellani, G.; Caetano Faria, A.M.; Franceschi, C.; Garagnani, P.; Nardini, C. Aberrant methylation patterns in colorectal cancer: A meta-analysis. Oncotarget 2017, 8, 12820–12830. [Google Scholar] [CrossRef]
- Romanenko, A.; Morell-Quadreny, L.; Lopez-Guerrero, J.A.; Pellin, A.; Nepomnyaschy, V.; Vozianov, A.; Llombart-Bosch, A. The INK4a/ARF locus: Role in cell cycle control for renal cell epithelial tumor growth after the Chernobyl accident. Virchows Arch. 2004, 445, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Little, J.B. Radiation carcinogenesis. Carcinogenesis 2000, 21, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Takai, D.; Jones, P.A. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc. Natl. Acad. Sci. USA 2002, 99, 3740–3745. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef]
- Seligson, D.B.; Horvath, S.; Shi, T.; Yu, H.; Tze, S.; Grunstein, M.; Kurdistani, S.K. Global histone modification patterns predict risk of prostate cancer recurrence. Nature 2005, 435, 1262–1266. [Google Scholar] [CrossRef]
- Jones, P.; Issa, J.; Baylin, S. Targeting the cancer epigenome for therapy. Nat. Rev. Genet. 2016, 17, 630–641. [Google Scholar] [CrossRef]
- Zhao, Z.; Shilatifard, A. Epigenetic modifications of histones in cancer. Genome Biol. 2019, 20, 245. [Google Scholar] [CrossRef]
- Dhall, A.; Zee, B.M.; Yan, F.; Blanco, M.A. Intersection of Epigenetic and Metabolic Regulation of Histone Modifications in Acute Myeloid Leukemia. Front. Oncol. 2019, 9, 432. [Google Scholar] [CrossRef]
- Reddy, K.B. MicroRNA (miRNA) in cancer. Cancer Cell Int. 2015, 15, 38. [Google Scholar] [CrossRef]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef]
- Chen, X.; Zhao, C.; Zhao, Z.; Wang, H.; Fang, Z. Specific Glioma Prognostic Subtype Distinctions Based on DNA Methylation Patterns. Front. Genet. 2019, 10, 786. [Google Scholar] [CrossRef] [PubMed]
- Tanno, B.; Babini, G.; Leonardi, S.; Giardullo, P.; De Stefano, I.; Pasquali, E.; Ottolenghi, A.; Atkinson, M.J.; Saran, A.; Mancuso, M. Ex vivo miRNome analysis in Ptch1+/− cerebellum granule cells reveals a subset of miRNAs involved in radiation-induced medulloblastoma. Oncotarget 2006, 7, 68253–68269. [Google Scholar] [CrossRef]
- Liu, C.; Li, B.; Cheng, Y.; Lin, J.; Hao, J.; Zhang, S.; Mitchel, R.E.J.; Sun, D.; Ni, J.; Zhao, L.; et al. MiR-21 plays an important role in radiation induced carcinogenesis in BALB/c mice by directly targeting the tumor suppressor gene Big-h3. Int. J. Biol. Sci. 2011, 7, 347–363. [Google Scholar] [CrossRef] [PubMed]
- Wilke, C.M.; Hess, J.; Klymenko, S.V.; Chumak, V.V.; Zakhartseva, L.M.; Bakhanova, E.V.; Feuchtinger, A.; Walch, A.K.; Selmansberger, M.; Braselmann, H.; et al. Expression of miRNA-26b-5p and its target TRPS1 is associated with radiation exposure in post-Chernobyl breast cancer. Int. J. Cancer 2018, 142, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef]
- Kamiya, K.; Yasukawa-Barnes, J.; Mitchen, J.M.; Gould, M.N.; Clifton, K.H. Evidence that carcinogenesis involves an imbalance between epigenetic high-frequency initiation and suppression of promotion. Proc. Natl. Acad. Sci. USA 1995, 92, 1332–1336. [Google Scholar] [CrossRef]
- Clifton, K.H. Comments on the evidence in support of the epigenetic nature of radiogenic initiation. Mutat. Res. 1996, 350, 77–80. [Google Scholar] [CrossRef]
