MicroRNA and ROS Crosstalk in Cardiac and Pulmonary Diseases

 ,
, 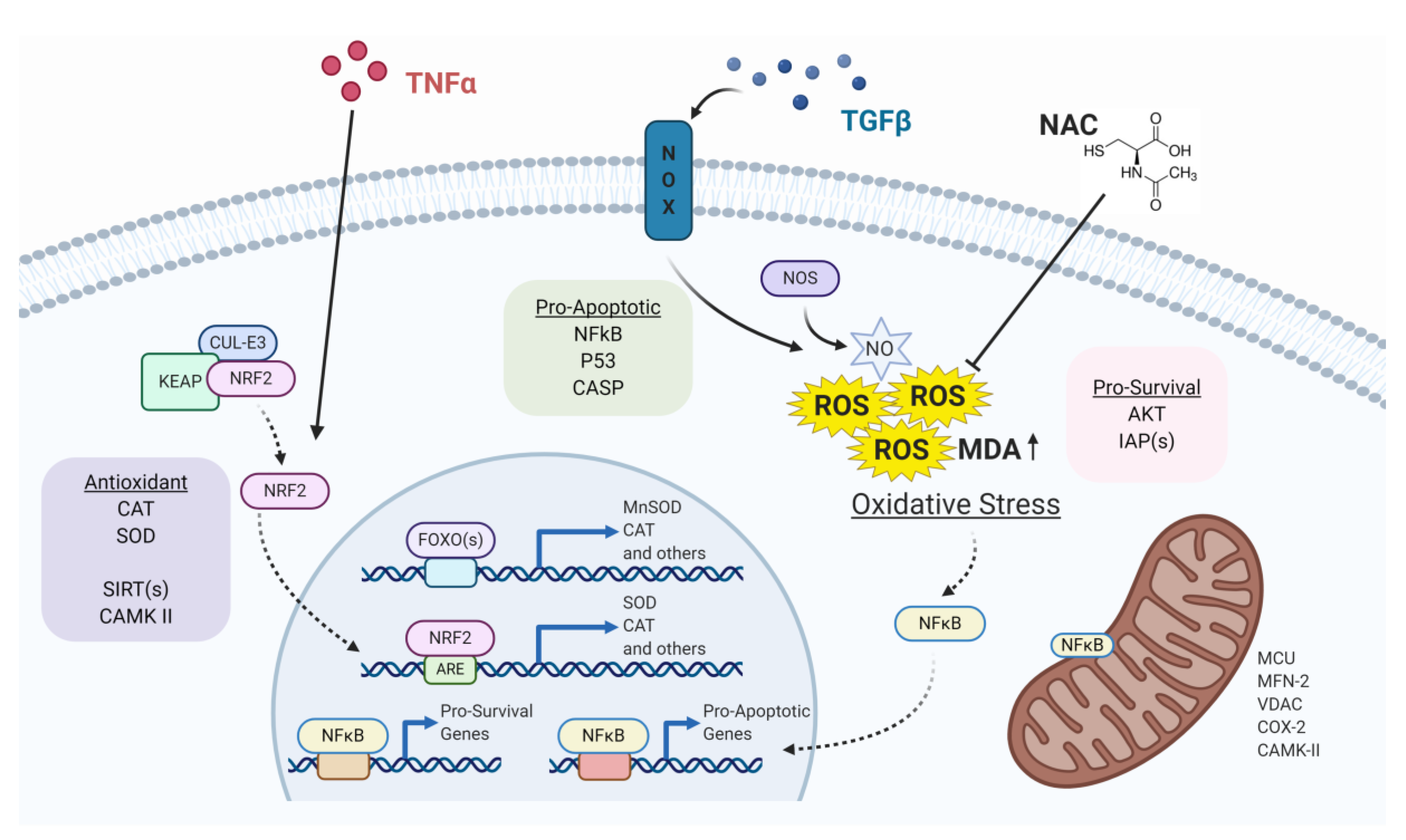{kind=link}
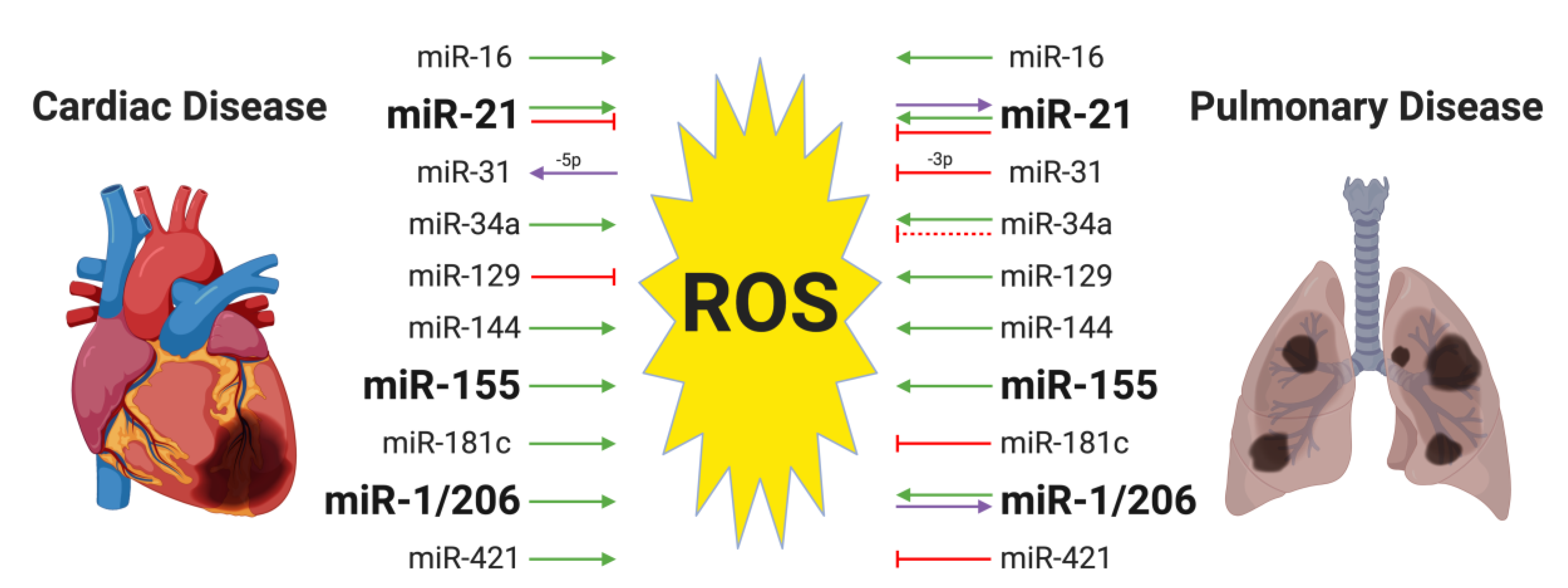{kind=link}
Abstract
1. Introduction
2. MiRNAs and Oxidative Stress in Cardiac Diseases
2.1. Cardiac Hypertrophy
2.1.1. MiRNA Induction of Oxidative Stress in Cardiac Hypertrophy
2.1.2. ROS Induction of MiRNAs in Cardiac Hypertrophy
2.1.3. Mitochondrial Oxidative Stress and MiRNAs in Cardiac Hypertrophy
2.2. Heart Failure
2.2.1. MiRNA Regulation of TNFα-Dependent Oxidative Stress in HF and CHF
2.2.2. MiRNA Induction of Oxidative Stress in HF
2.3. Myocardial Infarction and Ischemia/Reperfusion
2.3.1. Crosstalk between miRNAs and the Sirtuin Family in MI
2.3.2. MiRNA Targeting of the CaMKII Signaling Pathway in MI
2.3.3. MiRNA Fine-Tuning of the Antioxidant Program in MI
2.3.4. MiRNA and ROS Crosstalk during MI
2.4. Diabetic Cardiomyopathy
2.4.1. MiRNA Cardioprotective and Antioxidative Role in DCM
2.4.2. Cardiotoxic Role of MiRNAs in DCM
3. MiRNAs and Oxidative Stress in Pulmonary Diseases
3.1. Idiopathic Pulmonary Fibrosis
Crosstalk between MiRNAs and ROS in IPF
3.2. Acute Lung Injury and Acute Respiratory Distress Syndrome
Crosstalk between MiRNAs and ROS in ARDS/ALI
3.3. Airway Diseases: Asthma and Chronic Obstructive Pulmonary Disease
Crosstalk between MiRNAs and ROS in Asthma and COPD
3.4. Lung Cancer
3.4.1. Crosstalk between MiRNAs and ROS to Support Lung Cancer Growth
3.4.2. Crosstalk between MiRNAs and ROS to Suppress Lung Cancer Growth
3.4.3. Crosstalk between MiRNAs and ROS to Induce Metastasis
3.4.4. MiRNA Induction of ROS Accumulation in Lung Cancer
3.4.5. Crosstalk between MiRNAs and ROS to Favor Chemoresistance
3.4.6. MiRNAs as Mediators of ‘Natural’ Antioxidant in Lung Cancer
4. MiRNAs and Oxidative Stress in Both Cardiac and Pulmonary Diseases
4.1. MiR-155 and Oxidative Stress in Cardiac and Pulmonary Diseases
4.2. MiR-21 and Oxidative Stress in Cardiac and Pulmonary Diseases
4.3. MiR-1/206 and Oxidative Stress in the Cardiac and Pulmonary Diseases
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Magenta, A.; Dellambra, E.; Ciarapica, R.; Capogrossi, M.C. Oxidative stress, microRNAs and cytosolic calcium homeostasis. Cell Calcium 2016, 60, 207–217. [Google Scholar] [CrossRef]
- Bu, H.; Wedel, S.; Cavinato, M.; Jansen-Dürr, P. MicroRNA Regulation of Oxidative Stress-Induced Cellular Senescence. Oxid. Med. Cell. Longev. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS Signaling in Organismal Homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013, 1, 304–312. [Google Scholar] [CrossRef]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef]
- Aziz, N.; Jamil, R.T. Biochemistry, Xanthine Oxidase. 2020. Available online: https://europepmc.org/books/n/statpearls/article-31434/?extid=29083631&src=med (accessed on 3 May 2020).
