MicroRNAs Dysregulation and Mitochondrial Dysfunction in Neurodegenerative Diseases


 ,
,  ,
, 
 and
and
Abstract
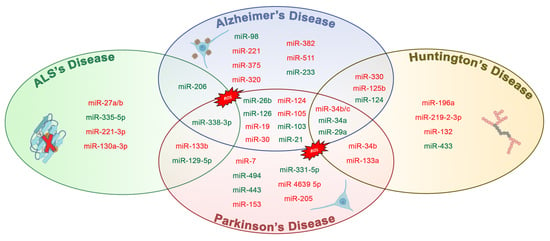
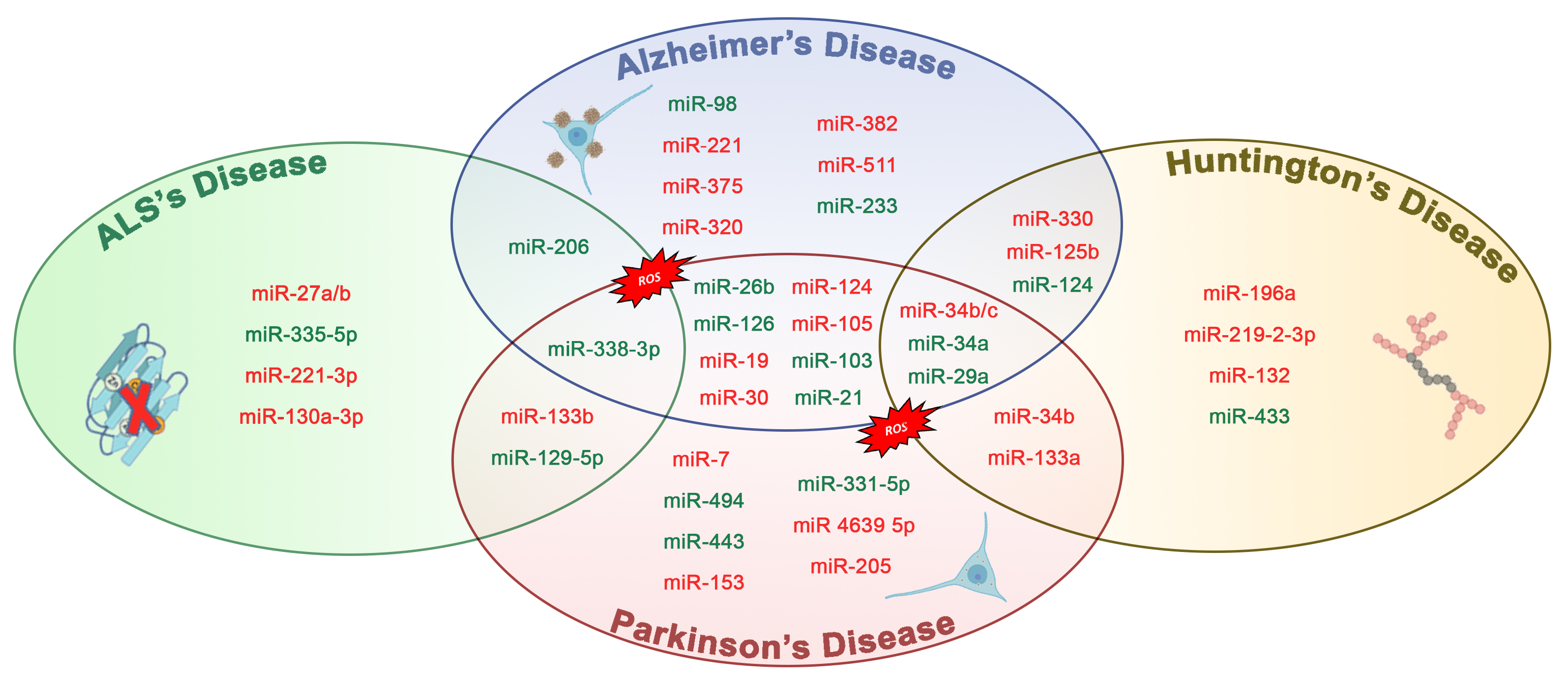
1. Introduction
2. miRNA Biology and Regulation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNAs | Role In | Target | Action | References |
|---|---|---|---|---|
| miR-26b ↑ | AD | Rb1 | Upregulation of Rb1/E2F cell cycle and proapoptotic transcriptional targets | [43] |
| miR-206 ↑ | AD | BDNF | Binds and downregulates specifically the 3-UTR region of BDNF | [44] |
| miR-34a ↑ | AD | VAMP2, SYT1, HCN1, NR2A, GLUR1, BCL2, | Targets genes linked to synaptic plasticity, energy metabolism, and resting state network activity | [45] |
| miR-126 ↑ | AD | IRS-1, PIK3R2 | Downregulated elements in the GF/PI3K/AKT and ERK signaling cascades | [46] |
| miR-9 ↓ | AD | Aβ | Increases production of γ-secretase and maintains neurons in sustaining Aβ production | [47] |
| miR-221 ↓ | AD | ADAM10 | Indirectly represses the 1 expression through the suppression of TIMP3 gene | [48] |
| miR-98 ↑ | AD | HEY2 | Reduces oxidative stress and mitochondrial dysfunction through the Notch signaling pathway via HEY2 protein | [49] |
| miR-330 ↓ | AD | VAV1 | Reduces oxidative stress and mitochondrial dysfunction through the MAPK pathway | [50] |
| miR-19 ↓ | AD | BACE1 | Alters PTEN/AKT/p53 pathway | [51] |
| miR-30 ↓ | AD | P53, DRP1 | Involved in regulating mitochondrial dynamics via Drp1 through p53 | [17] |
| miR-375 ↓ | AD | p53 | Increases the expression of TP53 gene through the 3-UTR region | [52] |
| miR-7 ↓ | PD | α-Synuclein, VDAC1 | Binds and downregulates specifically the 3-UTR region of synuclein | [53] |
| miR-124 ↓ | PD | FOXOa2 | Binds and downregulates specifically to the 3-UTR region of FOXO gene its | [54] |
| miR-153 ↓ | PD | α-Synuclein | Bind specifically and downregulates the 3-UTR region of synuclein | [55] |
| miR-443 ↑ | PD | FGF20 | Increases FGF20 mRNA translation that increases α-synuclein expression | [56] |
| miR-494 ↑, miR-34b/c ↓ | PD | DJ-1 | Binds and downregulates specifically the 3-UTR region of DJ.1 | [57,58] |
| miR-205 ↓ | PD | LRKK2 | Suppresses the expression of LRRK2 protein through the 3-UTR region | [59] |
| miR-27a ↓, miR-105 ↓ | PD | ATP5G3 (complex V) | TNF-α regulates the expression of miR-27a which regulates the transcript levels of ATP5G3 | [60] |
| miR-103 ↑ | PD | Complex I | TNF-α regulates the expression of mir-103 which regulates the transcript levels of complex I | [60] |
| miR 4639 5p ↓ | PD | DJ-1 | Suppresses the expression of PARK7 protein through the 3-UTR region | [61] |
| miR-21 ↑ | PD | BCL-2, PTEN | Suppresses the expression of BCL-2 and PTEN protein through the 3-UTR region | [62] |
| miR-331-5p ↑ | PD | NRP2 | Downregulation through the NRP2 gene | [63] |
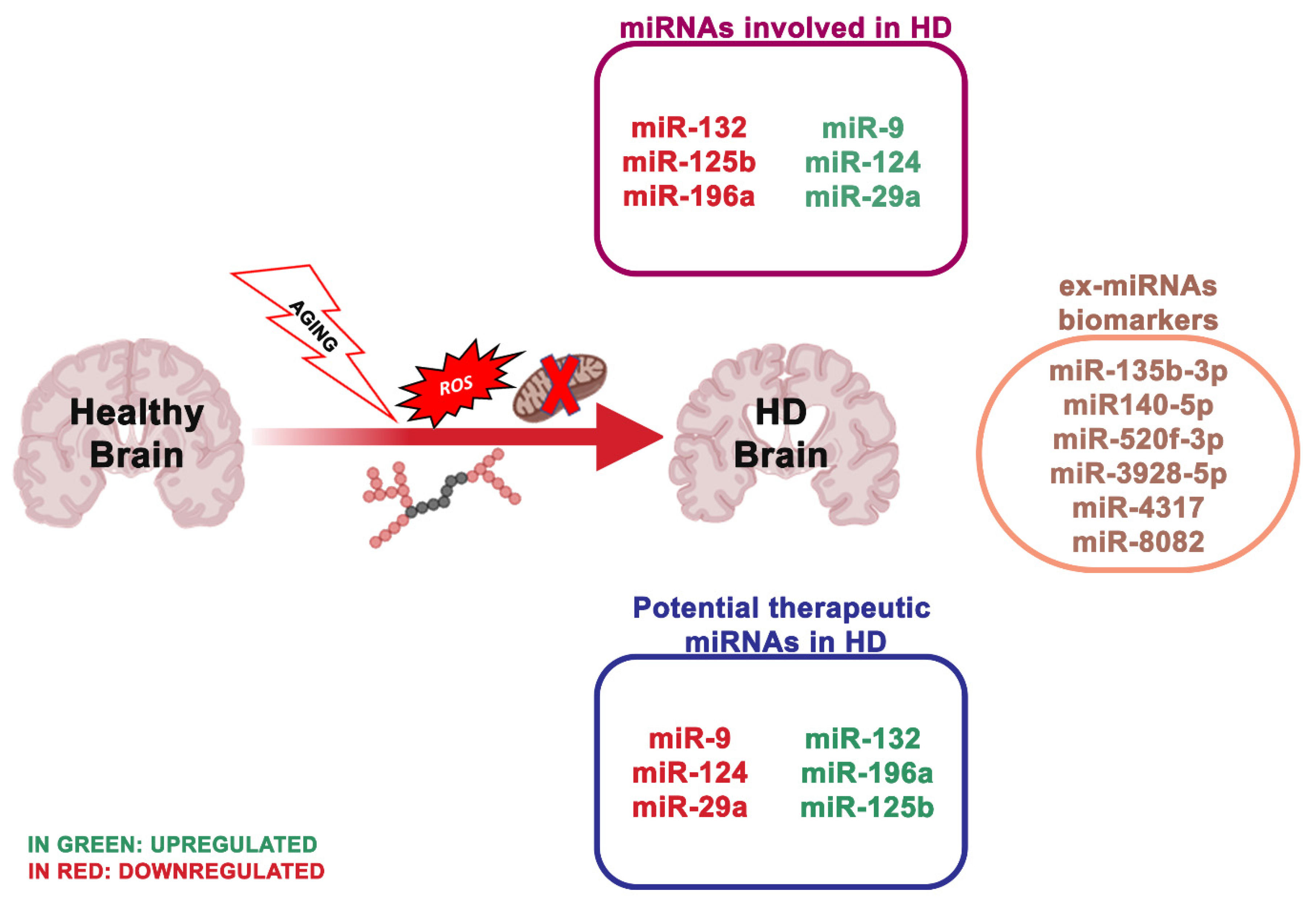
| miR-132 ↓ | HD | p250GAP, ACHE | Enhances neurite outgrowth and breakdown of the neurotransmitter acetylcholine through the dysregulation by p250GAP/ACHE | [64] |
| miR-125b ↓, miR-196a ↓, miR-146a ↓ | HD | mHTT | CAG length-dependent changes in miRNA expression in brain | [65,66,67] |
| miR-9 ↑, miR-124 ↑, miR-29a ↑ | HD | REST | Suppresses the expression of REST protein through the 3-UTR region | [68,69] |
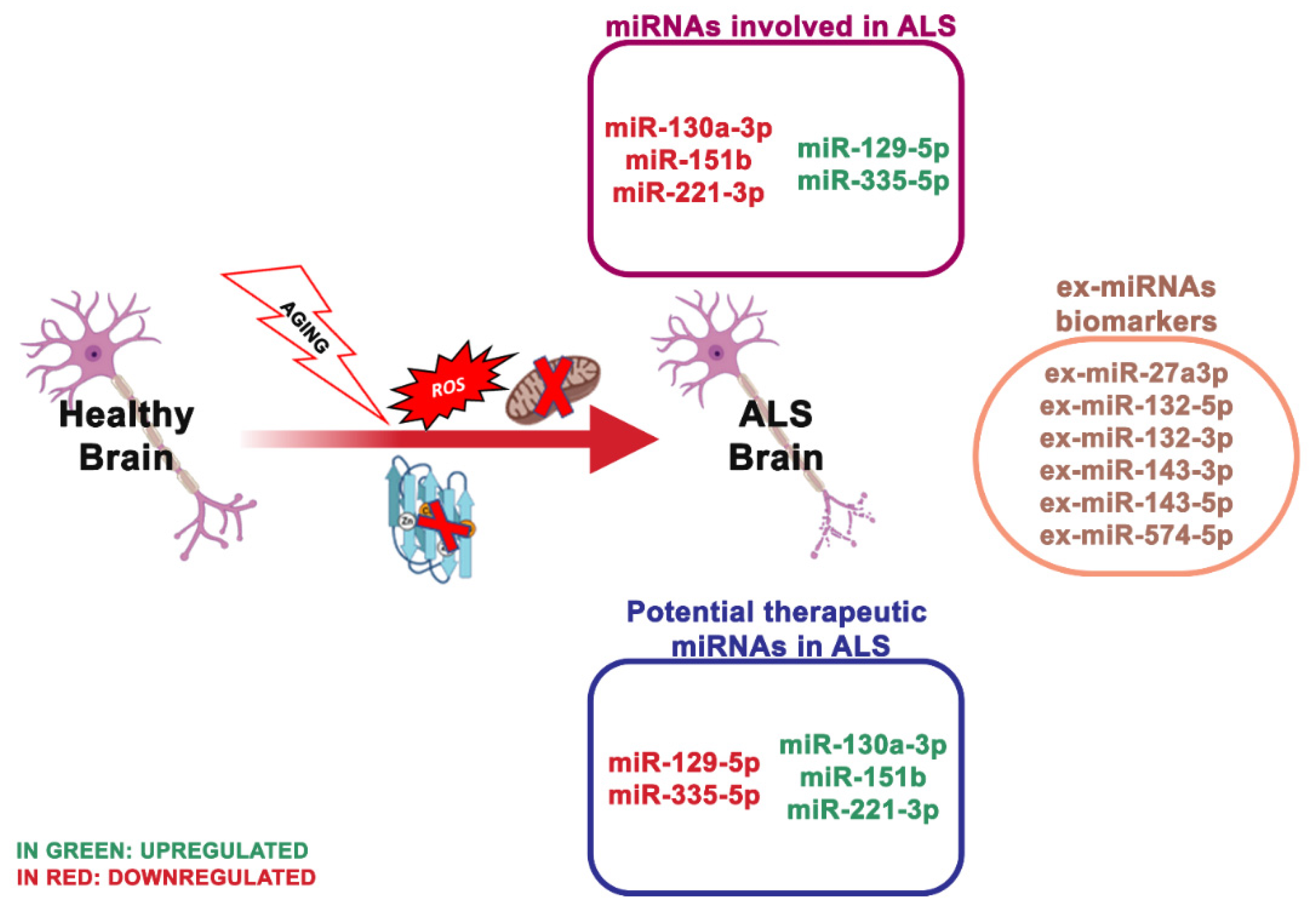
| miR-129-5p ↑, miR-133a ↓ | ALS | SOD1 | Suppresses the expression of SOD1 protein through the 3-UTR region | [70,71] |
| miR-27a-b, miR-335-5p ↑ | ALS | Caspase 3/7 | Alters mitochondrial dynamics and activates caspase 3/7 | [72] |
| miR-151b ↓ | ALS | PINK1, UCP2, PKM | Determine the downregulation of the expression of these genes | [73] |
| miR-221-3p ↓ | ALS | BAX, ITGA5, PRKCD, | Determine the downregulation of the expression of these genes | |
| miR-130a-3p ↓ | ALS | ABCG1, LGALS3, CTDSP1 | Determine the downregulation of the expression of these genes |
3. miRNA and Aging
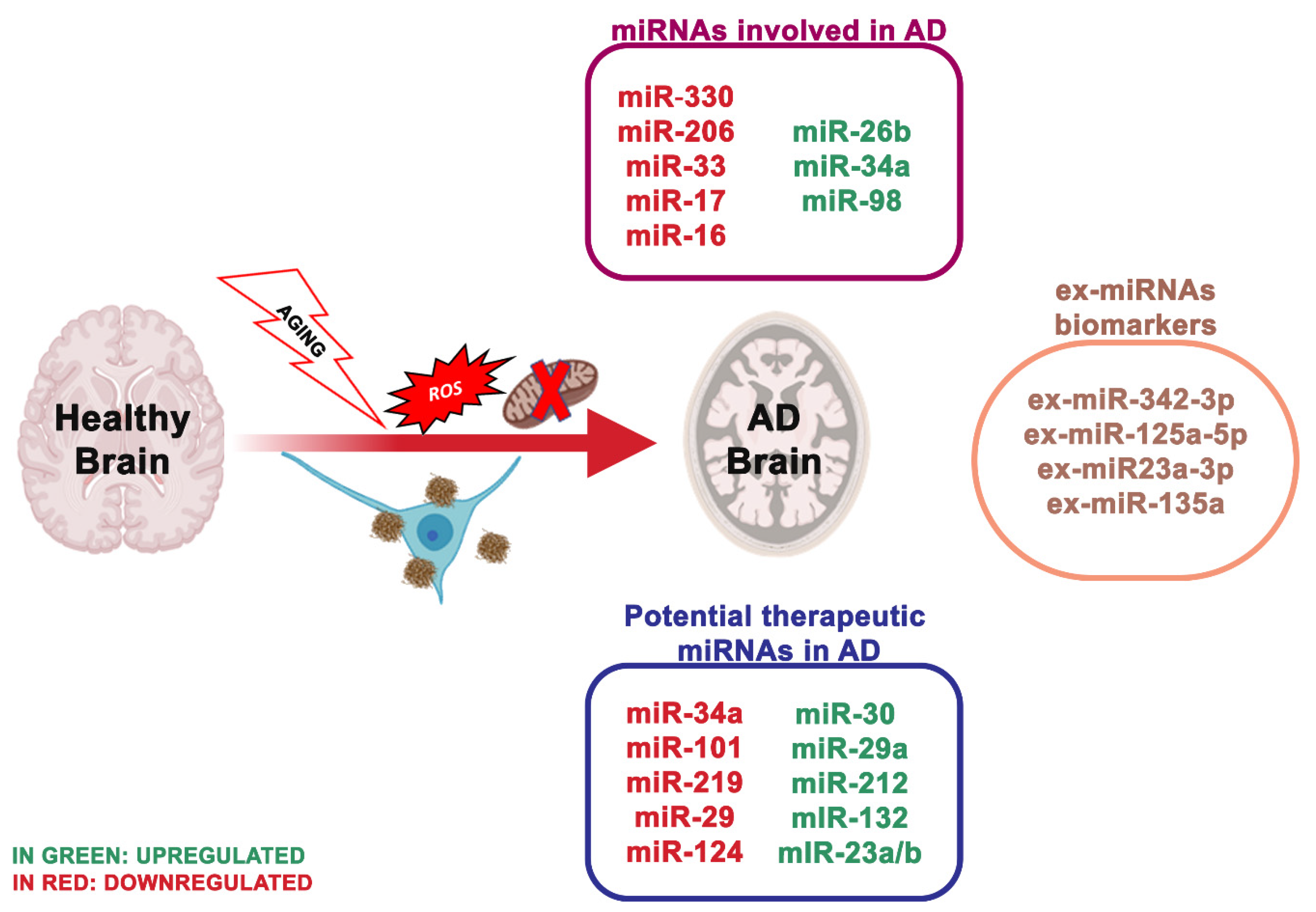
4. miRNA Dysregulation and Mitochondrial Dysfunction in Alzheimer’s
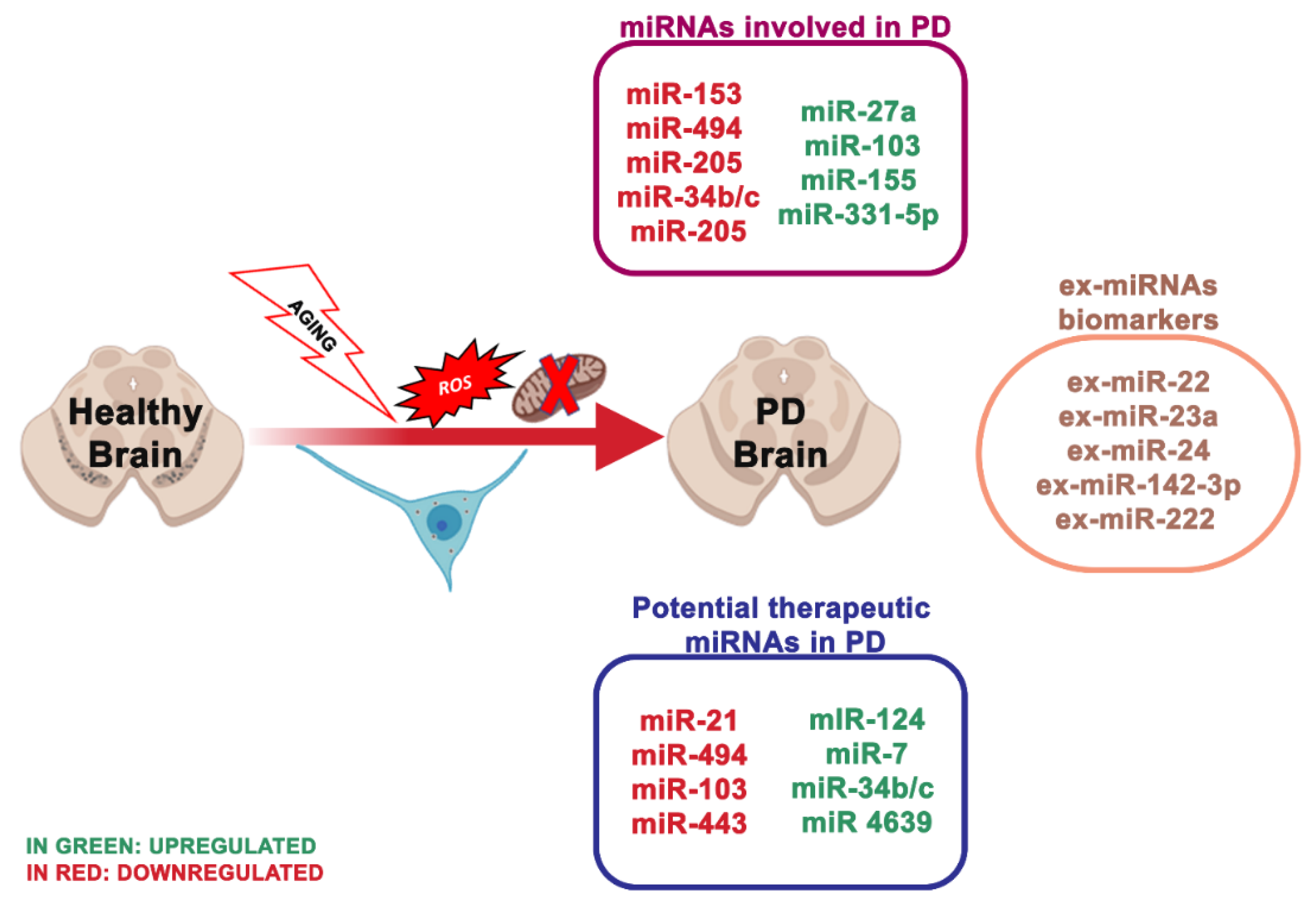
4.1. miRNA Dysregulation and Mitochondrial Dysfunction in Parkinson
- miR-133b that controls the transcriptional activator Pitx3, which is a crucial component in the development of the DA neuronal phenotype in vivo [74];
- miR-7, which suppressed α-synuclein in human neuroblastoma cells, and it may be inhibited by oxidative stress in vitro and in vivo [53].
- miR153, conserved across vertebrate species, and inhibited from α-synuclein [55];
- miR-433, related to a mutation of its binding region in the 3′UTR region of the FGF20 gene. miR-433 prevented the translation of the FGF 20 gene in vitro. Single nucleotide polymorphism in FGF20 shows that the genetic variability of FGF20 may represent a PD risk [56].
- miR-205, transfecting miR-205 in the neurons expressing a PD-related LRKK2 R1441G mutant avoided the neural development defects [123].
4.2. miRNA Dysregulation and Mitochondrial Dysfunction in Huntington’s Disease
4.3. miRNA Dysregulation and Mitochondrial Dysfunction in ALS
5. Discussion and Future Perspective
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| (qRT)-PCR | Real-Time Quantitative Reverse Transcription PCR |
| 3-NPA | Beta-Nitropropionic acid |
| ACHE | Acetylcholinesterase |
| AD | Alzheimer’s disease |
| AGO | Argonaute protein |
| ALS | Amyotrophic lateral sclerosis |
| APP | Amyloid precursor protein |
| ASO | Antisense oligonucleotide |
| ATP5G3 | ATP synthase membrane subunit c locus 3 |
| Aβ1–42 | Amyloid β-Peptide 1-42 |
| BAX | bcl-2-like protein 4 |
| BCl2 | B-cell lymphoma 2 |
| BIM | bcl-2-like protein 11 |
| C9ORF72 | Chromosome 9 open reading frame 72 |
| COX | Cyclooxygenase |
| CpG | Regions with a high frequency of CpG sites |
| CSF | Cerebrospinal fluid |
| CNS | central nervous system |
| DA | Dopaminergic neuron |
| DAT | Dopamine active transporter |
| DRP1 | dynamin-related protein 1 |
| EEs | Early endosomes |
| ERK1 | Extracellular Signal-Regulated Kinase |
| ETC | electrons transport chain |
| ex-miRNA | Exosome miRNA |
| FALS | Familial forms of ALS |
| FGF20 | Fibroblast Growth Factor 20 |
| FOXA2 | Forkhead box protein A2 |