- Feinberg, A.; Ohlsson, R.; Henikoff, S. The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 2006, 7, 21–33. [Google Scholar] [CrossRef]
- Jordan, C.T.; Guzman, M.L.; Noble, M. Cancer stem cells. N. Engl. J. Med. 2006, 355, 1253–1261. [Google Scholar] [CrossRef]
- Seibold, P.; Auvinen, A.; Averbeck, D.; Bourguignon, M.; Hartikainen, J.M.; Hoeschen, C.; Laurent, O.; Noël, G.; Sabatier, L.; Salomaa, S.; et al. Clinical and epidemiological observations on individual radiation sensitivity and susceptibility. Int. J. Radiat. Biol. 2020, 96, 324–339. [Google Scholar] [CrossRef] [PubMed]
- D’Urso, A.; Brickner, J.H. Mechanisms of epigenetic memory. Trends Genet. 2014, 30, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Little, M.P.; Goodhead, D.T.; Bridges, B.A.; Bouffler, S.D. Evidence relevant to untargeted and transgenerational effects in the offspring of irradiated parents. Mutat. Res. Rev. Mutat. Res. 2013, 753, 50–67. [Google Scholar] [CrossRef] [PubMed]
- Heard, E.; Martienssen, R.A. Transgenerational Epigenetic Inheritance: Myths and mechanisms. Cell 2014, 157, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Nelson, V.R.; Nadeau, J.H. Transgenerational genetic effects. Epigenomics 2010, 2, 797–806. [Google Scholar] [CrossRef]
- Reik, W.; Dean, W.; Walter, J. Epigenetic Reprogramming in Mammalian Development. Science 2001, 293, 1089. [Google Scholar] [CrossRef]
- Zeng, Y.; Chen, T. DNA Methylation Reprogramming during Mammalian Development. Genes 2019, 10, 257. [Google Scholar] [CrossRef]
- Luning, K.G.; Frolen, H.; Nilsson, A. Genetic effects of 239Pu salt injections in male mice. Mutat. Res. 1976, 34, 539–542. [Google Scholar] [CrossRef]
- Dubrova, Y.E. Radiation-induced transgenerational instability. Oncogene 2003, 22, 7087–7709. [Google Scholar] [CrossRef]
- Dubrova, Y.E.; Plumb, M.A. Ionising radiation and mutation induction at mouse minisatellite loci. The story of the two generations. Mutat. Res. 2002, 499, 143–150. [Google Scholar] [CrossRef]
- Kamstra, J.H.; Hurem, S.; Martin, L.M.; Lindeman, L.C.; Legler, J.; Oughton, D.; Salbu, B.; Brede, D.A.; Lyche, J.L.; Aleström, P. Ionizing radiation induces transgenerational effects of DNA methylation in zebrafish. Sci. Rep. 2018, 8, 15373. [Google Scholar] [CrossRef] [PubMed]
- Gardner, M.J.; Snee, M.P.; Hall, A.J.; Powell, C.A.; Downes, S.; Terrell, J.D. Results of case-control study of leukaemia and lymphoma among young people near Sellafield nuclear plant in West Cumbria. BMJ 1990, 300, 423–429. [Google Scholar] [CrossRef]
- Dubrova, Y.E.; Grant, G.; Chumak, A.A.; Stezhka, V.A.; Karakasian, A.N. Elevated minisatellite mutation rate in the post-Chernobyl families from Ukraine. Am. J. Hum. Genet. 2002, 71, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Dubrova, Y.E.; Bersimbaev, R.I.; Djansugurova, L.B.; Tankimanova, M.K.; Mamyrbaeva, Z.Z.; Mustonen, R.; Lindholm, C.; Hulten, M.; Salomaa, S. Nuclear weapons tests and human germline mutation rate. Science 2002, 295, 1037. [Google Scholar] [CrossRef]
- Dubrova, Y.E.; Ploshchanskaya, O.G.; Kozionova, O.S.; Akleyev, A.V. Minisatellite germline mutation rate in the Techa River population. Mutat. Res. 2006, 602, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Ozasa, K.; Cullings, K.M.; Ohishi, W.; Hida, A.; Grant, E.J. Epidemiological studies of atomic bomb radiation at the Radiation Effects Research Foundation. Int. J. Radiat. Biol. 2019, 95, 879–891. [Google Scholar] [CrossRef]