- Wu, M.Y.; Yiang, G.T.; Liao, W.T.; Tsai, A.P.Y.; Cheng, Y.L.; Cheng, P.W.; Li, C.Y.; Li, C.J. Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell. Physiol. Biochem. 2018, 46, 1650–1667. [Google Scholar] [CrossRef]
- Mailloux, R.J.; Gardiner, D.; O’Brien, M. 2-Oxoglutarate dehydrogenase is a more significant source of O2·−/H2O2 than pyruvate dehydrogenase in cardiac and liver tissue. Free Radic. Biol. Med. 2016, 97, 501–512. [Google Scholar] [CrossRef]
- Tretter, L.; Adam-Vizi, V. Alpha-ketoglutarate dehydrogenase: A target and generator of oxidative stress. Philos. Trans. R. Soc. B Biol. Sci. 2005, 360, 2335–2345. [Google Scholar] [CrossRef]
- Ambrus, A.; Adam-Vizi, V. Human dihydrolipoamide dehydrogenase (E3) deficiency: Novel insights into the structural basis and molecular pathomechanism. Neurochem. Int. 2018, 117, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.-J.; Sumien, N.; Thangthaeng, N.; Forster, M.J. Reversible inactivation of dihydrolipoamide dehydrogenase by mitochondrial hydrogen peroxide. Free Radic. Res. 2013, 47, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Ambrus, A.; Torocsik, B.; Tretter, L.; Ozohanics, O.; Adam-Vizi, V. Stimulation of reactive oxygen species generation by disease-causing mutations of lipoamide dehydrogenase. Hum. Mol. Genet. 2011, 20, 2984–2995. [Google Scholar] [CrossRef] [PubMed]
- Ambrus, A.; Tretter, L.; Adam-Vizi, V. Inhibition of the alpha-ketoglutarate dehydrogenase-mediated reactive oxygen species generation by lipoic acid. J. Neurochem. 2009, 109, 222–229. [Google Scholar] [CrossRef]
- Nagano, T.; Yamao, S.; Terachi, A.; Yarimizu, H.; Itoh, H.; Katasho, R.; Kawai, K.; Nakashima, A.; Iwasaki, T.; Kikkawa, U.; et al. D-amino acid oxidase promotes cellular senescence via the production of reactive oxygen species. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef]
- Matlashov, M.E.; Belousov, V.V.; Enikolopov, G. How much H2O2 is produced by recombinant D-Amino acid oxidase in mammalian cells? Antioxid. Redox Signal. 2014, 20, 1039–1044. [Google Scholar] [CrossRef]
- Sacchi, S.; Cappelletti, P.; Murtas, G. Biochemical properties of human D-amino acid oxidase variants and their potential significance in pathologies. Front. Mol. Biosci. 2018, 5, 55. [Google Scholar] [CrossRef]
- Müller, J.M.; Rupec, R.A.; Baeuerle, P.A. Study of gene regulation by NF-κB and AP-1 in response to reactive oxygen intermediates. Methods A Companion Methods Enzymol. 1997, 11, 301–312. [Google Scholar] [CrossRef]
- Sen, C.; Packer, L. Antioxidant and redox regulation of gene transcription. Faseb J. 1996, 10, 709–720. [Google Scholar] [CrossRef]
- Lee, E.K.; Kim, J.A.; Park, S.J.; Kim, J.K.; Heo, K.; Yang, K.M.; Son, T.G. Low-dose radiation activates Nrf1/2 through reactive species and the Ca2+/ERK1/2 signaling pathway in human skin fibroblast cells. BMB Rep. 2013, 46, 258–263. [Google Scholar] [CrossRef]
- Fourtounis, J.; Wang, I.M.; Mathieu, M.C.; Claveau, D.; Loo, T.; Jackson, A.L.; Peters, M.A.; Therien, A.G.; Boie, Y.; Crackower, M.A. Gene expression profiling following NRF2 and KEAP1 siRNA knockdown in human lung fibroblasts identifies CCL11/Eotaxin-1 as a novel NRF2 regulated gene. Respir. Res. 2012, 13, 92. [Google Scholar] [CrossRef] [PubMed]
- Szoltysek, K.; Walaszczyk, A.; Janus, P.; Kimmel, M.; Widlak, P. Irradiation with UV-C inhibits TNF-α-dependent activation of the NF-κB pathway in a mechanism potentially mediated by reactive oxygen species. Genes Cells 2017, 22, 45–58. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Spadaro, D.; Yun, B.W.; Spoel, S.H.; Chu, C.; Wang, Y.Q.; Loake, G.J. The redox switch: Dynamic regulation of protein function by cysteine modifications. Physiol. Plant. 2010, 138, 360–371. [Google Scholar] [CrossRef]
- Gào, X.; Schöttker, B. Reduction-oxidation pathways involved in cancer development: A systematic review of literature reviews. Oncotarget 2017, 8, 51888–51906. [Google Scholar] [CrossRef] [PubMed]
- Jaspers, I.; Flescher, E.; Chen, L.C. Ozone-induced IL-8 expression and transcription factor binding in respiratory epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 1997, 272, 504–511. [Google Scholar] [CrossRef]
- Lee, R.; Feinbaum, R.; Ambros, V. The C. elegans Heterochronic Gene lin-4 Encodes Small RNAs with Antisense Complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. MiRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- He, J.; Jiang, B.-H. Interplay between Reactive oxygen Species and MicroRNAs in Cancer Jun. Curr. Pharmacol. Rep. 2016, 2, 82–90. [Google Scholar] [CrossRef]
- Liu, Y.; Qiang, W.; Xu, X.; Dong, R.; Karst, A.M.; Liu, Z.; Kong, B.; Drapkin, R.I.; Wei, J.J. Role of miR-182 in response to oxidative stress in the cell fate of human fallopian tube epithelial cells. Oncotarget 2015, 6, 38983–38998. [Google Scholar] [CrossRef]
- Magenta, A.; Greco, S.; Gaetano, C.; Martelli, F. Oxidative stress and microRNAs in vascular diseases. Int. J. Mol. Sci. 2013, 14, 17319–17346. [Google Scholar] [CrossRef]
- Moris, D.; Spartalis, M.; Spartalis, E.; Karachaliou, G.S.; Karaolanis, G.I.; Tsourouflis, G.; Tsilimigras, D.I.; Tzatzaki, E.; Theocharis, S. The role of reactive oxygen species in the pathophysiology of cardiovascular diseases and the clinical significance of myocardial redox. Ann. Transl. Med. 2017, 5, 326. [Google Scholar] [CrossRef]
- Roy, J.; Galano, J.M.; Durand, T.; Le Guennec, J.Y.; Lee, J.C.Y. Physiological role of reactive oxygen species as promoters of natural defenses. FASEB J. 2017, 31, 3729–3745. [Google Scholar] [CrossRef] [PubMed]
- Kar, D.; Bandyopadhyay, A. Targeting Peroxisome Proliferator Activated Receptor α (PPAR α) for the Prevention of Mitochondrial Impairment and Hypertrophy in Cardiomyocytes. Cell. Physiol. Biochem. 2018, 49, 245–259. [Google Scholar] [CrossRef] [PubMed]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 2019, 51, 162. [Google Scholar] [CrossRef] [PubMed]
- Kura, B.; Bacova, B.S.; Kalocayova, B.; Sykora, M.; Slezak, J. Oxidative stress-responsive microRNAs in heart injury. Int. J. Mol. Sci. 2020, 21, 358. [Google Scholar] [CrossRef]
- Latronico, M.V.G.; Condorelli, G. microRNAs in hypertrophy and heart failure. Exp. Biol. Med. 2011, 236, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Latronico, M.V.G.; Catalucci, D.; Condorelli, G. MicroRNA and cardiac pathologies. Physiol. Genom. 2008, 34, 239–242. [Google Scholar] [CrossRef]
- Wang, Q.; Yu, X.; Dou, L.; Huang, X.; Zhu, K.; Guo, J.; Yan, M.; Wang, S.; Man, Y.; Tang, W.; et al. MIR-154-5p functions as an important regulator of angiotensin II-mediated heart remodeling. Oxid. Med. Cell. Longev. 2019. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Feferman, L.; Tobacman, J.K. Restriction of Aerobic Metabolism by Acquired or Innate Arylsulfatase B Deficiency: A New Approach to the Warburg Effect. Sci. Rep. 2016, 6, 32885. [Google Scholar] [CrossRef]
- Greer, E.L.; Brunet, A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 2005, 24, 7410–7425. [Google Scholar] [CrossRef]
- Song, G.; Zhu, L.; Ruan, Z.; Wang, R.; Shen, Y. MicroRNA-122 promotes cardiomyocyte hypertrophy via targeting FoxO3. Biochem. Biophys. Res. Commun. 2019, 519, 682–688. [Google Scholar] [CrossRef]