| FUS | Fused in Sarcoma |
| HD | Huntington’s disease |
| HEY2 | Hairy/enhancer-of-split related with YRPW motif protein 1 |
| IGF-1 | Insulin-like growth factor 1 |
| iPD | Idiopathic Parkinson’s disease |
| JNK1 | c-Jun N-terminal kinase |
| LES | Late endosomes |
| LOAD | Late onset Alzheimer’s disease |
| LRP-1 | Low density lipoprotein receptor-related protein 1 |
| LRRK2 | Leucine-rich repeat kinase 2 |
| MAPK | mitogen-activated protein kinase |
| mHTT | Mutant human huntingtin |
| miRISC | miRNA RISC complex |
| MPP+ | 1-Methyl-4-phenylpyridine |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| MVBs | Multivesicular bodies |
| NADPH | Nicotinamide adenine dinucleotide phosphate (reduced form) |
| NOTCH1 | Notch homolog 1, translocation-associated |
| OMM | Outer mitochondrial membrane |
| P38 | P38 mitogen-activated protein kinases |
| PARIS | Zinc finger protein 746 |
| PBMCs | Peripheral blood mononuclear cells |
| PD | Parkinson’s disease |
| PGC1α | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| PI3K/Akt | Phosphatidylinositol-3-Kinase and Protein Kinase B |
| PINK1 | PTEN-induced kinase 1 |
| PITX3 | Pituitary homeobox 3 |
| polyQ | Poly glutamine |
| pri-miRNA | Primary transcript |
| R1441G | disease-linked leucine-rich repeat kinase 2 |
| RAB | Ras-associated binding |
| RISC | RNA Induced Silencing Complex |
| SALS | Sporadic form of ALS |
| SASP | Senescence-associated secretory phenotype |
| SIRT4 | Sirtuin 4 |
| SNpc | substantia nigra pars compacta |
| SNX6 | Sorting Nexin 6 |
| SOD1 | Superoxide dismutase [Cu-Zn] |
| TARDBP | TAR DNA-binding protein 43 |
| TLR | Toll-like receptor |
| TNFα | Tumor necrosis factor-α |
| UTR | Untranslated region |
| VAV1 | Proto-oncogene vav |
| VDAC1 | Voltage-dependent anion-selective channel 1 |
| YOAD | Young onset Alzheimer’s disease |
References
- Castelli, V.; Benedetti, E.; Antonosante, A.; Catanesi, M.; Pitari, G.; Ippoliti, R.; Cimini, A.; d’Angelo, M. Neuronal Cells Rearrangement During Aging and Neurodegenerative Disease: Metabolism, Oxidative Stress and Organelles Dynamic. Front. Mol. Neurosci. 2019, 12, 132. [Google Scholar] [CrossRef] [PubMed]
- Castelli, V.; Melani, F.; Ferri, C.; d’Angelo, M.; Catanesi, M.; Grassi, D.; Benedetti, E.; Giordano, A.; Cimini, A.; Desideri, G. Neuroprotective activities of bacopa, lycopene, astaxanthin, and vitamin B12 combination on oxidative stress-dependent neuronal death. J. Cell. Biochem. 2020. [Google Scholar] [CrossRef] [PubMed]
- GBD 2016 Neurology Collaborators. Global, regional, and national burden of neurological disorders, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480. [CrossRef]
- Beal, M.F. Bioenergetic approaches for neuroprotection in Parkinson’s disease. Ann. Neurol. 2003, 53 (Suppl. 3), S39–S48. [Google Scholar] [CrossRef]
- Catanesi, M.; d’Angelo, M.; Antonosante, A.; Castelli, V.; Alfonsetti, M.; Benedetti, E.; Desideri, G.; Ferri, C.; Cimini, A. Neuroprotective potential of choline alfoscerate against β-amyloid injury: Involvement of neurotrophic signals. Cell Biol. Int. 2020. [Google Scholar] [CrossRef] [PubMed]
- d’Angelo, M.; Castelli, V.; Catanesi, M.; Antonosante, A.; Dominguez-Benot, R.; Ippoliti, R.; Benedetti, E.; Cimini, A. PPARγ and Cognitive Performance. Int. J. Mol. Sci. 2019, 20, 5068. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Llave, C.; Xie, Z.; Kasschau, K.D.; Carrington, J.C. Cleavage of Scarecrow-like mRNA targets directed by a class of Arabidopsis miRNA. Science 2002, 297, 2053–2056. [Google Scholar] [CrossRef]
- Williams, J.; Smith, F.; Kumar, S.; Vijayan, M.; Reddy, P.H. Are microRNAs true sensors of ageing and cellular senescence? Ageing Res. Rev. 2017, 35, 350–363. [Google Scholar] [CrossRef]
- Puisségur, M.-P.; Mazure, N.M.; Bertero, T.; Pradelli, L.; Grosso, S.; Robbe-Sermesant, K.; Maurin, T.; Lebrigand, K.; Cardinaud, B.; Hofman, V.; et al. miR-210 is overexpressed in late stages of lung cancer and mediates mitochondrial alterations associated with modulation of HIF-1 activity. Cell Death Differ. 2011, 18, 465–478. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.Y.; Datta, S.; Bandeira, E.; Cano, M.; Mallick, E.; Rai, U.; Powell, B.; Tian, J.; Witwer, K.W.; Handa, J.T.; et al. Release of extracellular vesicle miR-494-3p by ARPE-19 cells with impaired mitochondria. Biochim. Biophys. Acta Gen. Subj. 2020, 129598. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Tchernyshyov, I.; Chang, T.-C.; Lee, Y.-S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [PubMed]
- Lang, A.; Grether-Beck, S.; Singh, M.; Kuck, F.; Jakob, S.; Kefalas, A.; Altinoluk-Hambüchen, S.; Graffmann, N.; Schneider, M.; Lindecke, A.; et al. MicroRNA-15b regulates mitochondrial ROS production and the senescence-associated secretory phenotype through sirtuin 4/SIRT4. Aging (Albany N. Y.) 2016, 8, 484–505. [Google Scholar] [CrossRef]
- Vasa-Nicotera, M.; Chen, H.; Tucci, P.; Yang, A.L.; Saintigny, G.; Menghini, R.; Mahè, C.; Agostini, M.; Knight, R.A.; Melino, G.; et al. miR-146a is modulated in human endothelial cell with aging. Atherosclerosis 2011, 217, 326–330. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, Q.; Niu, J.; Lu, K.; Xie, B.; Cui, D.; Xu, S. Screening of microRNAs associated with Alzheimer’s disease using oxidative stress cell model and different strains of senescence accelerated mice. J. Neurol. Sci. 2014, 338, 57–64. [Google Scholar] [CrossRef]
- Reddy, P.H.; Williams, J.; Smith, F.; Bhatti, J.S.; Kumar, S.; Vijayan, M.; Kandimalla, R.; Kuruva, C.S.; Wang, R.; Manczak, M.; et al. MicroRNAs, Aging, Cellular Senescence, and Alzheimer’s Disease. Prog. Mol. Biol. Transl. Sci. 2017, 146, 127–171. [Google Scholar] [CrossRef]
- Kumar, S.; Reddy, P.H. Are circulating microRNAs peripheral biomarkers for Alzheimer’s disease? Biochim. Biophys. Acta 2016, 1862, 1617–1627. [Google Scholar] [CrossRef]
- Jung, H.J.; Suh, Y. MicroRNA in Aging: From Discovery to Biology. Curr. Genom. 2012, 13, 548–557. [Google Scholar] [CrossRef]
- Cullen, B.R. Transcription and processing of human microRNA precursors. Mol. Cell 2004, 16, 861–865. [Google Scholar] [CrossRef]