- Nomura, T.; Baleva, L.S.; Ryo, H.; Adachi, S.; Sipyagina, A.E.; Karakhan, N.M. Transgenerational effects of radiation on cancer and other disorders in mice and humans. J. Radiat. Cancer Res. 2017, 8, 123–134. [Google Scholar] [CrossRef]
- Beresford, N.A.; Horemans, N.; Copplestone, D.; Raines, K.E.; Orizaola, G.; Wood, M.D.; Laanen, P.; Whitehead, H.C.; Burrows, J.E.; Tinsley, M.C.; et al. Towards solving a scientific controversy—The effects of ionising radiation on the environment. J. Environ. Radioact. 2020, 211. [Google Scholar] [CrossRef]
- Horemans, N.; Spurgeon, D.J.; Lecomte-Pradines, C.; Saenen, E.; Bradshaw, C.; Oughton, D.; Rasnaca, I.; Kamstra, J.H.; Adam-Guillermin, C. Current evidence for a role of epigenetic mechanisms in response to ionizing radiation in an ecotoxicological context. Environ. Pollut. 2019, 251, 469–483. [Google Scholar] [CrossRef]
- United Nations Scientific Committee on the Effects of Atomic Radiation (UNSCEAR). UNSCEAR 2010 Report; United Nations: New York, NY, USA, 2011; pp. 1–14. [Google Scholar]
- United Nations Scientific Committee on the Effects of Atomic Radiation (UNSCEAR). Non-Targeted and Delayed Effects of Exposure to Ionizing Radiation. In UNSCEAR 2006 Report; United Nations: New York, NY, USA, 2006; Volume 2, pp. 1–79. [Google Scholar]
- ICRP. ICRP Publication 118: ICRP Statement on Tissue Reactions/Early and Late Effects of Radiation in Normal Tissues and Organs-Threshold Doses for Tissue Reactions in a Radiation Protection Context. Ann. ICRP 2012, 41, 1–322. [Google Scholar] [CrossRef]
- Meyers, C.A. Neurocognitive dysfunction in cancer patients. Oncology 2000, 14, 75–79. Available online: https://pubmed.ncbi.nlm.nih.gov/10680150/ (accessed on 18 August 2020).
- Syndikus, I.; Tait, D.; Ashley, S.; Jannoun, L. Long-term follow-up of young children with brain tumors after irradiation. Int. J. Radiat. Oncol. Biol. Phys. 1994, 30, 781–787. [Google Scholar] [CrossRef]
- Hall, P.; Adami, H.O.; Trichopoulos, D.; Pedersen, N.L.; Lagiou, P.; Ekbom, A.; Ingvar, M.; Lundell, M.; Granath, F. Effect of low doses of ionising radiation in infancy on cognitive function in adulthood: Swedish population based cohort study. BMJ 2004, 328, 19. [Google Scholar] [CrossRef]
- Otake, M.; Schull, W.J. Radiation-related brain damage and growth retardation among the prenatally exposed atomic bomb survivors. Int. J. Radia. T Biol. 1998, 74, 159–171. [Google Scholar] [CrossRef]
- Schull, W.J.; Otake, M. Cognitive function and prenatal exposure to ionizing radiation. Teratology 1999, 59, 222–226. [Google Scholar] [CrossRef]
- Little, M.P. A review of non-cancer effects, especially circulatory and ocular diseases. Radiat. Environ. Biophys. 2013, 52, 435–449. [Google Scholar] [CrossRef]
- Cucinotta, F.A.; Alp, M.; Sulzman, F.M.; Wang, M. Space radiation risks to the central nervous system. Life Sci. Space Res. 2014, 2, 54–69. [Google Scholar] [CrossRef]
- Parihar, V.K.; Allen, B.; Tran, K.K.; Macaraeg, T.G.; Chu, E.M.; Kwok, S.F.; Chmielewski, N.N.; Craver, B.M.; Baulch, J.E.; Acharya, M.M.; et al. What happens to your brain on the way to Mars. Sci. Adv. 2015, 1, e1400256. [Google Scholar] [CrossRef]