- Hu, S.; Cheng, M.; Guo, X.; Wang, S.; Liu, B.; Jiang, H.; Huang, C.; Wu, G. Down-regulation of miR-200c attenuates AngII-induced cardiac hypertrophy via targeting the MLCK-mediated pathway. J. Cell. Mol. Med. 2019, 23, 2505–2516. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Kyrychenko, S.; Kyrychenko, V.; Badr, M.A.; Ikeda, Y.; Sadoshima, J.; Shirokova, N. Pivotal role of MIR-448 in the development of ROS-induced cardiomyopathy. Cardiovasc. Res. 2015, 108, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.; Mushtaq, I.; Maryam, S.; Farhan, A.; Saba, K.; Jan, M.I.; Sultan, A.; Anees, M.; Duygu, B.; Hamera, S.; et al. Interplay of N– acetyl cysteine and melatonin in regulating oxidative stress-induced cardiac hypertrophic factors and microRNAs. Arch. Biochem. Biophys. 2019, 661, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Granatiero, V.; Pacifici, M.; Raffaello, A.; De Stefani, D.; Rizzuto, R. Overexpression of Mitochondrial Calcium Uniporter Causes Neuronal Death. Oxid. Med. Cell. Longev. 2019, 2019. [Google Scholar] [CrossRef]
- Pan, L.; Huang, B.J.; Ma, X.E.; Wang, S.Y.; Feng, J.; Lv, F.; Liu, Y.; Liu, Y.; Li, C.M.; Liang, D.D.; et al. MiR-25 protects cardiomyocytes against oxidative damage by targeting the mitochondrial calcium uniporter. Int. J. Mol. Sci. 2015, 16, 5420–5433. [Google Scholar] [CrossRef]
- Shen, T.; Zheng, M.; Cao, C.; Chen, C.; Tang, J.; Zhang, W.; Cheng, H.; Chen, K.H.; Xiao, R.P. Mitofusin-2 is a major determinant of oxidative stress-mediated heart muscle cell apoptosis. J. Biol. Chem. 2007, 282, 23354–23361. [Google Scholar] [CrossRef]
- Guan, X.; Wang, L.; Liu, Z.; Guo, X.; Jiang, Y.; Lu, Y.; Peng, Y.; Liu, T.; Yang, B.; Shan, H.; et al. miR-106a promotes cardiac hypertrophy by targeting mitofusin 2. J. Mol. Cell. Cardiol. 2016, 99, 207–217. [Google Scholar] [CrossRef]
- Tian, C.; Gao, L.; Zimmerman, M.C.; Zucker, I.H. Myocardial infarction-induced microRNA-enriched exosomes contribute to cardiac Nrf2 dysregulation in chronic heart failure. Am. J. Physiol. Hear. Circ. Physiol. 2018, 314, H928–H939. [Google Scholar] [CrossRef]
- Münzel, T.; Camici, G.G.; Maack, C.; Bonetti, N.R.; Fuster, V.; Kovacic, J.C. Impact of Oxidative Stress on the Heart and Vasculature. J. Am. Coll. Cardiol. 2017, 70, 212–229. [Google Scholar] [CrossRef] [PubMed]
- Shang, F.; Zhao, L.; Zheng, Q.; Wang, J.; Xu, Z.; Liang, W.; Liu, H.; Liu, S.; Zhang, L. Simvastatin inhibits lipopolysaccharide-induced tumor necrosis factor- a expression in neonatal rat cardiomyocytes: The role of reactive oxygen species. Biochem. Biophys. Res. Commun. 2006, 351, 947–952. [Google Scholar] [CrossRef] [PubMed]
- Rushworth, S.A.; Shah, S.; Macewan, D.J. TNF Mediates the Sustained Activation of Nrf2 in Human Monocytes. J. Immunol. 2011, 187. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Zeh, H.J.; Lotze, M.T. High-mobility group box 1, oxidative stress, and disease. Antioxid. Redox Signal. 2011, 14, 1315–1335. [Google Scholar] [CrossRef]
- Hickson-Bick, D.L.M.; Sparagna, G.C.; Buja, L.M.; McMillin, J.B. Palmitate-induced apoptosis in neonatal cardiomyocytes is not dependent on the generation of ROS. Am. J. Physiol. Hear. Circ. Physiol. 2002, 282, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Yang, Y.; Wang, Y.; Li, J.; Schiller, P.W.; Peng, T. MicroRNA-195 promotes palmitate-induced apoptosis in cardiomyocytes by down-regulating Sirt1. Cardiovasc. Res. 2011, 92, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Ren, J.; Ren, H.; Fu, J.; Yu, D. MicroRNA-132 suppresses cell proliferation in human breast cancer by directly targeting FOXA1. Acta Pharmacol. Sin. 2018, 39, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Tong, Z.; Chen, K.; Hu, X.; Jin, H.; Hou, M. The Role of miRNA-132 against Apoptosis and Oxidative Stress in Heart Failure. Biomed. Res. Int. 2018, 2018. [Google Scholar] [CrossRef]
- Wang, X.; Song, C.; Zhou, X.; Han, X.; Li, J.; Wang, Z.; Shang, H.; Liu, Y.; Cao, H. Mitochondria Associated MicroRNA Expression Profiling of Heart Failure. Biomed. Res. Int. 2017, 2017. [Google Scholar] [CrossRef]
- Cong, B.H.; Zhu, X.Y.; Ni, X. The roles of microRNA-22 in myocardial infarction. Sheng Li Xue Bao 2017, 69, 571–578. [Google Scholar] [CrossRef]
- Liu, Y.; Qian, X.M.; He, Q.C.; Weng, J.K. MiR-421 inhibition protects H9c2 cells against hypoxia/reoxygenation-induced oxidative stress and apoptosis by targeting Sirt3. Perfusion 2020, 35, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhou, J.; Yang, B.; Yu, Y. microRNA-340-5p inhibits hypoxia/reoxygenation-induced apoptosis and oxidative stress in cardiomyocytes by regulating the Act1/NF-κB pathway. J. Cell. Biochem. 2019, 120, 14618–14627. [Google Scholar] [CrossRef]
- Xuan, Y.T.; Tang, X.L.; Banerjee, S.; Takano, H.; Li, R.C.X.; Han, H.; Qiu, Y.; Li, J.J.; Bolli, R. Nuclear factor-κB plays an essential role in the late phase of ischemic preconditioning in conscious rabbits. Circ. Res. 1999, 84, 1095–1109. [Google Scholar] [CrossRef] [PubMed]
- Singh, C.K.; Chhabra, G.; Ndiaye, M.A.; Garcia-Peterson, L.M.; MacK, N.J.; Ahmad, N. The Role of Sirtuins in Antioxidant and Redox Signaling. Antioxid. Redox Signal. 2018, 28, 643–661. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, S.; Li, Y.; Yu, S.; Zhao, Y. SIRT1/PGC-1α signaling promotes mitochondrial functional recovery and reduces apoptosis after intracerebral hemorrhage in rats. Front. Mol. Neurosci. 2018, 10, 443. [Google Scholar] [CrossRef] [PubMed]
- Du, J.K.; Cong, B.H.; Yu, Q.; Wang, H.; Wang, L.; Wang, C.N.; Tang, X.L.; Lu, J.Q.; Zhu, X.Y.; Ni, X. Upregulation of microRNA-22 contributes to myocardial ischemia-reperfusion injury by interfering with the mitochondrial function. Free Radic. Biol. Med. 2016, 96, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Chong, M.S.K.; Ng, W.K.; Chan, J.K.Y. Concise Review: Endothelial Progenitor Cells in Regenerative Medicine: Applications and Challenges. Stem Cells Transl. Med. 2016, 5, 530–538. [Google Scholar] [CrossRef]
- Bianconi, V.; Sahebkar, A.; Kovanen, P.; Bagaglia, F.; Ricciuti, B.; Calabrò, P.; Patti, G.; Pirro, M. Endothelial and cardiac progenitor cells for cardiovascular repair: A controversial paradigm in cell therapy. Pharmacol. Ther. 2018, 181, 156–168. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Qi, B.; Duan, X.; Ming, X.; Yan, F.; He, Y.; Bu, X.; Sun, S.; Zhu, H. MicroRNA-126 enhances the biological function of endothelial progenitor cells under oxidative stress via PI3K/Akt/GSK-3β and ERK1/2 signaling pathways. Bosn. J. Basic Med. Sci. 2020. [Google Scholar] [CrossRef]
- Shimizu, I.; Minamino, T. Physiological and pathological cardiac hypertrophy. J. Mol. Cell. Cardiol. 2016, 97, 245–262. [Google Scholar] [CrossRef]
- Anderson, M.E. Oxidant stress promotes disease by activating CaMKII. J. Mol. Cell. Cardiol. 2015, 89, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, R.; Liu, D.; Deng, W.; Xu, G.; Liu, W.; Rong, J.; Long, X.; Ge, J.; Shi, B. Exosomes Derived from miR-214-Enriched Bone Marrow-Derived Mesenchymal Stem Cells Regulate Oxidative Damage in Cardiac Stem Cells by Targeting CaMKII. Oxid. Med. Cell. Longev. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Xu, Z.; Li, C.; Xu, C.; Lei, Z.; Zhang, H.T.; Zhao, J. MiR-145 and miR-203 represses TGF-β-induced epithelial-mesenchymal transition and invasion by inhibiting SMAD3 in non-small cell lung cancer cells. Lung Cancer 2016, 97, 87–94. [Google Scholar] [CrossRef]