- Lee, Y.S.; Nakahara, K.; Pham, J.W.; Kim, K.; He, Z.; Sontheimer, E.J.; Carthew, R.W. Distinct roles for Drosophila Dicer-1 and Dicer-2 in the siRNA/miRNA silencing pathways. Cell 2004, 117, 69–81. [Google Scholar] [CrossRef]
- Wu, J.; Yang, J.; Cho, W.C.; Zheng, Y. Argonaute proteins: Structural features, functions and emerging roles. J. Adv. Res. 2020, 24, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Okamura, K. Distinct roles for Argonaute proteins in small RNA-directed RNA cleavage pathways. Genes Dev. 2004, 18, 1655–1666. [Google Scholar] [CrossRef]
- Li, D.; Li, Y.-P.; Li, Y.-X.; Zhu, X.-H.; Du, X.-G.; Zhou, M.; Li, W.-B.; Deng, H.-Y. Effect of Regulatory Network of Exosomes and microRNAs on Neurodegenerative Diseases. Chin. Med. J. 2018, 131, 2216–2225. [Google Scholar] [CrossRef]
- Kourembanas, S. Exosomes: Vehicles of Intercellular Signaling, Biomarkers, and Vectors of Cell Therapy. Annu. Rev. Physiol. 2015, 77, 13–27. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef]
- Kowal, J.; Tkach, M.; Théry, C. Biogenesis and secretion of exosomes. Curr. Opin. Cell Biol. 2014, 29, 116–125. [Google Scholar] [CrossRef]
- Pant, S.; Hilton, H.; Burczynski, M.E. The multifaceted exosome: Biogenesis, role in normal and aberrant cellular function, and frontiers for pharmacological and biomarker opportunities. Biochem. Pharmacol. 2012, 83, 1484–1494. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, L. Circulating Exosomal miRNA as Diagnostic Biomarkers of Neurodegenerative Diseases. Front. Mol. Neurosci. 2020, 13, 53. [Google Scholar] [CrossRef]
- Ricci, C.; Marzocchi, C.; Battistini, S. MicroRNAs as Biomarkers in Amyotrophic Lateral Sclerosis. Cells 2018, 7, 219. [Google Scholar] [CrossRef] [PubMed]
- Roser, A.E.; Caldi Gomes, L.; Schünemann, J.; Maass, F.; Lingor, P. Circulating miRNAs as Diagnostic Biomarkers for Parkinson’s Disease. Front. Neurosci. 2018, 12, 625. [Google Scholar] [CrossRef] [PubMed]
- Mohr, A.; Mott, J. Overview of MicroRNA Biology. Semin. Liver Dis. 2015, 35, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Gantier, M.P.; McCoy, C.E.; Rusinova, I.; Saulep, D.; Wang, D.; Xu, D.; Irving, A.T.; Behlke, M.A.; Hertzog, P.J.; Mackay, F.; et al. Analysis of microRNA turnover in mammalian cells following Dicer1 ablation. Nucleic Acids Res. 2011, 39, 5692–5703. [Google Scholar] [CrossRef]
- Shao, N.-Y.; Hu, H.; Yan, Z.; Xu, Y.; Hu, H.; Menzel, C.; Li, N.; Chen, W.; Khaitovich, P. Comprehensive survey of human brain microRNA by deep sequencing. BMC Genom. 2010, 11, 409. [Google Scholar] [CrossRef] [PubMed]
- Adlakha, Y.K.; Saini, N. Brain microRNAs and insights into biological functions and therapeutic potential of brain enriched miRNA-128. Mol. Cancer 2014, 13, 33. [Google Scholar] [CrossRef]
- Nelson, P.T.; Wang, W.-X.; Rajeev, B.W. MicroRNAs (miRNAs) in Neurodegenerative Diseases. Brain Pathol. 2008, 18, 130–138. [Google Scholar] [CrossRef]
- Bian, S.; Sun, T. Functions of Noncoding RNAs in Neural Development and Neurological Diseases. Mol. Neurobiol. 2011, 44, 359–373. [Google Scholar] [CrossRef]
- Tan, L.; Yu, J.-T.; Tan, L. Causes and Consequences of MicroRNA Dysregulation in Neurodegenerative Diseases. Mol. Neurobiol. 2015, 51, 1249–1262. [Google Scholar] [CrossRef]
- Lagos-Quintana, M. Identification of Novel Genes Coding for Small Expressed RNAs. Science 2001, 294, 853–858. [Google Scholar] [CrossRef]
- Kapsimali, M.; Kloosterman, W.P.; de Bruijn, E.; Rosa, F.; Plasterk, R.H.; Wilson, S.W. MicroRNAs show a wide diversity of expression profiles in the developing and mature central nervous system. Genome Biol. 2007, 8, R173. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.-B. Posttranscriptional control of neuronal development by microRNA networks. Trends Neurosci. 2008, 31, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Absalon, S.; Kochanek, D.M.; Raghavan, V.; Krichevsky, A.M. MiR-26b, Upregulated in Alzheimer’s Disease, Activates Cell Cycle Entry, Tau-Phosphorylation, and Apoptosis in Postmitotic Neurons. J. Neurosci. 2013, 33, 14645–14659. [Google Scholar] [CrossRef] [PubMed]
- Tian, N.; Cao, Z.; Zhang, Y. MiR-206 decreases brain-derived neurotrophic factor levels in a transgenic mouse model of Alzheimer’s disease. Neurosci. Bull. 2014, 30, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Raymick, J.; Imam, S. Neuroprotective and Therapeutic Strategies against Parkinson’s Disease: Recent Perspectives. Int. J. Mol. Sci. 2016, 17, 904. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Lee, Y.; McKenna, N.D.; Yi, M.; Simunovic, F.; Wang, Y.; Kong, B.; Rooney, R.J.; Seo, H.; Stephens, R.M.; et al. miR-126 contributes to Parkinson’s disease by dysregulating the insulin-like growth factor/phosphoinositide 3-kinase signaling. Neurobiol. Aging 2014, 35, 1712–1721. [Google Scholar] [CrossRef]
- Krichevsky, A.M. A microRNA array reveals extensive regulation of microRNAs during brain development. RNA 2003, 9, 1274–1281. [Google Scholar] [CrossRef]
- Manzine, P.R.; Pelucchi, S.; Horst, M.A.; Vale, F.A.C.; Pavarini, S.C.I.; Audano, M.; Mitro, N.; Di Luca, M.; Marcello, E.; Cominetti, M.R. microRNA 221 Targets ADAM10 mRNA and is Downregulated in Alzheimer’s Disease. JAD 2017, 61, 113–123. [Google Scholar] [CrossRef]
- Chen, F.; Zhao, Y.; Chen, H. MicroRNA-98 reduces amyloid β-protein production and improves oxidative stress and mitochondrial dysfunction through the Notch signaling pathway via HEY2 in Alzheimer’s disease mice. Int. J. Mol. Med. 2018. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, Z.-F.; Li, W.; Hong, H.; Chen, J.; Tian, Y.; Liu, Z.-Y. Protective effects of microRNA-330 on amyloid β-protein production, oxidative stress, and mitochondrial dysfunction in Alzheimer’s disease by targeting VAV1 via the MAPK signaling pathway. J. Cell. Biochem. 2018, 119, 5437–5448. [Google Scholar] [CrossRef]
- Zhu, M.; Huang, C.; Ma, X.; Wu, R.; Zhu, W.; Li, X.; Liang, Z.; Deng, F.; Zhu, J.; Xie, W.; et al. Modulation of miR-19 in Aluminum-Induced Neural Cell Apoptosis. JAD 2016, 50, 1149–1162. [Google Scholar] [CrossRef] [PubMed]
- De Santis, R.; Santini, L.; Colantoni, A.; Peruzzi, G.; de Turris, V.; Alfano, V.; Bozzoni, I.; Rosa, A. FUS Mutant Human Motoneurons Display Altered Transcriptome and microRNA Pathways with Implications for ALS Pathogenesis. Stem Cell Rep. 2017, 9, 1450–1462. [Google Scholar] [CrossRef] [PubMed]