- Impey, S.; Jopson, T.; Pelz, C.; Tafessu, A.; Fareh, F.; Zuloaga, D.; Marzulla, T.; Riparip, L.K.; Stewart, B.; Rosi, S.; et al. Short- and long-term effects of 56Fe irradiation on cognition and hippocampal DNA methylation and gene expression. BMC Genom. 2016, 17, 825. [Google Scholar] [CrossRef]
- Acharya, M.M.; Baddour, A.A.D.; Kawashita, T.; Allen, B.D.; Syage, A.R.; Nguyen, T.H.; Yoon, N.; Giedzinski, E.; Yu, L.; Parihar, V.K.; et al. Epigenetic determinants of space radiation-induced cognitive dysfunction. Sci. Rep. 2017, 7, 42885. [Google Scholar] [CrossRef]
- Schultz-Hector, S.; Trott, K.R. Radiation-induced cardiovascular diseases: Is the epidemiologic evidence compatible with the radiobiologic data? Int. J. Radiat. Oncol. Biol. Phys. 2007, 67, 10–18. [Google Scholar] [CrossRef]
- Little, M.P.; Tawn, E.J.; Tzoulaki, I.; Wakeford, R.; Hildebrandt, G. A systematic review of epidemiological associations between low and moderate doses of ionizing radiation and late cardiovascular effects, and their possible mechanisms. Radiat. Res. 2008, 169, 99–109. [Google Scholar] [CrossRef]
- Mitchel, R.E.; Hasu, M.; Bugden, M.; Wyatt, H.; Little, M.P.; Gola, A.; Hildebrandt, G.; Priest, N.D.; Whitman, S.C. Low-dose radiation exposure and atherosclerosis in ApoE−/− mice. Radiat. Res. 2011, 175, 665–676. [Google Scholar] [CrossRef]
- Lowe, D.; Raj, K. Premature aging induced by radiation exhibits pro-atherosclerotic effects mediated by epigenetic activation of CD44 expression. Aging Cell 2014, 13, 900–910. [Google Scholar] [CrossRef]
- Koturbash, I.; Miousse, I.R.; Sridharan, V.; Nzabarushimana, E.; Skinner, C.M.; Melnyk, S.B.; Pavliv, O.; Hauer-Jensen, M.; Nelson, G.; Boerma, M. Radiation-induced changes in DNA methylation of repetitive elements in the mouse heart. Mutat. Res. 2016, 787, 43–53. [Google Scholar] [CrossRef]
- Liu, Y.C.; Wilkins, M.; Kim, T.; Malyugin, B.; Mehta, J.S. Cataracts. Lancet 2017, 390, 600–612. [Google Scholar] [CrossRef]
- Truscott, R.J.W. Age-related nuclear cataract—Oxidation is the key. Exp. Eye Res. 2005, 80, 709–725. [Google Scholar] [CrossRef]
- Blakely, E.A.; Kleiman, N.J.; Neriishi, K.; Chodick, G.; Chylack, L.T.; Cucinotta, F.A.; Minamoto, A.; Nakashima, E.; Kumagami, T.; Kitaoka, T.; et al. Radiation Cataractogenesis: Epidemiology and Biology. Radiat. Res. 2010, 173, 709–717. [Google Scholar] [CrossRef]
- Ainsbury, E.A.; Barnard, S.; Bright, S.; Dalke, C.; Jarrin, M.; Kunze, S.; Tanner, R.; Dynlacht, J.R.; Quinlan, R.; Graw, J.; et al. Ionizing radiation induced cataracts: Recent biological and mechanistic developments and perspectives for future research. Mutat Res. 2016, 770, 238–261. [Google Scholar] [CrossRef]
- Lanza, M.; Benincasa, G.; Costa, D.; Napoli, C. Clinical Role of Epigenetics and Network Analysis in Eye Diseases: A Translational Science Review. J. Ophthalmol. 2019, 2019, 2424956. [Google Scholar] [CrossRef]
- Alkozi, H.A.; Franco, R.; Pintor, J.J. Epigenetics in the Eye: An Overview of the Most Relevant Ocular Diseases. Front. Genet. 2017, 8, 144. [Google Scholar] [CrossRef] [PubMed]
- HLEG. High Level and Expert Group Report on European Low Dose Risk Research-Radiation Protection. European Commission EUR 23884; Luxembourg: Office for Official Publications of the European Communities. 2009. Available online: https://cordis.europa.eu/docs/publications/1070/107087891-6_en.pdf (accessed on 18 August 2020).