- Climent, M.; Quintavalle, M.; Miragoli, M.; Chen, J.; Condorelli, G.; Elia, L. TGFβ triggers miR-143/145 transfer from smooth muscle cells to endothelial cells, thereby modulating vessel stabilization. Circ. Res. 2015, 116, 1753–1764. [Google Scholar] [CrossRef] [PubMed]
- Cha, M.; Jang, J.; Ham, O.; Song, B.; Lee, S.; Yeon, C.; Park, J.; Lee, J.; Seo, H.; Choi, E.; et al. Biochemical and Biophysical Research Communications MicroRNA-145 suppresses ROS-induced Ca2+ overload of cardiomyocytes by targeting CaMKII d. Biochem. Biophys. Res. Commun. 2013, 435, 720–726. [Google Scholar] [CrossRef]
- Li, R.; Yan, G.; Li, Q.; Sun, H.; Hu, Y.; Sun, J.; Xu, B. MicroRNA-145 Protects Cardiomyocytes against Hydrogen Peroxide (H2O2) -Induced Apoptosis through Targeting the Mitochondria Apoptotic Pathway. PLoS ONE 2012, 7, e44907. [Google Scholar] [CrossRef]
- Li, X.; Commane, M.; Nie, H.; Hua, X.; Chatterjee-kishore, M.; Wald, D.; Haag, M.; Stark, G.R. Act1, an NF-k-activating protein. Proc. Natl. Acad. Sci. USA 2000, 97, 10489–10493. [Google Scholar] [CrossRef]
- Long, B.; Gan, T.Y.; Zhang, R.C.; Zhang, Y.H. miR-23a regulates cardiomyocyte apoptosis by targeting manganese superoxide dismutase. Mol. Cells 2017, 40, 542–549. [Google Scholar] [CrossRef]
- Cramer-morales, K.; Heer, C.; Mapuskar, K.; Domann, F.E.; City, I. SOD2 targeted gene editing by CRISPR/Cas9 yields human cells devoid of MnSOD. Free Radic. Biol. Med. 2015, 89, 379–386. [Google Scholar] [CrossRef]
- Li, S.; Ren, J.; Sun, Q. The expression of microRNA-23a regulates acute myocardial infarction in patients and in vitro through targeting PTEN. Mol. Med. Rep. 2018, 17, 6866–6872. [Google Scholar] [CrossRef]
- Tian, K.; Di, R.; Wang, L. MicroRNA-23a enhances migration and invasion through PTEN in osteosarcoma. Cancer Gene Ther. 2015, 22, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Leslie, N.R.; Bennett, D.; Lindsay, Y.E.; Stewart, H.; Gray, A.; Downes, C.P. Redox regulation of PI 3-kinase signalling via PTEN-7595424a.pdf. EMBO J. 2003, 22, 5501–5510. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sano, M.; Shinmura, K.; Tamaki, K.; Katsumata, Y.; Matsuhashi, T.; Morizane, S.; Ito, H.; Hishiki, T.; Endo, J.; et al. 4-Hydroxy-2-nonenal protects against cardiac ischemia-reperfusion injury via the Nrf2-dependent pathway. J. Mol. Cell. Cardiol. 2010, 49, 576–586. [Google Scholar] [CrossRef]
- Xiao, X.; Lu, Z.; Lin, V.; May, A.; Shaw, D.H.; Wang, Z.; Che, B.; Tran, K.; Du, H.; Shaw, P.X. MicroRNA miR-24-3p reduces apoptosis and regulates Keap1-Nrf2 pathway in mouse cardiomyocytes responding to ischemia/reperfusion injury. Oxid. Med. Cell. Longev. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Carè, A.; Catalucci, D.; Felicetti, F.; Bonci, D.; Addario, A.; Gallo, P.; Bang, M.L.; Segnalini, P.; Gu, Y.; Dalton, N.D.; et al. MicroRNA-133 controls cardiac hypertrophy. Nat. Med. 2007, 13, 613–618. [Google Scholar] [CrossRef]
- Matkovich, S.J.; Wang, W.; Tu, Y.; Eschenbacher, W.H.; Dorn, L.E.; Condorelli, G.; Diwan, A.; Nerbonne, J.M.; Ii, G.W.D. MicroRNA-133a protects against myocardial fibrosis and modulates electrical repolarization without affecting hypertrophy in pressure overloaded adult hearts. Circ. Res. 2010, 106, 166–175. [Google Scholar] [CrossRef]
- Castaldi, A.; Zaglia, T.; Di Mauro, V.; Carullo, P.; Viggiani, G.; Borile, G.; Di Stefano, B.; Schiattarella, G.G.; Gualazzi, M.G.; Elia, L.; et al. MicroRNA-133 modulates the β1-adrenergic receptor transduction cascade. Circ. Res. 2014, 115, 273–283. [Google Scholar] [CrossRef]
- Izarra, A.; Moscoso, I.; Levent, E.; Cañón, S.; Cerrada, I.; Díez-Juan, A.; Blanca, V.; Núñez-Gil, I.J.; Valiente, I.; Ruíz-Sauri, A.; et al. MiR-133a enhances the protective capacity of cardiac progenitors cells after myocardial infarction. Stem Cell Rep. 2014, 3, 1029–1042. [Google Scholar] [CrossRef]
- Yu, Y.; Liu, H.; Yang, D.; He, F.; Yuan, Y.; Guo, J.; Hu, J.; Yu, J.; Yan, X.; Wang, S.; et al. Aloe-emodin attenuates myocardial infarction and apoptosis via up-regulating miR-133 expression. Pharmacol. Res. 2019, 146. [Google Scholar] [CrossRef]
- Sun, C.; Liu, H.; Guo, J.; Yu, Y.; Yang, D.; He, F.; Du, Z. MicroRNA-98 negatively regulates myocardial infarction-induced apoptosis by down-regulating Fas and caspase-3. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Qiu, Z.; Wang, L.; Mao, H.; Xu, F.; Sun, B.; Lian, X.; Wang, J.; Kong, F.; Wang, L.; Chen, Y. MiR-370 inhibits the oxidative stress and apoptosis of cardiac myocytes induced by hydrogen peroxide by targeting FOXO1. Exp. Ther. Med. 2019, 3025–3031. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Li, G.; Chen, L.; Guo, J.; Liu, Y. Downregulation of microRNA-100 protects H2O2-induced apoptosis in neonatal cardiomyocytes. Int. J. Clin. Exp. Pathol. 2015, 8, 5491–5496. [Google Scholar] [PubMed]
- Gogiraju, R.; Schroeter, M.R.; Bochenek, M.L.; Hubert, A.; Münzel, T.; Hasenfuss, G.; Schäfer, K. Endothelial deletion of protein tyrosine phosphatase-1B protects against pressure overload-induced heart failure in mice. Cardiovasc. Res. 2016, 111, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Sun, Y.; Yu, B. MicroRNA-208a Correlates Apoptosis and Oxidative Stress Induced by H 2 O 2 through Protein Tyrosine Kinase/Phosphatase Balance in Cardiomyocytes. Int. Hear. J. Assoc. 2018, 59, 829–836. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Li, M.Y.; Su, Y.F.; Fu, J.; Zou, Z.Y.; Wang, Y.; Li, S.N. Absence of miR-223-3p ameliorates hypoxia-induced injury through repressing cardiomyocyte apoptosis and oxidative stress by targeting KLF15. Eur. J. Pharmacol. 2018, 841, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Zhang, D.; Zhu, M.; Wang, Y.; Guo, S.; Xu, B.; Hou, G.; Feng, Y.; Liu, G. Liver X receptor α is targeted by microRNA-1 to inhibit cardiomyocyte apoptosis through a ROS-mediated mitochondrial pathway. Biochem. Cell Biol. 2018, 96, 11–18. [Google Scholar] [CrossRef]
- Qadir, M.M.F.; Klein, D.; Álvarez-Cubela, S.; Domínguez-Bendala, J.; Pastori, R.L. The role of microRNAs in diabetes-related oxidative stress. Int. J. Mol. Sci. 2019, 20, 5423. [Google Scholar] [CrossRef]
- Dludla, P.V.; Nkambule, B.B.; Dias, S.C.; Johnson, R. Cardioprotective potential of N-acetyl cysteine against hyperglycaemia-induced oxidative damage: A protocol for a systematic review. Syst. Rev. 2017, 6, 96. [Google Scholar] [CrossRef]
- Yang, X.; Li, X.; Lin, Q.; Xu, Q. Up-regulation of microRNA-203 inhibits myocardial fibrosis and oxidative stress in mice with diabetic cardiomyopathy through the inhibition of PI3K/Akt signaling pathway via PIK3CA. Gene 2019, 715, 143995. [Google Scholar] [CrossRef]
- Li, H.; Dai, B.; Fan, J.; Chen, C.; Nie, X.; Yin, Z.; Zhao, Y.; Zhang, X.; Wang, D.W. The Different Roles of miRNA-92a-2-5p and let-7b-5p in Mitochondrial Translation in db/db Mice. Mol. Ther. Nucleic Acids 2019, 17, 424–435. [Google Scholar] [CrossRef]