- Junn, E.; Lee, K.-W.; Jeong, B.S.; Chan, T.W.; Im, J.-Y.; Mouradian, M.M. Repression of -synuclein expression and toxicity by microRNA-7. Proc. Natl. Acad. Sci. USA 2009, 106, 13052–13057. [Google Scholar] [CrossRef] [PubMed]
- Kittappa, R.; Chang, W.W.; Awatramani, R.B.; McKay, R.D.G. The foxa2 Gene Controls the Birth and Spontaneous Degeneration of Dopamine Neurons in Old Age. PLoS Biol. 2007, 5, e325. [Google Scholar] [CrossRef]
- Doxakis, E. Post-transcriptional Regulation of α-Synuclein Expression by mir-7 and mir-153. J. Biol. Chem. 2010, 285, 12726–12734. [Google Scholar] [CrossRef]
- Itoh, N.; Ohta, H. Roles of FGF20 in dopaminergic neurons and Parkinson’s disease. Front. Mol. Neurosci. 2013, 6, 15. [Google Scholar] [CrossRef]
- Xiong, R.; Wang, Z.; Zhao, Z.; Li, H.; Chen, W.; Zhang, B.; Wang, L.; Wu, L.; Li, W.; Ding, J.; et al. MicroRNA-494 reduces DJ-1 expression and exacerbates neurodegeneration. Neurobiol. Aging 2014, 35, 705–714. [Google Scholar] [CrossRef]
- Miñones-Moyano, E.; Porta, S.; Escaramís, G.; Rabionet, R.; Iraola, S.; Kagerbauer, B.; Espinosa-Parrilla, Y.; Ferrer, I.; Estivill, X.; Martí, E. MicroRNA profiling of Parkinson’s disease brains identifies early downregulation of miR-34b/c which modulate mitochondrial function. Hum. Mol. Genet. 2011, 20, 3067–3078. [Google Scholar] [CrossRef]
- Cho, H.J.; Liu, G.; Jin, S.M.; Parisiadou, L.; Xie, C.; Yu, J.; Sun, L.; Ma, B.; Ding, J.; Vancraenenbroeck, R.; et al. MicroRNA-205 regulates the expression of Parkinson’s disease-related leucine-rich repeat kinase 2 protein. Hum. Mol. Genet. 2013, 22, 608–620. [Google Scholar] [CrossRef]
- Prajapati, P.; Sripada, L.; Singh, K.; Bhatelia, K.; Singh, R.; Singh, R. TNF-α regulates miRNA targeting mitochondrial complex-I and induces cell death in dopaminergic cells. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 451–461. [Google Scholar] [CrossRef]
- Chen, C.-M.; Orefice, L.L.; Chiu, S.-L.; LeGates, T.A.; Hattar, S.; Huganir, R.L.; Zhao, H.; Xu, B.; Kuruvilla, R. Wnt5a is essential for hippocampal dendritic maintenance and spatial learning and memory in adult mice. Proc. Natl. Acad. Sci. USA 2017, 114, E619–E628. [Google Scholar] [CrossRef]
- Gumireddy, K.; Young, D.D.; Xiong, X.; Hogenesch, J.B.; Huang, Q.; Deiters, A. Small-Molecule Inhibitors of MicroRNA miR-21 Function. Angew. Chem. Int. Ed. 2008, 47, 7482–7484. [Google Scholar] [CrossRef]
- Hunsberger, H.C.; Weitzner, D.S.; Rudy, C.C.; Hickman, J.E.; Libell, E.M.; Speer, R.R.; Gerhardt, G.A.; Reed, M.N. Riluzole rescues glutamate alterations, cognitive deficits, and tau pathology associated with P301L tau expression. J. Neurochem. 2015, 135, 381–394. [Google Scholar] [CrossRef]
- Vo, N.; Klein, M.E.; Varlamova, O.; Keller, D.M.; Yamamoto, T.; Goodman, R.H.; Impey, S. From The Cover: A cAMP-response element binding protein-induced microRNA regulates neuronal morphogenesis. Proc. Natl. Acad. Sci. USA 2005, 102, 16426–16431. [Google Scholar] [CrossRef]
- Cui, L.; Jeong, H.; Borovecki, F.; Parkhurst, C.N.; Tanese, N.; Krainc, D. Transcriptional Repression of PGC-1α by Mutant Huntingtin Leads to Mitochondrial Dysfunction and Neurodegeneration. Cell 2006, 127, 59–69. [Google Scholar] [CrossRef]
- Sinha, M.; Ghose, J.; Bhattarcharyya, N.P. Micro RNA -214,-150,-146a and-125b target Huntingtin gene. RNA Biol. 2011, 8, 1005–1021. [Google Scholar] [CrossRef]
- Cheng, P.-H.; Li, C.-L.; Chang, Y.-F.; Tsai, S.-J.; Lai, Y.-Y.; Chan, A.W.S.; Chen, C.-M.; Yang, S.-H. miR-196a Ameliorates Phenotypes of Huntington Disease in Cell, Transgenic Mouse, and Induced Pluripotent Stem Cell Models. Am. J. Hum. Genet. 2013, 93, 306–312. [Google Scholar] [CrossRef]
- Martí, E.; Pantano, L.; Bañez-Coronel, M.; Llorens, F.; Miñones-Moyano, E.; Porta, S.; Sumoy, L.; Ferrer, I.; Estivill, X. A myriad of miRNA variants in control and Huntington’s disease brain regions detected by massively parallel sequencing. Nucleic Acids Res. 2010, 38, 7219–7235. [Google Scholar] [CrossRef]
- Johnson, R.; Zuccato, C.; Belyaev, N.D.; Guest, D.J.; Cattaneo, E.; Buckley, N.J. A microRNA-based gene dysregulation pathway in Huntington’s disease. Neurobiol. Dis. 2008, 29, 438–445. [Google Scholar] [CrossRef]
- Loffreda, A.; Nizzardo, M.; Arosio, A.; Ruepp, M.-D.; Calogero, R.A.; Volinia, S.; Galasso, M.; Bendotti, C.; Ferrarese, C.; Lunetta, C.; et al. miR-129-5p: A key factor and therapeutic target in amyotrophic lateral sclerosis. Prog. Neurobiol. 2020, 190, 101803. [Google Scholar] [CrossRef]
- Dobrowolny, G.; Bernardini, C.; Martini, M.; Baranzini, M.; Barba, M.; Musarò, A. Muscle Expression of SOD1G93A Modulates microRNA and mRNA Transcription Pattern Associated with the Myelination Process in the Spinal Cord of Transgenic Mice. Front. Cell. Neurosci. 2015, 9. [Google Scholar] [CrossRef] [PubMed]
- De Luna, N.; Turon-Sans, J.; Cortes-Vicente, E.; Carrasco-Rozas, A.; Illán-Gala, I.; Dols-Icardo, O.; Clarimón, J.; Lleó, A.; Gallardo, E.; Illa, I.; et al. Downregulation of miR-335-5P in Amyotrophic Lateral Sclerosis Can Contribute to Neuronal Mitochondrial Dysfunction and Apoptosis. Sci. Rep. 2020, 10, 4308. [Google Scholar] [CrossRef] [PubMed]
- Liguori, M.; Nuzziello, N.; Introna, A.; Consiglio, A.; Licciulli, F.; D’Errico, E.; Scarafino, A.; Distaso, E.; Simone, I.L. Dysregulation of MicroRNAs and Target Genes Networks in Peripheral Blood of Patients With Sporadic Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2018, 11, 288. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Inoue, K.; Ishii, J.; Vanti, W.B.; Voronov, S.V.; Murchison, E.; Hannon, G.; Abeliovich, A. A MicroRNA Feedback Circuit in Midbrain Dopamine Neurons. Science 2007, 317, 1220–1224. [Google Scholar] [CrossRef]
- Singh, T.; Yadav, S. Role of microRNAs in neurodegeneration induced by environmental neurotoxicants and aging. Ageing Res. Rev. 2020, 60, 101068. [Google Scholar] [CrossRef]
- Caravia, X.M.; López-Otín, C. Regulatory Roles of miRNAs in Aging. Adv. Exp. Med. Biol. 2015, 887, 213–230. [Google Scholar] [CrossRef]