- Morgan, W.F.; Bair, W.J. Issues in Low Dose Radiation Biology: The Controversy Continues. A Perspective. Radiat. Res. 2013, 179, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Belli, M.; Tabocchini, M.A.; Jourdain, J.R.; Salomaa, S.; Repussard, J. The European initiative on low-dose risk research: From the HLEG to MELODI. Radiat. Prot. Dosimetry. 2015, 16, 178–181. [Google Scholar] [CrossRef] [PubMed]
- MELODI SRA Working Group. Strategic Research Agenda of the Multidisciplinary European Low Dose Initiative (MELODI)-2019. Available online: http://www.melodi-online.eu/m_docs_sra.html (accessed on 18 August 2020).
- Goodhead, D.T. Spatial and temporal distribution of energy. Health Phys. 1988, 55, 231–240. [Google Scholar] [CrossRef] [PubMed]
- United Nations Scientific Committee on the Effects of Atomic Radiation (UNSCEAR). Sources and Effects of Ionizing Radiation. In 1993 Report to the General Assembly, with Scientific Annexes; United Nations: New York, NY, USA, 1993. [Google Scholar]
- Wakeford, R.; Tawn, E.J. The meaning of low dose and low dose-rate. J. Radiol. Prot. 2010, 30, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Rothkamm, K.; Löbrich, M. Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses. Proc. Natl. Acad. Sci. USA 2003, 100, 5057–5062. [Google Scholar] [CrossRef]
- Grudzenski, S.; Raths, A.; Conrad, S.; Rübe, C.E.; Löbrich, M. Inducible response required for repair of low-dose radiation damage in human fibroblasts. Proc. Natl. Acad. Sci. USA 2010, 107, 14205–14210. [Google Scholar] [CrossRef]
- Amundson, S.A.; Do, K.T.; Fornace, A.J. Induction of Stress Genes by Low Doses of Gamma Rays. Radiat. Res. 1999, 152, 225. [Google Scholar] [CrossRef]
- Amundson, S.A.; Lee, R.A.; Koch-Paiz, C.A.; Bittner, M.L.; Meltzer, P.; Trent, J.M.; Fornace, A.J., Jr. Differential Responses of Stress Genes to Low Dose-Rate γ Irradiation. Mol. Cancer Res. 2003, 1, 445–452. [Google Scholar]
- Ding, L.H.; Shingyoji, M.; Chen, F.; Hwang, J.J.; Burma, S.; Lee, C.; Cheng, J.F.; Chen, D.J. Gene Expression Profiles of Normal Human Fibroblasts after Exposure to Ionizing Radiation: A Comparative Study of Low and High Doses. Radiat. Res. 2005, 164, 17–26. [Google Scholar] [CrossRef]
- Sokolov, M.; Neumann, R. Global Gene Expression Alterations as a Crucial Constituent of Human Cell Response to Low Doses of Ionizing Radiation Exposure. Int. J. Mol. Sci. 2016, 17, 55. [Google Scholar] [CrossRef] [PubMed]
- Mezentsev, A.; Amundson, S.A. Global Gene Expression Responses to Low- or High-Dose Radiation in a Human Three-Dimensional Tissue Model. Radiat. Res. 2011, 175, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, Z.; Rocke, D.M.; Schwietert, C.; Berglund, S.R.; Santana, A.; Jones, A.; Lehmann, J.; Stern, R.; Lu, R.; Siantar, C.H. Human in vivo dose-response to controlled, low-dose low linear energy transfer ionizing radiation exposure. Clin. Cancer Res. 2006, 12, 3723–3729. [Google Scholar] [CrossRef] [PubMed]
- Mitchel, R.E.; Jackson, J.S.; Morrison, D.P.; Carlisle, S.M. Low doses of radiation increase the latency of spontaneous lymphomas and spinal osteosarcomas in cancer-prone, radiation-sensitive Trp53 heterozygous mice. Radiat. Res. 2003, 159, 320–327. [Google Scholar] [CrossRef]