- Cantó, C.; Auwerx, J. PGC-1α, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Zhao, Y.; He, M.; Li, H.; Fan, J.; Nie, X.; Yan, M.; Chen, C.; Wang, D.W. MiR-30c/PGC-1β protects against diabetic cardiomyopathy via PPARα. Cardiovasc. Diabetol. 2019, 18, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Wan, Q.; Liu, X.; Wang, Y.; Luo, Y.; Liu, D.; Lin, N.; Zhou, H.; Zhong, J. miR-503 is involved in the protective effect of Phase II enzyme inducer (CPDT) in diabetic cardiomyopathy via Nrf2/ARE signaling pathway. Biomed Res. Int. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Ma, J.; Yu, Y.; Li, M.; Ni, R.; Wang, G.; Chen, R.; Li, J.; Fan, G.C.; Lacefield, J.C.; et al. Silencing of miR-195 reduces diabetic cardiomyopathy in C57BL/6 mice. Diabetologia 2015, 58, 1949–1958. [Google Scholar] [CrossRef]
- Castaldi, A.; Horie, M.; Rieger, M.E.; Dubourd, M.; Sunohara, M.; Pandit, K.; Zhou, B.; Offringa, I.A.; Marconett, C.; Borok, Z. Genome-wide integration of microRNA and transcriptomic profiles of differentiating human alveolar epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, published. [Google Scholar] [CrossRef]
- Das, S.; Kumar, M.; Negi, V.; Pattnaik, B.; Prakash, Y.S.; Agrawal, A.; Ghosh, B. MicroRNA-326 regulates profibrotic functions of transforming growth factor-β in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2014, 50, 882–892. [Google Scholar] [CrossRef]
- Pandit, K.V.; Corcoran, D.; Yousef, H.; Yarlagadda, M.; Tzouvelekis, A.; Gibson, K.F.; Konishi, K.; Yousem, S.A.; Singh, M.; Handley, D.; et al. Inhibition and role of let-7d in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2010, 182, 220–229. [Google Scholar] [CrossRef]
- Yang, S.; Banerjee, S.; De Freitas, A.; Sanders, Y.Y.; Ding, Q.; Matalon, S.; Thannickal, V.J.; Abraham, E.; Liu, G. Participation of miR-200 in pulmonary fibrosis. Am. J. Pathol. 2012, 180, 484–493. [Google Scholar] [CrossRef]
- Liang, C.; Li, X.; Zhang, L.; Cui, D.; Quan, X.; Yang, W. The anti-fibrotic effects of microRNA-153 by targeting TGFBR-2 in pulmonary fibrosis. Exp. Mol. Pathol. 2015, 99, 279–285. [Google Scholar] [CrossRef]
- Dakhlallah, D.; Batte, K.; Wang, Y.; Cantemir-Stone, C.Z.; Yan, P.; Nuovo, G.; Mikhail, A.; Hitchcock, C.L.; Wright, V.P.; Nana-Sinkam, S.P.; et al. Epigenetic regulation of mir-17∼92 contributes to the pathogenesis of pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2013, 187, 397–405. [Google Scholar] [CrossRef]
- Liu, G.; Friggeri, A.; Yang, Y.; Milosevic, J.; Ding, Q.; Thannickal, V.J.; Kaminski, N.; Abraham, E. miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J. Exp. Med. 2010, 207, 1589–1597. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Xu, Q.; Zhang, Q.; Wang, Z.; Guan, S. A novel molecular mechanism of microRNA-21 inducing pulmonary fibrosis and human pulmonary fibroblast extracellular matrix through transforming growth factor β1–mediated SMADs activation. J. Cell. Biochem. 2018, 119, 7834–7843. [Google Scholar] [CrossRef]
- Cameli, P.; Carleo, A.; Bergantini, L.; Landi, C.; Prasse, A.; Bargagli, E. Oxidant/Antioxidant Disequilibrium in Idiopathic Pulmonary Fibrosis Pathogenesis. Inflammation 2020, 43. [Google Scholar] [CrossRef] [PubMed]
- Borok, Z.; Horie, M.; Flodby, P.; Wang, H.; Liu, Y.; Ganesh, S.; Firth, A.L.; Minoo, P.; Li, C.; Beers, M.F.; et al. Grp78 loss in epithelial progenitors reveals an age-linked role for endoplasmic reticulum stress in pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2020, 201, 198–211. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Hu, H.; Jiang, Z.; Tang, S.; Zhou, Y.; Sheng, J.; Chen, J.; Cao, Y. APACHE score, severity index of paraquat poisoning, and serum lactic acid concentration in the prognosis of paraquat poisoning of Chinese patients. Pediatr. Emerg. Care 2015, 31, 117–121. [Google Scholar] [CrossRef]
- Liu, M.W.; Su, M.X.; Tang, D.Y.; Hao, L.; Xun, X.H.; Huang, Y.Q. Ligustrazin increases lung cell autophagy and ameliorates paraquat-induced pulmonary fibrosis by inhibiting PI3K/Akt/mTOR and hedgehog signalling via increasing miR-193a expression. BMC Pulm. Med. 2019, 19, 35. [Google Scholar] [CrossRef]
- Kang, J.Y.; Kim, J.J.; Jang, S.Y.; Bae, Y.S. The p53-p21Cip1/WAF1 pathway is necessary for cellular senescence induced by the inhibition of protein kinase CKII in human colon cancer cells. Mol. Cells 2009, 28, 489–494. [Google Scholar] [CrossRef]
- Lee, Y.H.; Kim, S.Y.; Bae, Y.S. Upregulation of miR-760 and miR-186 is associated with replicative senescence in human lung fibroblast cells. Mol. Cells 2014, 37, 620–627. [Google Scholar] [CrossRef]
- Fierro-Fernández, M.; Busnadiego, Ó.; Sandoval, P.; Espinosa-Díez, C.; Blanco-Ruiz, E.; Rodríguez, M.; Pian, H.; Ramos, R.; López-Cabrera, M.; García-Bermejo, M.L.; et al. miR-9-5p suppresses pro-fibrogenic transformation of fibroblasts and prevents organ fibrosis by targeting NOX 4 and TGFBR 2. EMBO Rep. 2015, 16, 1358–1377. [Google Scholar] [CrossRef]
- Wynn, T.A. Integrating mechanisms of pulmonary fibrosis. J. Exp. Med. 2011, 208, 1339–1350. [Google Scholar] [CrossRef]
- Amara, N.; Goven, D.; Prost, F.; Muloway, R.; Crestani, B.; Boczkowski, J. NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGFβ1-induced fibroblast differentiation into myofibroblasts. Thorax 2010, 65, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Cucoranu, I.; Clempus, R.; Dikalova, A.; Phelan, P.J.; Ariyan, S.; Dikalov, S.; Sorescu, D. NAD(P)H oxidase 4 mediates transforming growth factor-β1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ. Res. 2005, 97, 900–907. [Google Scholar] [CrossRef]
- Hecker, L.; Vittal, R.; Jones, T.; Jagirdar, R.; Luckhardt, T.R.; Horowitz, J.C.; Pennathur, S.; Martinez, F.J.; Thannickal, V.J. NADPH Oxidase-4 Mediates Myofibroblast Activation and Fibrogenic Responses to Lung Injury. Nat. Med. 2009, 15, 1077–1081. [Google Scholar] [CrossRef]
- Kellner, M.; Noonepalle, S.; Lu, Q.; Srivastava, A.; Zemskov, E.; Black, S.M. ROS Signaling in the Pathogenesis of Acute Lung Injury (ALI) and Acute Respiratory Distress Syndrome (ARDS). Adv. Exp. Med. Biol. 2017, 967, 105–137. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wu, C.; Gu, W.; Ji, H.; Zhu, L. Serum Exosomal MicroRNAs Predict Acute Respiratory Distress Syndrome Events in Patients with Severe Community-Acquired Pneumonia. Biomed Res. Int. 2019, 2019. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-M.; Fan, L.M.; Christie, M.R.; Shah, A.M. Acute Tumor Necrosis Factor Alpha Signaling via NADPH Oxidase in Microvascular Endothelial Cells: Role of p47phox Phosphorylation and Binding to TRAF4. Mol. Cell. Biol. 2005, 25, 2320–2330. [Google Scholar] [CrossRef]
- Di, A.; Kawamura, T.; Gao, X.P.; Tang, H.; Berdyshev, E.; Vogel, S.M.; Zhao, Y.Y.; Sharma, T.; Bachmaier, K.; Xu, J.; et al. A novel function of sphingosine kinase 1 suppression of JNK activity in preventing inflammation and injury. J. Biol. Chem. 2010, 285, 15848–15857. [Google Scholar] [CrossRef]
- Wang, T.; Liu, Y.P.; Wang, T.; Xu, B.Q.; Xu, B. ROS feedback regulates the microRNA-19-targeted inhibition of the p47phox-mediated LPS-induced inflammatory response. Biochem. Biophys. Res. Commun. 2017, 489, 361–368. [Google Scholar] [CrossRef]
- Lee, H.; Zhang, D.; Zhu, Z.; Dela Cruz, C.S.; Jin, Y. Epithelial cell-derived microvesicles activate macrophages and promote inflammation via microvesicle-containing microRNAs. Sci. Rep. 2016, 6, 35250. [Google Scholar] [CrossRef]
- Zhang, D.; Lee, H.; Cao, Y.; Dela Cruz, C.S.; Jin, Y. miR-185 mediates lung epithelial cell death after oxidative stress. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L700–L710. [Google Scholar] [CrossRef]