- Borgdorff, V.; Lleonart, M.E.; Bishop, C.L.; Fessart, D.; Bergin, A.H.; Overhoff, M.G.; Beach, D.H. Multiple microRNAs rescue from Ras-induced senescence by inhibiting p21(Waf1/Cip1). Oncogene 2010, 29, 2262–2271. [Google Scholar] [CrossRef]
- Overhoff, M.G.; Garbe, J.C.; Koh, J.; Stampfer, M.R.; Beach, D.H.; Bishop, C.L. Cellular senescence mediated by p16INK4A-coupled miRNA pathways. Nucleic Acids Res. 2014, 42, 1606–1618. [Google Scholar] [CrossRef]
- de Lencastre, A.; Pincus, Z.; Zhou, K.; Kato, M.; Lee, S.S.; Slack, F.J. MicroRNAs both promote and antagonize longevity in C. elegans. Curr. Biol. 2010, 20, 2159–2168. [Google Scholar] [CrossRef]
- Quesada, A.; Lee, B.Y.; Micevych, P.E. PI3 kinase/Akt activation mediates estrogen and IGF-1 nigral DA neuronal neuroprotection against a unilateral rat model of Parkinson’s disease. Dev. Neurobiol. 2008, 68, 632–644. [Google Scholar] [CrossRef]
- Sonntag, K.C.; Woo, T.-U.W.; Krichevsky, A.M. Converging miRNA functions in diverse brain disorders: A case for miR-124 and miR-126. Exp. Neurol. 2012, 235, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kim, M.S.; Jia, B.; Yan, J.; Zuniga-Hertz, J.P.; Han, C.; Cai, D. Hypothalamic stem cells control ageing speed partly through exosomal miRNAs. Nature 2017, 548, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, E.; Cristiano, L.; Antonosante, A.; d’Angelo, M.; D’Angelo, B.; Selli, S.; Castelli, V.; Ippoliti, R.; Giordano, A.; Cimini, A. PPARs in Neurodegenerative and Neuroinflammatory Pathways. CAR 2018, 15, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Pandey, A.; Shukla, A.; Talwelkar, S.S.; Kumar, A.; Pant, A.B.; Parmar, D. miR-497 and miR-302b regulate ethanol-induced neuronal cell death through BCL2 protein and cyclin D2. J. Biol. Chem. 2011, 286, 37347–37357. [Google Scholar] [CrossRef] [PubMed]
- Balaraman, S.; Winzer-Serhan, U.H.; Miranda, R.C. Opposing actions of ethanol and nicotine on microRNAs are mediated by nicotinic acetylcholine receptors in fetal cerebral cortical-derived neural progenitor cells. Alcohol. Clin. Exp. Res. 2012, 36, 1669–1677. [Google Scholar] [CrossRef]
- Weber, J.A.; Baxter, D.H.; Zhang, S.; Huang, D.Y.; How Huang, K.; Jen Lee, M.; Galas, D.J.; Wang, K. The MicroRNA Spectrum in 12 Body Fluids. Clin. Chem. 2010, 56, 1733–1741. [Google Scholar] [CrossRef]
- Kumar, S.; Vijayan, M.; Bhatti, J.S.; Reddy, P.H. MicroRNAs as Peripheral Biomarkers in Aging and Age-Related Diseases. Prog. Mol. Biol. Transl. Sci. 2017, 146, 47–94. [Google Scholar] [CrossRef]
- Sawada, S.; Akimoto, T.; Takahashi, M.; Sakurai, R.; Shinkai, S.; Ushida, T.; Fujiwara, Y.; Suzuki, K. Effect of Aging and Sex on Circulating MicroRNAs in Humans. AAR 2014, 3, 152–159. [Google Scholar] [CrossRef]
- Olivieri, F.; Bonafè, M.; Spazzafumo, L.; Gobbi, M.; Prattichizzo, F.; Recchioni, R.; Marcheselli, F.; Sala, L.L.; Galeazzi, R.; Rippo, M.R.; et al. Age- and glycemia-related miR-126-3p levels in plasma and endothelial cells. Aging 2014, 6, 771–786. [Google Scholar] [CrossRef]
- Ameling, S.; Kacprowski, T.; Chilukoti, R.K.; Malsch, C.; Liebscher, V.; Suhre, K.; Pietzner, M.; Friedrich, N.; Homuth, G.; Hammer, E.; et al. Associations of circulating plasma microRNAs with age, body mass index and sex in a population-based study. BMC Med. Genom. 2015, 8, 61. [Google Scholar] [CrossRef]
- Machida, T.; Tomofuji, T.; Ekuni, D.; Maruyama, T.; Yoneda, T.; Kawabata, Y.; Mizuno, H.; Miyai, H.; Kunitomo, M.; Morita, M. MicroRNAs in Salivary Exosome as Potential Biomarkers of Aging. Int. J. Mol. Sci. 2015, 16, 21294–21309. [Google Scholar] [CrossRef] [PubMed]
- Hooten, N.N.; Fitzpatrick, M.; Wood, W.H.; De, S.; Ejiogu, N.; Zhang, Y.; Mattison, J.A.; Becker, K.G.; Zonderman, A.B.; Evans, M.K. Age-related changes in microRNA levels in serum. Aging 2013, 5, 725–740. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.N. Simultaneous activation of Nrf2 and elevation of antioxidant compounds for reducing oxidative stress and chronic inflammation in human Alzheimer’s disease. Mech. Ageing Dev. 2016, 153, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Bonet-Costa, V.; Pomatto, L.C.-D.; Davies, K.J.A. The Proteasome and Oxidative Stress in Alzheimer’s Disease. Antioxid. Redox Signal. 2016, 25, 886–901. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Boyd-Kimball, D. Oxidative Stress, Amyloid-β Peptide, and Altered Key Molecular Pathways in the Pathogenesis and Progression of Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 1345–1367. [Google Scholar] [CrossRef] [PubMed]
- García-Escudero, V.; Martín-Maestro, P.; Perry, G.; Avila, J. Deconstructing mitochondrial dysfunction in Alzheimer disease. Oxid. Med. Cell. Longev. 2013, 2013, 162152. [Google Scholar] [CrossRef]
- Šimić, G.; Babić Leko, M.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; de Silva, R.; Di Giovanni, G.; et al. Tau Protein Hyperphosphorylation and Aggregation in Alzheimer’s Disease and Other Tauopathies, and Possible Neuroprotective Strategies. Biomolecules 2016, 6, 6. [Google Scholar] [CrossRef]
- Williamson, J.; Goldman, J.; Marder, K.S. Genetic aspects of Alzheimer disease. Neurologist 2009, 15, 80–86. [Google Scholar] [CrossRef]
- Blass, J.P. Brain metabolism and brain disease: Is metabolic deficiency the proximate cause of Alzheimer dementia? J. Neurosci. Res. 2001, 66, 851–856. [Google Scholar] [CrossRef]
- Malik, A.N.; Czajka, A. Is mitochondrial DNA content a potential biomarker of mitochondrial dysfunction? Mitochondrion 2013, 13, 481–492. [Google Scholar] [CrossRef]
- Brai, E.; Alina Raio, N.; Alberi, L. Notch1 hallmarks fibrillary depositions in sporadic Alzheimer’s disease. Acta Neuropathol. Commun. 2016, 4, 64. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Li, X.; Wang, L.; Zhang, Y.; Chen, L. miR-98-5p Acts as a Target for Alzheimer’s Disease by Regulating Aβ Production Through Modulating SNX6 Expression. J. Mol. Neurosci. 2016, 60, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.-K.; Wang, X.; Li, L.; Du, Y.-H.; Ye, H.-T.; Li, C.-Y. MicroRNA-98 induces an Alzheimer’s disease-like disturbance by targeting insulin-like growth factor 1. Neurosci. Bull. 2013, 29, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zhou, H.; Jiang, L.; Mao, Y.; Cui, X.; Xie, B.; Cui, D.; Wang, H.; Zhang, Q.; Xu, S. MiR-195 dependent roles of mitofusin2 in the mitochondrial dysfunction of hippocampal neurons in SAMP8 mice. Brain Res. 2016, 1652, 135–143. [Google Scholar] [CrossRef]