- Bernal, A.J.; Dolinoy, D.C.; Huang, D.; Skaar, D.A.; Weinhouse, C.; Jirtle, R.L. Adaptive radiation-induced epigenetic alterations mitigated by antioxidants. FASEB J. 2013, 27, 665–671. [Google Scholar] [CrossRef]
- Vaiserman, A.M. Hormesis and epigenetics: Is there a link? Ageing Res. Rev. 2011, 10, 413–421. [Google Scholar] [CrossRef]
- Kovalchuk, O.; Burke, P.; Besplug, J.; Slovack, M.; Filkowski, J.; Pogribny, I. Methylation changes in muscle and liver tissues of male and female mice exposed to acute and chronic low-dose X-ray-irradiation. Mutat. Res. 2004, 548, 75–84. [Google Scholar] [CrossRef]
- Taki, K.; Wang, B.; Nakajima, T.; Wu, J.; Ono, T.; Uehara, Y.; Matsumoto, T.; Oghiso, Y.; Tanaka, K.; Ichinohe, K.; et al. Microarray Analysis of Differentially Expressed Genes in the Kidneys and Testes of Mice after Long-term Irradiation with Low-dose-rate γ-rays. J. Radiat. Res. 2009, 50, 241–252. [Google Scholar] [CrossRef]
- Ye, S.; Yuan, D.; Xie, Y.; Pan, Y.; Shao, C. Role of DNA methylation in long-term low-dose γ-rays induced adaptive response in human B lymphoblast cells. Int. J. Radiat. Biol. 2013, 89, 898–906. [Google Scholar] [CrossRef]
- United Nations Scientific Committee on the Effects of Atomic Radiation (UNSCEAR). Sources and Effects of Ionizing Radiation. In 2008 Report to the General Assembly; United Nations: New York, NY, USA, 2010; Volume 1. [Google Scholar]
- Planel, H.; Soleilhavoup, J.P.; Tixador, R.; Richoilley, G.; Conter, A.; Croute, F.; Caratero, C.; Gaubin, Y. Influence on cell proliferation of background radiation or exposure to very low, chronic gamma radiation. Health Phys. 1987, 52, 571–578. [Google Scholar] [CrossRef]
- Smith, G.B.; Grof, Y.; Navarrette, A.; Guilmette, R.A. Exploring biological effects of low background radiation from the other side of the background. Health Phys. 2011, 100, 263–265. [Google Scholar] [CrossRef] [PubMed]
- Castillo, H.; Schoderbek, D.; Dulal, S.; Escobar, G.; Wood, J.; Nelson, R.; Smith, G. Stress induction in the bacteria Shewanella Oneidensis and Deinococcus Radiodurans in response to below-background ionizing radiation. Int. J. Radiat. Biol. 2015, 3002, 1–33. [Google Scholar] [CrossRef]
- Satta, L.; Augusti-Tocco, G.; Ceccarelli, R.; Esposito, A.; Fiore, M.; Paggi, P.; Poggesi, I.; Ricordy, R.; Scarsella, G.; Cundari, E. Low environmental radiation background impairs biological defence of the yeast Saccharomyces cerevisiae to chemical radiomimetic agents. Mutat. Res. 1995, 347, 129–133. [Google Scholar] [CrossRef]
- Satta, L.; Antonelli, F.; Belli, M.; Sapora, O.; Simone, G.; Sorrentino, E.; Tabocchini, M.A.; Amicarelli, F.; Ara, C.; Nisi, S. Influence of a low background radiation environment on biochemical and biological responses in V79 cells. Radiat. Environ. Biophys. 2002, 41, 217–224. [Google Scholar] [CrossRef]
- Carbone, M.C.; Pinto, M.; Antonelli, F.; Amicarelli, F.; Balata, M.; Belli, M.; Conti Devirgiliis, L.; Sapora, O.; Simone, G.; Sorrentino, E.; et al. Effects of deprivation of background environmental radiation on cultured human cells. II Nuovo Cim. 2010, 4, 469–477. [Google Scholar] [CrossRef]