- Mach, W.J.; Thimmesch, A.R.; Pierce, J.T.; Pierce, J.D. Consequences of Hyperoxia and the Toxicity of Oxygen in the Lung. Nurs. Res. Pract. 2011, 2011, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Han, C.; Zhang, J.; Song, K.; Wang, Y.; Wu, T. MiR-150 deficiency protects against FASInduced acute liver injury in mice through regulation of AKT. PLoS ONE 2015, 10, e0132734. [Google Scholar] [CrossRef]
- Zhang, D.; Lee, H.; Haspel, J.A.; Jin, Y. Long noncoding RNA FOXD3-AS1 regulates oxidative stress-induced apoptosis via sponging microRNA-150. FASEB J. 2017, 31, 4472–4481. [Google Scholar] [CrossRef] [PubMed]
- Mishra, V.; Banga, J.; Silveyra, P. Oxidative stress and cellular pathways of asthma and inflammation: Therapeutic strategies and pharmacological targets. Pharmacol. Ther. 2018, 181, 169–182. [Google Scholar] [CrossRef]
- Barnes, P.J. Cellular and molecular mechanisms of asthma and COPD. Clin. Sci. 2017, 131, 1541–1558. [Google Scholar] [CrossRef]
- Kirkham, P.A.; Barnes, P.J. Oxidative stress in COPD. Chest 2013, 144, 266–273. [Google Scholar] [CrossRef]
- Qian, Y.; Mao, Z.D.; Shi, Y.J.; Liu, Z.G.; Cao, Q.; Zhang, Q. Comprehensive Analysis of miRNA-mRNA-lncRNA Networks in Non-Smoking and Smoking Patients with Chronic Obstructive Pulmonary Disease. Cell. Physiol. Biochem. 2018, 50, 1154–1163. [Google Scholar] [CrossRef]
- Ba, Q.; Cui, C.; Wen, L.; Feng, S.; Zhou, J.; Yang, K. Schisandrin B shows neuroprotective effect in 6-OHDA-induced Parkinson’s disease via inhibiting the negative modulation of miR-34a on Nrf2 pathway. Biomed. Pharmacother. 2015, 75, 165–172. [Google Scholar] [CrossRef]
- Srinoun, K.; Sathirapongsasuti, N.; Paiboonsukwong, K.; Sretrirutchai, S.; Wongchanchailert, M.; Fucharoen, S. miR-144 regulates oxidative stress tolerance of thalassemic erythroid cell via targeting NRF2. Ann. Hematol. 2019, 98, 2045–2052. [Google Scholar] [CrossRef]
- Ho, C.Y.; Lu, C.C.; Weng, C.J.; Yen, G.C. Protective Effects of Diallyl Sulfide on Ovalbumin-Induced Pulmonary Inflammation of Allergic Asthma Mice by MicroRNA-144, -34a, and -34b/c-Modulated Nrf2 Activation. J. Agric. Food Chem. 2016, 64, 151–160. [Google Scholar] [CrossRef]
- Mizuno, S.; Bogaard, H.J.; Gomez-Arroyo, J.; Alhussaini, A.; Kraskauskas, D.; Cool, C.D.; Voelkel, N.F. MicroRNA-199a-5p is associated with hypoxia-inducible factor-1α expression in lungs from patients with COPD. Chest 2012, 142, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, K.; Loridas, S. Pulmonary oxidative stress, inflammation and cancer: Respirable particulate matter, fibrous dusts and ozone as major causes of lung carcinogenesis through reactive oxygen species mechanisms. Int. J. Environ. Res. Public Health 2013, 10, 3886–3907. [Google Scholar] [CrossRef]
- Lu, Y.; Thomson, J.M.; Wang, H.Y.F.; Hammond, S.M.; Hogan, B.L.M. Transgenic over-expression of the microRNA miR-17-92 cluster promotes proliferation and inhibits differentiation of lung epithelial progenitor cells. Dev. Biol. 2007, 310, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Ventura, A.; Young, A.G.; Winslow, M.M.; Lintault, L.; Meissner, A.; Erkel, S.J.; Newman, J.; Bronson, R.T.; Crowley, D.; Stone, J.R.; et al. Targeted Deletion Reveals Essential and Overlapping Functions of the miR-17-92 Family of miRNA Clusters. Cell 2008, 132, 875–886. [Google Scholar] [CrossRef]
- Ebi, H.; Sato, T.; Sugito, N.; Hosono, Y.; Yatabe, Y.; Matsuyama, Y.; Yamaguchi, T.; Osada, H.; Suzuki, M.; Takahashi, T. Counterbalance between RB inactivation and miR-17-92 overexpression in reactive oxygen species and DNA damage induction in lung cancers. Oncogene 2009, 28, 3371–3379. [Google Scholar] [CrossRef] [PubMed]
- Tort, F.; Bartkova, J.; Sehested, M.; Oørntoft, T.; Lukas, J.; Bartek, J. Retinoblastoma pathway defects show differential ability to activate the constitutive DNA damage response in human tumorigenesis. Cancer Res. 2006, 66, 10258–10263. [Google Scholar] [CrossRef]
- O’Donnell, K.A.; Wentzel, E.A.; Zeller, K.I.; Dang, C.V.; Mendell, J.T. c-Myc-regulated microRNAs modulate E2F1 expression. Nature 2005, 435, 839–843. [Google Scholar] [CrossRef]
- He, J.; Wang, M.; Jiang, Y.; Chen, Q.; Xu, S.; Xu, Q.; Jiang, B.H.; Liu, L.Z. Chronic arsenic exposure and angiogenesis in human bronchial epithelial Cells via the ROS/miR-199a-5p/HIF-1α/COX-2 pathway. Environ. Health Perspect. 2014, 122, 255–261. [Google Scholar] [CrossRef]
- Wang, Q.; Ye, B.; Wang, P.; Yao, F.; Zhang, C.; Yu, G. Overview of microRNA-199a regulation in cancer. Cancer Manag. Res. 2019, 11, 10327–10335. [Google Scholar] [CrossRef]
- Mishra, R.; Benlhabib, H.; Guo, W.; Lerma Cervantes, C.B.; Mendelson, C.R. Developmental Decline in the MicroRNA 199a (miR-199a)/miR-214 Cluster in Human Fetal Lung Promotes Type II Cell Differentiation by Upregulating Key Transcription Factors. Mol. Cell. Biol. 2018, 38, e00037-18. [Google Scholar] [CrossRef] [PubMed]
- Lino Cardenas, C.L.; Henaoui, I.S.; Courcot, E.; Roderburg, C.; Cauffiez, C.; Aubert, S.; Copin, M.C.; Wallaert, B.; Glowacki, F.; Dewaeles, E.; et al. miR-199a-5p Is Upregulated during Fibrogenic Response to Tissue Injury and Mediates TGFbeta-Induced Lung Fibroblast Activation by Targeting Caveolin-1. PLoS Genet. 2013, 9. [Google Scholar] [CrossRef]
- Carpenter, R.L.; Jiang, Y.; Jing, Y.; He, J.; Rojanasakul, Y.; Liu, L.Z.; Jiang, B.H. Arsenite induces cell transformation by reactive oxygen species, AKT, ERK1/2, and p70S6K1. Biochem. Biophys. Res. Commun. 2011, 414, 533–538. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Xu, Q.; Jing, Y.; Agani, F.; Qian, X.; Carpenter, R.; Li, Q.; Wang, X.R.; Peiper, S.S.; Lu, Z.; et al. Reactive oxygen species regulate ERBB2 and ERBB3 expression via miR-199a/125b and DNA methylation. EMBO Rep. 2012, 13, 1116–1122. [Google Scholar] [CrossRef]
- Gately, S.; Li, W.W. Multiple roles of COX-2 in tumor angiogenesis: A target for antiangiogenic therapy. Semin. Oncol. 2004, 31, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Peng, Y.; Wang, W.; Su, B. Rapid evolution of an X-linked microRNA cluster in primates. Genome Res. 2007, 17, 612–617. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Ren, X.; Zhang, X.; Luo, Y.; Wang, G.; Huang, K.; Feng, S.; Bao, X.; Huang, K.; He, X.; et al. Selective killing of lung cancer cells by miRNA-506 molecule through inhibiting NF-κB p65 to evoke reactive oxygen species generation and p53 activation. Oncogene 2015, 34, 691–703. [Google Scholar] [CrossRef]
- Sun, M.; Hong, S.; Li, W.; Wang, P.; You, J.; Zhang, X.; Tang, F.; Wang, P.; Zhang, C. MIR-99a regulates ROS-mediated invasion and migration of lung adenocarcinoma cells by targeting NOX4. Oncol. Rep. 2016, 35, 2755–2766. [Google Scholar] [CrossRef]
- Wang, H.; Zhu, Y.; Zhao, M.; Wu, C.; Zhang, P.; Tang, L.; Zhang, H.; Chen, X.; Yang, Y.; Liu, G. miRNA-29c Suppresses Lung Cancer Cell Adhesion to Extracellular Matrix and Metastasis by Targeting Integrin β1 and Matrix Metalloproteinase2 (MMP2). PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Kogo, R.; How, C.; Chaudary, N.; Bruce, J.; Shi, W.; Hill, R.P.; Zahedi, P.; Yip, K.W.; Liu, F.F. The microRNA-218~Survivin axis regulates migration, invasion, and lymph node metastasis in cervical cancer. Oncotarget 2015, 6, 1090–1100. [Google Scholar] [CrossRef]