- Li, J.; Donath, S.; Li, Y.; Qin, D.; Prabhakar, B.S.; Li, P. miR-30 regulates mitochondrial fission through targeting p53 and the dynamin-related protein-1 pathway. PLoS Genet. 2010, 6, e1000795. [Google Scholar] [CrossRef]
- Ryan, M.M.; Guévremont, D.; Mockett, B.G.; Abraham, W.C.; Williams, J.M. Circulating Plasma microRNAs are Altered with Amyloidosis in a Mouse Model of Alzheimer’s Disease. JAD 2018, 66, 835–852. [Google Scholar] [CrossRef]
- Reddy, P.H.; Tonk, S.; Kumar, S.; Vijayan, M.; Kandimalla, R.; Kuruva, C.S.; Reddy, A.P. A critical evaluation of neuroprotective and neurodegenerative MicroRNAs in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2017, 483, 1156–1165. [Google Scholar] [CrossRef]
- Ravanidis, S.; Bougea, A.; Papagiannakis, N.; Maniati, M.; Koros, C.; Simitsi, A.; Bozi, M.; Pachi, I.; Stamelou, M.; Paraskevas, G.P.; et al. Circulating Brain-Enriched MicroRNAs for Detection and Discrimination of Idiopathic and Genetic Parkinson’s Disease. Mov. Disord. 2020, 35, 457–467. [Google Scholar] [CrossRef]
- Lugli, G.; Cohen, A.M.; Bennett, D.A.; Shah, R.C.; Fields, C.J.; Hernandez, A.G.; Smalheiser, N.R. Plasma Exosomal miRNAs in Persons with and without Alzheimer Disease: Altered Expression and Prospects for Biomarkers. PLoS ONE 2015, 10, e0139233. [Google Scholar] [CrossRef]
- Rani, A.; O’Shea, A.; Ianov, L.; Cohen, R.A.; Woods, A.J.; Foster, T.C. miRNA in Circulating Microvesicles as Biomarkers for Age-Related Cognitive Decline. Front. Aging Neurosci. 2017, 9, 323. [Google Scholar] [CrossRef]
- Gámez-Valero, A.; Campdelacreu, J.; Vilas, D.; Ispierto, L.; Reñé, R.; Álvarez, R.; Armengol, M.P.; Borràs, F.E.; Beyer, K. Exploratory study on microRNA profiles from plasma-derived extracellular vesicles in Alzheimer’s disease and dementia with Lewy bodies. Transl. Neurodegener. 2019, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.T.; Liu, C.G.; Gao, S.C.; Zhang, Y.; Wang, P.C. The Serum Exosome Derived MicroRNA-135a, -193b, and -384 Were Potential Alzheimer’s Disease Biomarkers. Biomed. Environ. Sci. 2018, 31, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Xu, Y.; Xu, W.; Zhou, Q.; Chen, Q.; Yang, M.; Feng, F.; Liu, Y.; Zhu, X.; Yu, M.; et al. Serum Exosomal miR-223 Serves as a Potential Diagnostic and Prognostic Biomarker for Dementia. Neuroscience 2018, 379, 167–176. [Google Scholar] [CrossRef]
- McKeever, P.M.; Schneider, R.; Taghdiri, F.; Weichert, A.; Multani, N.; Brown, R.A.; Boxer, A.L.; Karydas, A.; Miller, B.; Robertson, J.; et al. MicroRNA Expression Levels Are Altered in the Cerebrospinal Fluid of Patients with Young-Onset Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 8826–8841. [Google Scholar] [CrossRef] [PubMed]
- Riancho, J.; Vázquez-Higuera, J.L.; Pozueta, A.; Lage, C.; Kazimierczak, M.; Bravo, M.; Calero, M.; Gonalezález, A.; Rodríguez, E.; Lleó, A.; et al. MicroRNA Profile in Patients with Alzheimer’s Disease: Analysis of miR-9-5p and miR-598 in Raw and Exosome Enriched Cerebrospinal Fluid Samples. JAD 2017, 57, 483–491. [Google Scholar] [CrossRef]
- Grasso, M.; Piscopo, P.; Crestini, A.; Confaloni, A.; Denti, M.A. Circulating microRNAs in Neurodegenerative Diseases. Exp. Suppl. 2015, 106, 151–169. [Google Scholar] [CrossRef]
- Sanders, L.H.; Timothy Greenamyre, J. Oxidative damage to macromolecules in human Parkinson disease and the rotenone model. Free Radic. Biol. Med. 2013, 62, 111–120. [Google Scholar] [CrossRef]
- Subramaniam, S.R.; Chesselet, M.-F. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog. Neurobiol. 2013, 106–107, 17–32. [Google Scholar] [CrossRef]
- Hsu, L.J.; Sagara, Y.; Arroyo, A.; Rockenstein, E.; Sisk, A.; Mallory, M.; Wong, J.; Takenouchi, T.; Hashimoto, M.; Masliah, E. α-Synuclein Promotes Mitochondrial Deficit and Oxidative Stress. Am. J. Pathol. 2000, 157, 401–410. [Google Scholar] [CrossRef]
- Tansey, M.G.; McCoy, M.K.; Frank-Cannon, T.C. Neuroinflammatory mechanisms in Parkinson’s disease: Potential environmental triggers, pathways, and targets for early therapeutic intervention. Exp. Neurol. 2007, 208, 1–25. [Google Scholar] [CrossRef]
- Buendia, I.; Michalska, P.; Navarro, E.; Gameiro, I.; Egea, J.; León, R. Nrf2–ARE pathway: An emerging target against oxidative stress and neuroinflammation in neurodegenerative diseases. Pharmacol. Ther. 2016, 157, 84–104. [Google Scholar] [CrossRef] [PubMed]
- Hoss, A.G.; Labadorf, A.; Beach, T.G.; Latourelle, J.C.; Myers, R.H. microRNA Profiles in Parkinson’s Disease Prefrontal Cortex. Front. Aging Neurosci. 2016, 8, 36. [Google Scholar] [CrossRef]
- Gui, Y.; Liu, H.; Zhang, L.; Lv, W.; Hu, X. Altered microRNA profiles in cerebrospinal fluid exosome in Parkinson disease and Alzheimer disease. Oncotarget 2015, 6, 37043–37053. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Metzakopian, E.; Mavromatakis, Y.E.; Gao, N.; Balaskas, N.; Sasaki, H.; Briscoe, J.; Whitsett, J.A.; Goulding, M.; Kaestner, K.H.; et al. Foxa1 and Foxa2 function both upstream of and cooperatively with Lmx1a and Lmx1b in a feedforward loop promoting mesodiencephalic dopaminergic neuron development. Dev. Biol. 2009, 333, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.D.; Parks, J.K.; Swerdlow, R.H. Complex I deficiency in Parkinson’s disease frontal cortex. Brain Res. 2008, 1189, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ye, Y.; Zhu, Z.; Mo, L.; Lin, C.; Wang, Q.; Wang, H.; Gong, X.; He, X.; Lu, G.; et al. MiR-124 Regulates Apoptosis and Autophagy Process in MPTP Model of Parkinson’s Disease by Targeting to Bim. Brain Pathol. 2016, 26, 167–176. [Google Scholar] [CrossRef]
- Hayashi, T.; Ishimori, C.; Takahashi-Niki, K.; Taira, T.; Kim, Y.; Maita, H.; Maita, C.; Ariga, H.; Iguchi-Ariga, S.M.M. DJ-1 binds to mitochondrial complex I and maintains its activity. Biochem. Biophys. Res. Commun. 2009, 390, 667–672. [Google Scholar] [CrossRef]