- Fratini, E.; Carbone, C.; Capece, D.; Esposito, G.; Simone, G.; Tabocchini, M.A.; Tomasi, M.; Belli, M.; Satta, L. Low-radiation environment affects the development of protection mechanisms in V79 cells. Radiat. Environ. Biophys. 2015. [Google Scholar] [CrossRef]
- Morciano, P.; Iorio, R.; Iovino, D.; Cipressa, F.; Esposito, G.; Porrazzo, A.; Satta, L.; Alesse, E.; Tabocchini, M.A.; Cenci, G. Effects of reduced natural background radiation on Drosophila melanogaster growth and development as revealed by the FLYINGLOW program. J. Cell Physiol. 2018, 233, 23–29. [Google Scholar] [CrossRef]
- Morciano, P.; Cipressa, F.; Porrazzo, A.; Esposito, G.; Tabocchini, M.A.; Cenci, G. Fruit flies provide new insights in low radiation background biology at the INFN underground Gran Sasso National Laboratory (LNGS). Radiat. Res. 2018, 190, 217–225. [Google Scholar] [CrossRef]
- Little, M.P.; Tawn, E.J.; Tzoulaki, I.; Hildebrandt, G.; Paris, F.; Tapio, S.; Elliott, P. Review and meta-analysis of epidemiological associations between low/moderate doses of ionizing radiation and circulatory disease risks, and their possible mechanisms. Radiat. Environ. Biophys. 2010, 49, 139–153. [Google Scholar] [CrossRef]
- Vaiserman, A.M. Hormesis, adaptive epigenetic reorganization, and implications for human health and longevity. Dose Response 2010, 8, 16–21. [Google Scholar] [CrossRef]
- Azzam, E.I.; Colangelo, N.W.; Domogauer, J.D.; Sharma, N.; de Toledo, S.M. Is Ionizing Radiation Harmful at any Exposure? An Echo That Continues to Vibrate. Health Phys. 2016, 110, 249–251. [Google Scholar] [CrossRef] [PubMed]
- Vaiserman, A.; Koliada, A.; Zabuga, O.; Socol, Y. Health Impacts of Low-Dose Ionizing Radiation: Current Scientific Debates and Regulatory Issues. Dose Response 2018, 16, 1559325818796331. [Google Scholar] [CrossRef] [PubMed]
- Feinendegen, L.E. Evidence for beneficial low level radiation effects and radiation hormesis. Br. J. Radiol. 2005, 78, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J.; O’Connor, M.K. Estimating risk of low radiation doses—A critical review of the BEIR VII report and its use of the linear no-threshold (LNT) hypothesis. Radiat. Res. 2014, 182, 463–474. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belli, M.; Tabocchini, M.A. Ionizing Radiation-Induced Epigenetic Modifications and Their Relevance to Radiation Protection. Int. J. Mol. Sci. 2020, 21, 5993. https://doi.org/10.3390/ijms21175993
Belli M, Tabocchini MA. Ionizing Radiation-Induced Epigenetic Modifications and Their Relevance to Radiation Protection. International Journal of Molecular Sciences. 2020; 21(17):5993. https://doi.org/10.3390/ijms21175993
Chicago/Turabian StyleBelli, Mauro, and Maria Antonella Tabocchini. 2020. "Ionizing Radiation-Induced Epigenetic Modifications and Their Relevance to Radiation Protection" International Journal of Molecular Sciences 21, no. 17: 5993. https://doi.org/10.3390/ijms21175993
APA StyleBelli, M., & Tabocchini, M. A. (2020). Ionizing Radiation-Induced Epigenetic Modifications and Their Relevance to Radiation Protection. International Journal of Molecular Sciences, 21(17), 5993. https://doi.org/10.3390/ijms21175993