- Li, G.; Li, M.; Hu, J.; Lei, R.; Xiong, H.; Ji, H.; Yin, H.; Wei, Q.; Hu, G. The microRNA-182-PDK4 axis regulates lung tumorigenesis by modulating pyruvate dehydrogenase and lipogenesis. Oncogene 2017, 36, 989–998. [Google Scholar] [CrossRef]
- Maione, P.; Rossi, A.; Sacco, P.C.; Bareschino, M.A.; Schettino, C.; Gridelli, C. Advances in chemotherapy in advanced non-small-cell lung cancer. Expert Opin. Pharmacother. 2010, 11, 2997–3007. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, Q.; Li, D.; Zhou, Y.; Zheng, X.; Sun, H.; Yan, J.; Zhang, L.; Lin, Y.; Wang, X. Wogonin enhances antitumor activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo through ROS-mediated downregulation of cFLIPL and IAP proteins. Apoptosis 2013, 18, 618–626. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, W.; Bai, L.; Chen, W.; Padilla, M.T.; Lin, A.S.; Shi, S.; Wang, X.; Lin, Y. Receptor-interacting protein 1 increases chemoresistance by maintaining inhibitor of apoptosis protein levels and reducing reactive oxygen species through a microRNA-146a-mediated catalase pathway. J. Biol. Chem. 2014, 289, 5654–5663. [Google Scholar] [CrossRef]
- Guddo, F.; Giatromanolaki, A.; Koukourakis, M.I.; Reina, C.; Vignola, A.; Chlouverakis, G.; Hilkens, J.; Gatter, K.C.; Harris, A.L.; Bonsignore, G. MUC1 (episialin) expression in non-small cell lung cancer is independent of EGFR and c-erbB-2 expression and correlates with poor. J. Clin. Pathol. 1998, 51, 667–671. [Google Scholar] [CrossRef]
- Xu, X.; Wells, A.; Padilla, M.T.; Kato, K.; Kim, K.C.; Lin, Y. A signaling pathway consisting of miR-551b, catalase and MUC1 contributes to acquired apoptosis resistance and chemoresistance. Carcinogenesis 2014, 35, 2457–2466. [Google Scholar] [CrossRef]
- Christofidou-Solomidou, M.; Pietrofesa, R.; Arguiri, E.; McAlexander, M.A.; Witwer, K.W. Dietary flaxseed modulates the miRNA profile in irradiated and non-irradiated murine lungs: A novel mechanism of tissue radioprotection by flaxseed. Cancer Biol. Ther. 2014, 15, 930–937. [Google Scholar] [CrossRef]
- Zhang, L.; Liao, Y.; Tang, L. MicroRNA-34 family: A potential tumor suppressor and therapeutic candidate in cancer. J. Exp. Clin. Cancer Res. 2019, 38, 53. [Google Scholar] [CrossRef]
- Tu, L.; Long, X.; Song, W.; Lv, Z.; Zeng, H.; Wang, T.; Liu, X.; Dong, J.; Xu, P. MiR-34c acts as a tumor suppressor in non-small cell lung cancer by inducing endoplasmic reticulum stress through targeting HMGB1. Onco Targets Ther. 2019, 12, 5729–5739. [Google Scholar] [CrossRef]
- Khan, M.A.; Tania, M.; Fu, S.; Fu, J. Thymoquinone, as an anticancer molecule: From basic research to clinical investigation. Oncotarget 2017, 8, 51907–51919. [Google Scholar] [CrossRef] [PubMed]
- Mostofa, A.G.M.; Hossain, M.K.; Basak, D.; Sayeed, M.S. Bin Thymoquinone as a potential adjuvant therapy for cancer treatment: Evidence from preclinical studies. Front. Pharmacol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, P.; Sarker, S.; Ghosh, A.; Gupta, P.; Das, S.; Ahir, M.; Bhattacharya, S.; Chattopadhyay, S.; Ghosh, S.; Adhikary, A. Transferrin-decorated thymoquinone-loaded PEG-PLGA nanoparticles exhibit anticarcinogenic effect in non-small cell lung carcinoma: Via the modulation of miR-34a and miR-16. Biomater. Sci. 2019, 7, 4325–4344. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Liu, T.; Xiao, Y.; Li, X.; Zhu, Y.; Zhao, Y.; Bao, J.; Wu, C. Polygonatum odoratum lectin induces apoptosis and autophagy by regulation of microRNA-1290 and microRNA-15a-3p in human lung adenocarcinoma A549 cells. Int. J. Biol. Macromol. 2016, 85, 217–226. [Google Scholar] [CrossRef]
- Li, J.; Zhou, Q.; Liang, Y.; Pan, W.; Bei, Y.; Zhang, Y.; Wang, J.; Jiao, Z. miR-486 inhibits PM2.5-induced apoptosis and oxidative stress in human lung alveolar epithelial A549 cells. Ann. Transl. Med. 2018, 6, 209. [Google Scholar] [CrossRef] [PubMed]
- Veerappan, I.; Sankareswaran, S.K.; Palanisamy, R. Morin protects human respiratory cells from PM2.5 induced genotoxicity by mitigating ROS and reverting altered miRNA expression. Int. J. Environ. Res. Public Health 2019, 16, 2389. [Google Scholar] [CrossRef] [PubMed]
- Lusha, E.; Jiang, H.; Lu, Z. MicroRNA—144 attenuates cardiac ischemia / reperfusion injury by targeting FOXO1. Exp. Ther. Med. 2019, 17, 2152–2160. [Google Scholar] [CrossRef]
- Liu, J.; Sun, F.; Wang, Y.; Yang, W.; Xiao, H.; Zhang, Y.; Lu, R.; Zhu, H.; Zhuang, Y.; Pan, Z.; et al. Suppression of microRNA-16 protects against acute myocardial infarction by reversing beta2-adrenergic receptor downregulation in rats. Oncotarget 2017, 8, 20122–20132. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhu, J.; Han, W.; Jiang, X.; Xu, M.; Zhao, Y.; Dong, Q.; Pang, Z.; Guan, Q.; Gao, L.; et al. Significance of serum microRNAs in pre-diabetes and newly diagnosed type 2 diabetes: A clinical study. Acta Diabetol. 2011, 48, 61–69. [Google Scholar] [CrossRef]
- Costantino, S.; Paneni, F.; Lüscher, T.F.; Cosentino, F. MicroRNA profiling unveils hyperglycaemic memory in the diabetic heart. Eur. Heart J. 2016, 37, 572–576. [Google Scholar] [CrossRef]
- Li, Q.; Kim, Y.R.; Vikram, A.; Kumar, S.; Kassan, M.; Gabani, M.; Lee, S.K.; Jacobs, J.S.; Irani, K. P66Shc-Induced MicroRNA-34a Causes Diabetic Endothelial Dysfunction by Downregulating Sirtuin1. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2394–2403. [Google Scholar] [CrossRef]
- Zhu, Y.; Qian, X.; Li, J.; Lin, X.; Luo, J.; Huang, J.; Jin, Z. Astragaloside-IV protects H9C2(2-1) cardiomyocytes from high glucose-induced injury via miR-34a-mediated autophagy pathway. Artif. Cells Nanomed. Biotechnol. 2019, 47, 4172–4181. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Duan, F.G.; Wang, M.F.; Cao, Y.B.; Li, D.; Li, R.Z.; Fan, X.X.; Khan, I.; Lai, H.L.; Zhang, Y.Z.; Hsiao, W.W.L.; et al. MicroRNA-421 confers paclitaxel resistance by binding to the KEAP1 3′UTR and predicts poor survival in non-small cell lung cancer. Cell Death Dis. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cui, X.; Li, Y.; Zhang, T.; Li, S. Upregulated expression of miR-421 is associated with poor prognosis in non-small-cell lung cancer. Cancer Manag. Res. 2018, 10, 2627–2633. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D.; Hannink, M. Distinct Cysteine Residues in Keap1 Are Required for Keap1-Dependent Ubiquitination of Nrf2 and for Stabilization of Nrf2 by Chemopreventive Agents and Oxidative Stress. Mol. Cell. Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Tang, D.; Kang, R. Oxidative stress-mediated HMGB1 biology. Front. Physiol. 2015, 6. [Google Scholar] [CrossRef]
- Xiao, N.; Zhang, J.; Chen, C.; Wan, Y.; Wang, N.; Yang, J. miR-129-5p improves cardiac function in rats with chronic heart failure through targeting HMGB1. Mamm. Genome 2019, 30, 276–288. [Google Scholar] [CrossRef]
- Sholler, G.S.; Currier, E.A.; Dutta, A.; Slavik, M.A.; Illenye, S.A.; Mendonca, M.C.F.; Dragon, J.; Roberts, S.S.; Bond, J.P. PCI-24781 (abexinostat), a novel histone deacetylase inhibitor, induces reactive oxygen species-dependent apoptosis and is synergistic with bortezomib in neuroblastoma Giselle. J. Cancer Res. Ther. 2014, 2, 21. [Google Scholar] [CrossRef]