- Irrcher, I.; Aleyasin, H.; Seifert, E.L.; Hewitt, S.J.; Chhabra, S.; Phillips, M.; Lutz, A.K.; Rousseaux, M.W.C.; Bevilacqua, L.; Jahani-Asl, A.; et al. Loss of the Parkinson’s disease-linked gene DJ-1 perturbs mitochondrial dynamics. Hum. Mol. Genet. 2010, 19, 3734–3746. [Google Scholar] [CrossRef]
- Mortiboys, H.; Johansen, K.K.; Aasly, J.O.; Bandmann, O. Mitochondrial impairment in patients with Parkinson disease with the G2019S mutation in LRRK2. Neurology 2010, 75, 2017–2020. [Google Scholar] [CrossRef]
- Wang, X.; Yan, M.H.; Fujioka, H.; Liu, J.; Wilson-Delfosse, A.; Chen, S.G.; Perry, G.; Casadesus, G.; Zhu, X. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum. Mol. Genet. 2012, 21, 1931–1944. [Google Scholar] [CrossRef]
- Chaudhuri, A.D.; Choi, D.C.; Kabaria, S.; Tran, A.; Junn, E. MicroRNA-7 Regulates the Function of Mitochondrial Permeability Transition Pore by Targeting VDAC1 Expression. J. Biol. Chem. 2016, 291, 6483–6493. [Google Scholar] [CrossRef]
- Zheng, B.; Liao, Z.; Locascio, J.J.; Lesniak, K.A.; Roderick, S.S.; Watt, M.L.; Eklund, A.C.; Zhang-James, Y.; Kim, P.D.; Hauser, M.A.; et al. PGC-1, A Potential Therapeutic Target for Early Intervention in Parkinson’s Disease. Sci. Transl. Med. 2010, 2, 52ra73. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, Z.; Le, W. Tiny But Mighty: Promising Roles of MicroRNAs in the Diagnosis and Treatment of Parkinson’s Disease. Neurosci. Bull. 2017, 33, 543–551. [Google Scholar] [CrossRef]
- Valdés, P.; Schneider, B.L. Gene Therapy: A Promising Approach for Neuroprotection in Parkinson’s Disease? Front. Neuroanat. 2016, 10, 123. [Google Scholar] [CrossRef]
- Serafin, A.; Foco, L.; Zanigni, S.; Blankenburg, H.; Picard, A.; Zanon, A.; Giannini, G.; Pichler, I.; Facheris, M.F.; Cortelli, P.; et al. Overexpression of blood microRNAs 103a, 30b, and 29a in L-dopa-treated patients with PD. Neurology 2015, 84, 645–653. [Google Scholar] [CrossRef]
- Ebert, M.S.; Neilson, J.R.; Sharp, P.A. MicroRNA sponges: Competitive inhibitors of small RNAs in mammalian cells. Nat. Methods 2007, 4, 721–726. [Google Scholar] [CrossRef]
- Yao, Y.-F.; Qu, M.-W.; Li, G.-C.; Zhang, F.-B.; Rui, H.-C. Circulating exosomal miRNAs as diagnostic biomarkers in Parkinson’s disease. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 5278–5283. [Google Scholar] [CrossRef]
- Barbagallo, C.; Mostile, G.; Baglieri, G.; Giunta, F.; Luca, A.; Raciti, L.; Zappia, M.; Purrello, M.; Ragusa, M.; Nicoletti, A. Specific Signatures of Serum miRNAs as Potential Biomarkers to Discriminate Clinically Similar Neurodegenerative and Vascular-Related Diseases. Cell. Mol. Neurobiol. 2020, 40, 531–546. [Google Scholar] [CrossRef]
- Xie, Y.; Chen, Y. microRNAs: Emerging Targets Regulating Oxidative Stress in the Models of Parkinson’s Disease. Front. Neurosci. 2016, 10. [Google Scholar] [CrossRef]
- Walker, F.O. Huntington’s disease. Lancet 2007, 369, 218–228. [Google Scholar] [CrossRef]
- Finkbeiner, S. Huntington’s Disease. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Andrew, S.E.; Goldberg, Y.P.; Kremer, B.; Telenius, H.; Theilmann, J.; Adam, S.; Starr, E.; Squitieri, F.; Lin, B.; Kalchman, M.A. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington’s disease. Nat. Genet. 1993, 4, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Franco-Iborra, S.; Vila, M.; Perier, C. Mitochondrial Quality Control in Neurodegenerative Diseases: Focus on Parkinson’s Disease and Huntington’s Disease. Front. Neurosci. 2018, 12, 342. [Google Scholar] [CrossRef] [PubMed]
- Gutekunst, C.-A.; Li, S.-H.; Yi, H.; Ferrante, R.J.; Li, X.-J.; Hersch, S.M. The Cellular and Subcellular Localization of Huntingtin-Associated Protein 1 (HAP1): Comparison with Huntingtin in Rat and Human. J. Neurosci. 1998, 18, 7674–7686. [Google Scholar] [CrossRef] [PubMed]
- Choo, Y.S. Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum. Mol. Genet. 2004, 13, 1407–1420. [Google Scholar] [CrossRef] [PubMed]
- Orr, A.L.; Li, S.; Wang, C.-E.; Li, H.; Wang, J.; Rong, J.; Xu, X.; Mastroberardino, P.G.; Greenamyre, J.T.; Li, X.-J. N-Terminal Mutant Huntingtin Associates with Mitochondria and Impairs Mitochondrial Trafficking. J. Neurosci. 2008, 28, 2783–2792. [Google Scholar] [CrossRef]
- Puigserver, P.; Spiegelman, B.M. Peroxisome Proliferator-Activated Receptor-γ Coactivator 1α (PGC-1α): Transcriptional Coactivator and Metabolic Regulator. Endocr. Rev. 2003, 24, 78–90. [Google Scholar] [CrossRef]
- Stanek, L.M.; Bu, J.; Shihabuddin, L.S. Astrocyte transduction is required for rescue of behavioral phenotypes in the YAC128 mouse model with AAV-RNAi mediated HTT lowering therapeutics. Neurobiol. Dis. 2019, 129, 29–37. [Google Scholar] [CrossRef]
- Bradford, J.; Shin, J.-Y.; Roberts, M.; Wang, C.-E.; Li, X.-J.; Li, S. Expression of mutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms. PNAS 2009, 106, 22480–22485. [Google Scholar] [CrossRef]
- Skotte, N.H.; Andersen, J.V.; Santos, A.; Aldana, B.I.; Willert, C.W.; Nørremølle, A.; Waagepetersen, H.S.; Nielsen, M.L. Integrative Characterization of the R6/2 Mouse Model of Huntington’s Disease Reveals Dysfunctional Astrocyte Metabolism. Cell Rep. 2018, 23, 2211–2224. [Google Scholar] [CrossRef]
- Gaughwin, P.M.; Ciesla, M.; Lahiri, N.; Tabrizi, S.J.; Brundin, P.; Björkqvist, M. Hsa-miR-34b is a plasma-stable microRNA that is elevated in pre-manifest Huntington’s disease. Hum. Mol. Genet. 2011, 20, 2225–2237. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Gao, F.; Wang, N.; Howland, D.; Kwak, S.; Vogt, T.F.; Aaronson, J.S.; Rosinski, J.; Coppola, G.; Horvath, S.; et al. MicroRNA signatures of endogenous Huntingtin CAG repeat expansion in mice. PLoS ONE 2018, 13, e0190550. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Standley, C.; Sapp, E.; Valencia, A.; Qin, Z.-H.; Kegel, K.B.; Yoder, J.; Comer-Tierney, L.A.; Esteves, M.; Chase, K.; et al. Mutant huntingtin impairs vesicle formation from recycling endosomes by interfering with Rab11 activity. Mol. Cell. Biol. 2009, 29, 6106–6116. [Google Scholar] [CrossRef] [PubMed]
- Hoss, A.G.; Labadorf, A.; Latourelle, J.C.; Kartha, V.K.; Hadzi, T.C.; Gusella, J.F.; MacDonald, M.E.; Chen, J.-F.; Akbarian, S.; Weng, Z.; et al. miR-10b-5p expression in Huntington’s disease brain relates to age of onset and the extent of striatal involvement. BMC Med. Genom. 2015, 8, 10. [Google Scholar] [CrossRef]
- Lee, S.-T.; Chu, K.; Im, W.-S.; Yoon, H.-J.; Im, J.-Y.; Park, J.-E.; Park, K.-H.; Jung, K.-H.; Lee, S.K.; Kim, M.; et al. Altered microRNA regulation in Huntington’s disease models. Exp. Neurol. 2011, 227, 172–179. [Google Scholar] [CrossRef]
- Miska, E.A.; Alvarez-Saavedra, E.; Townsend, M.; Yoshii, A.; Sestan, N.; Rakic, P.; Constantine-Paton, M.; Horvitz, H.R. Microarray analysis of microRNA expression in the developing mammalian brain. Genome Biol. 2004, 5, R68. [Google Scholar] [CrossRef]
- McGleenon, B.M.; Dynan, K.B.; Passmore, A.P. Acetylcholinesterase inhibitors in Alzheimer’s disease: Acetylcholinesterase inhibitors in Alzheimer’s disease. Br. J. Clin. Pharmacol. 2001, 48, 471–480. [Google Scholar] [CrossRef]
- Lee, S.T.; Im, W.; Ban, J.J.; Lee, M.; Jung, K.H.; Lee, S.K.; Chu, K.; Kim, M. Exosome-Based Delivery of miR-124 in a Huntington’s Disease Model. JMD 2017, 10, 45–52. [Google Scholar] [CrossRef]
- Reed, E.R.; Latourelle, J.C.; Bockholt, J.H.; Bregu, J.; Smock, J.; Paulsen, J.S.; Myers, R.H.; PREDICT-HD CSF ancillary study investigators. MicroRNAs in CSF as prodromal biomarkers for Huntington disease in the PREDICT-HD study. Neurology 2018, 90, e264–e272. [Google Scholar] [CrossRef]
- Chia, R.; Chiò, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef]
- Ajroud-Driss, S.; Siddique, T. Sporadic and hereditary amyotrophic lateral sclerosis (ALS). Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, Y.; Mieda-Sato, A. TDP-43 promotes microRNA biogenesis as a component of the Drosha and Dicer complexes. Proc. Natl. Acad. Sci. USA 2012, 109, 3347–3352. [Google Scholar] [CrossRef] [PubMed]
- Buratti, E.; De Conti, L.; Stuani, C.; Romano, M.; Baralle, M.; Baralle, F. Nuclear factor TDP-43 can affect selected microRNA levels: TDP-43 and miRNA regulation. FEBS J. 2010, 277, 2268–2281. [Google Scholar] [CrossRef] [PubMed]
- Morgan, S.; Orrell, R.W. Pathogenesis of amyotrophic lateral sclerosis. Br. Med. Bull. 2016, 119, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Hamzeiy, H.; Suluyayla, R.; Brinkrolf, C.; Janowski, S.J.; Hofestädt, R.; Allmer, J. Visualization and Analysis of miRNAs Implicated in Amyotrophic Lateral Sclerosis Within Gene Regulatory Pathways. Stud. Health Technol. Inform. 2018, 253, 183–187. [Google Scholar]
- Capitano, F.; Camon, J.; Licursi, V.; Ferretti, V.; Maggi, L.; Scianni, M.; Del Vecchio, G.; Rinaldi, A.; Mannironi, C.; Limatola, C.; et al. MicroRNA-335-5p modulates spatial memory and hippocampal synaptic plasticity. Neurobiol. Learn. Mem. 2017, 139, 63–68. [Google Scholar] [CrossRef]
- Freischmidt, A.; Müller, K.; Zondler, L.; Weydt, P.; Mayer, B.; von Arnim, C.A.F.; Hübers, A.; Dorst, J.; Otto, M.; Holzmann, K.; et al. Serum microRNAs in sporadic amyotrophic lateral sclerosis. Neurobiol. Aging 2015, 36, 2660.e15–2660.e20. [Google Scholar] [CrossRef]
- Xu, Z.; Henderson, R.D.; David, M.; McCombe, P.A. Neurofilaments as Biomarkers for Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0164625. [Google Scholar] [CrossRef]
- Freischmidt, A.; Müller, K.; Ludolph, A.C.; Weishaupt, J.H. Systemic dysregulation of TDP-43 binding microRNAs in amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 2013, 1, 42. [Google Scholar] [CrossRef]




| miRNAs | Specificity | Localization | References |
|---|---|---|---|
| miR-26b ↑ | AD, PD | CNS, Blood | [43] |
| miR-206 ↑ | AD, ALS | CSF | [44] |
| miR-34a ↑ | AD, PD, HD | CNS, Blood | [45] |
| miR-126 ↑ | AD, PD | CNS | [46] |
| miR-9 ↓ | AD | Cortex | [47] |
| miR-221 ↓ | AD | Cortex | [48] |
| miR-98 ↑ | AD | Cortex | [49] |
| miR-330 ↓ | AD, HD | CNS | [50] |
| miR-19 ↓ | AD, PD | CNS | [51] |
| miR-30 ↓ | AD, PD | Blood, CSF | [17] |
| miR-375 ↓ | AD | CSF | [53] |
| miR-7 ↓ | PD | CNS | [52] |
| miR-124 ↓ | PD, AD | CNS | [54] |
| miR-133b ↓ | PD, HD, ALS | CNS, Cortex | [74] |
| miR-153 ↓ | PD | CNS | [55] |
| miR-443 ↑ | PD | CNS | [56] |
| miR-494 ↑ | PD | CSF | [57] |
| miR-34b/c ↓ | PD, AD, HD | CNS, Blood | [58] |
| miR-205 ↓ | PD | CNS | [59] |
| miR-27a ↓ | PD, ALS | CNS | [60] |
| miR-105 ↓ | PD, AD | CSF | [60] |
| miR-103 ↑ | PD, AD | CNS, Blood | [60] |
| miR 4639 5p ↓ | PD | CNS | [61] |
| miR-21 ↑ | PD, AD | Lymphocytes, CSF, Blood | [62] |
| miR-331-5p ↑ | PD | CNS, Blood | [63] |
| miR-132 ↓ | HD, ALS, AD | CNS, CSF, Cortex, Cerebellum | [64] |
| miR-125b ↓ | HD, AD | CNS, Hippocampus | [65] |
| miR-146a ↓ | HD, AD, ALS | CSF, Spinal cord | [66] |
| miR-196a ↓ | HD | Cortex | [67] |
| miR-9 ↑ | HD, AD, ALS | CNS | [68] |
| miR-124 ↑ | HD, AD | CSF, Hippocampus, Cortex | [69] |
| miR-29a ↑ | HD, AD, PD | Cortex, Blood | [69] |
| miR-129-5p ↑ | ALS. PD | Cerebellum, CSF | [70] |
| miR-27a/b ↓ | ALS | Monocytes CD14 + CD16- | [72] |
| miR-335-5p ↑ | ALS | CNS | [72] |
| miR-151b ↓ | ALS, HD, AD | Cortex, CNS | [73] |
| miR-221-3p ↓ | ALS | CNS | [73] |
| miR-130a-3p ↓ | ALS | CNS | [73] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Catanesi, M.; d’Angelo, M.; Tupone, M.G.; Benedetti, E.; Giordano, A.; Castelli, V.; Cimini, A. MicroRNAs Dysregulation and Mitochondrial Dysfunction in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 5986. https://doi.org/10.3390/ijms21175986
Catanesi M, d’Angelo M, Tupone MG, Benedetti E, Giordano A, Castelli V, Cimini A. MicroRNAs Dysregulation and Mitochondrial Dysfunction in Neurodegenerative Diseases. International Journal of Molecular Sciences. 2020; 21(17):5986. https://doi.org/10.3390/ijms21175986
Chicago/Turabian StyleCatanesi, Mariano, Michele d’Angelo, Maria Grazia Tupone, Elisabetta Benedetti, Antonio Giordano, Vanessa Castelli, and Annamaria Cimini. 2020. "MicroRNAs Dysregulation and Mitochondrial Dysfunction in Neurodegenerative Diseases" International Journal of Molecular Sciences 21, no. 17: 5986. https://doi.org/10.3390/ijms21175986
APA StyleCatanesi, M., d’Angelo, M., Tupone, M. G., Benedetti, E., Giordano, A., Castelli, V., & Cimini, A. (2020). MicroRNAs Dysregulation and Mitochondrial Dysfunction in Neurodegenerative Diseases. International Journal of Molecular Sciences, 21(17), 5986. https://doi.org/10.3390/ijms21175986