- Zhu, Y.; Das, K.; Wu, J.; Lee, M.H.; Tan, P. RNH1 regulation of reactive oxygen species contributes to histone deacetylase inhibitor resistance in gastric cancer cells. Oncogene 2014, 33, 1527–1537. [Google Scholar] [CrossRef]
- You, B.R.; Park, W.H. Suberoylanilide hydroxamic acid induces thioredoxin1-mediated apoptosis in lung cancer cells via up-regulation of miR-129-5p. Mol. Carcinog. 2017, 56, 2566–2577. [Google Scholar] [CrossRef]
- Li, G.; Xie, J.; Wang, J. Tumor suppressor function of mir-129-5p in lung cancer. Oncol. Lett. 2019, 17, 5777–5783. [Google Scholar] [CrossRef]
- Wu, Y.F.; Ou, C.C.; Chien, P.J.; Chang, H.Y.; Ko, J.L.; Wang, B.Y. Chidamide-induced ROS accumulation and miR-129-3p-dependent cell cycle arrest in non-small lung cancer cells. Phytomedicine 2019, 56, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Bourens, M.; Fontanesi, F.; Soto, I.C.; Liu, J.; Barrientos, A. Redox and reactive oxygen species regulation of mitochondrial cytochrome C oxidase biogenesis. Antioxid. Redox Signal. 2013, 19, 1940–1952. [Google Scholar] [CrossRef] [PubMed]
- Dennerlein, S.; Rehling, P. Human mitochondrial COX1 assembly into cytochrome c oxidase at a glance. J. Cell Sci. 2015, 128, 833–837. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Bedja, D.; Campbell, N.; Dunkerly, B.; Chenna, V.; Maitra, A.; Steenbergen, C. miR-181c regulates the mitochondrial genome, bioenergetics, and propensity for heart failure in vivo. PLoS ONE 2014, 9, e96820. [Google Scholar] [CrossRef]
- Du, Y.; Ding, Y.; Chen, X.; Mei, Z.; Ding, H.; Wu, Y.; Jie, Z. MicroRNA-181c inhibits cigarette smoke-induced chronic obstructive pulmonary disease by regulating CCN1 expression. Respir. Res. 2017, 18, 155. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; Guo, S.; Zhang, T.; Yang, Y.; Zhao, G.; Shaukat, A.; Wu, H.; Deng, G. Downregulation of TLR4 by miR-181a provides negative feedback regulation to lipopolysaccharide-induced inflammation. Front. Pharmacol. 2018, 9, 142. [Google Scholar] [CrossRef]
- Martinez, E.C.; Lilyanna, S.; Wang, P.; Vardy, L.A.; Jiang, X.; Armugam, A.; Jeyaseelan, K.; Richards, A.M. MicroRNA-31 promotes adverse cardiac remodeling and dysfunction in ischemic heart disease. J. Mol. Cell. Cardiol. 2017, 112, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Zhang, L.; Meng, J. Circular RNA ANKRD36 attends to lipopolysaccharide-aroused MRC-5 cell injury via regulating microRNA-31-3p. BioFactors 2019. [Google Scholar] [CrossRef]
- Gulei, D.; Raduly, L.; Broseghini, E.; Ferracin, M.; Berindan-Neagoe, I. The extensive role of miR-155 in malignant and non-malignant diseases. Mol. Asp. Med. 2019, 70, 33–56. [Google Scholar] [CrossRef]
- Tam, W.; Ben-Yehuda, D.; Hayward, W.S. Bic, a novel gene activated by proviral insertions in avian leukosis virus-induced lymphomas, is likely to function through its noncoding RNA. Mol. Cell. Biol. 1997, 17, 1490–1502. [Google Scholar] [CrossRef]
- Gulei, D.; Irimie, A.I.; Cojocneanu-Petric, R.; Schultze, J.L.; Berindan-Neagoe, I. Exosomes—Small Players, Big Sound. Bioconjug. Chem. 2018, 29, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Bayraktar, R.; Van Roosbroeck, K. miR-155 in cancer drug resistance and as target for miRNA-based therapeutics. Cancer Metastasis Rev. 2018, 37, 33–44. [Google Scholar] [CrossRef]
- Tan, W.Q.; Wang, K.; Lv, D.Y.; Li, P.F. Foxo3a inhibits cardiomyocyte hypertrophy through transactivating catalase. J. Biol. Chem. 2008, 283, 29730–29739. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.L.; Zhang, H.G.; Xu, Y.L.; Gao, Y.H.; Yang, X.J.; Hao, X.Q.; Li, X.H. Resveratrol inhibits right ventricular hypertrophy induced by monocrotaline in rats. Clin. Exp. Pharmacol. Physiol. 2010, 37, 150–155. [Google Scholar] [CrossRef]
- Fan, Y.; Liu, L.; Fang, K.; Huang, T.; Wan, L.; Liu, Y.; Zhang, S.; Yan, D.; Li, G.; Gao, Y.; et al. Resveratrol ameliorates cardiac hypertrophy by down-regulation of miR-155 through activation of breast cancer type 1 susceptibility protein. J. Am. Heart Assoc. 2016, 5, e002648. [Google Scholar] [CrossRef] [PubMed]
- Rosell, R.; Skrzypski, M.; Jassem, E.; Taron, M.; Bartolucci, R.; Sanchez, J.J.; Mendez, P.; Chaib, I.; Perez-Roca, L.; Szymanowska, A.; et al. BRCA1: A novel prognostic factor in resected non-small-cell lung cancer. PLoS ONE 2007, 2. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Chen, J.; Zheng, Y.; Wu, C. Critical role of miR-155/FoxO1/ROS axis in the regulation of non-small cell lung carcinomas. Tumor Biol. 2016, 37, 5185–5192. [Google Scholar] [CrossRef] [PubMed]
- Cittadini, A.; Monti, M.G.; Iaccarino, G.; Castiello, M.C.; Baldi, A.; Bossone, E.; Longobardi, S.; Marra, A.M.; Petrillo, V.; Saldamarco, L.; et al. SOCS1 gene transfer accelerates the transition to heart failure through the inhibition of the gp130/JAK/STAT pathway. Cardiovasc. Res. 2012, 96, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Eisenhardt, S.U.; Weiss, J.B.W.; Smolka, C.; Maxeiner, J.; Pankratz, F.; Bemtgen, X.; Kustermann, M.; Thiele, J.R.; Schmidt, Y.; Bjoern Stark, G.; et al. MicroRNA-155 aggravates ischemia–reperfusion injury by modulation of inflammatory cell recruitment and the respiratory oxidative burst. Basic Res. Cardiol. 2015, 110, s00395–s015. [Google Scholar] [CrossRef]
- Qiu, L.; Zhang, Y.; Do, D.C.; Ke, X.; Zhang, S.; Lambert, K.; Kumar, S.; Hu, C.; Zhou, Y.; Ishmael, F.T.; et al. miR-155 Modulates Cockroach Allergen and Oxidative Stress- Induced Cyclooxygenase-2 in Asthma. J. Immunol. 2018, 201, 916–929. [Google Scholar] [CrossRef]
- Kurosu, T.; Fukuda, T.; Miki, T.; Miura, O. BCL6 overexpression prevents increase in reactive oxygen species and inhibits apoptosis induced by chemotherapeutic reagents in B-cell lymphoma cells. Oncogene 2003, 22, 4459–4468. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Liu, Y.; Guo, S.; Xiao, L.; Wu, L.; Wang, Z.; Liang, C.; Yao, R.; Zhang, Y. LAZ3 protects cardiac remodeling in diabetic cardiomyopathy via regulating miR-21/PPARa signaling. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3322–3338. [Google Scholar] [CrossRef] [PubMed]
- Tochhawng, L.; Deng, S.; Pugalenthi, G.; Prem, A.; Hon Lim, K.; Zea Tan, T.; Yang, H.; Chuan Hooi, S.; Chong Goh, Y.; Maciver, S.K.; et al. Gelsolin-Cu/ZnSOD interaction alters intracellular reactive oxygen species levels to promote cancer cell invasion. Oncotarget 2016, 7, 52832. [Google Scholar] [CrossRef] [PubMed]
- Weisser-Thomas, J.; Kempelmann, H.; Nickenig, G.; Grohe, C.; Djoufack, P.; Fink, K.; Meyer, R. Influence of gelsolin deficiency on excitation contraction coupling in adult murine cardiomyocytes. J. Physiol. Pharmacol. 2015, 66, 373–383. [Google Scholar] [PubMed]
- Dai, B.; Li, H.; Fan, J.; Zhao, Y.; Yin, Z.; Nie, X.; Wang, D.W.; Chen, C. MiR-21 protected against diabetic cardiomyopathy induced diastolic dysfunction by targeting gelsolin. Cardiovasc. Diabetol. 2018, 17, 1–17. [Google Scholar] [CrossRef]
- Cheng, Y.; Liu, X.; Zhang, S.; Lin, Y.; Yang, J.; Zhang, C. MicroRNA-21 protects against the H2O2 -induced injury on cardiac myocytes via its target gene PDCD4. J. Mol. Cell. Cardiol. 2009, 47, 5–14. [Google Scholar] [CrossRef]
- Rippe, C.; Blimline, M.; Magerko, K.A.; Lawson, B.R.; Donato, A.J.; Seals, D.R. MicroRNA Changes in Human Arterial Endothelial Cells with Senescence: Relation to Apoptosis, eNOS and Inflammation. Exp. Gerontol. 2012, 47, 45–51. [Google Scholar] [CrossRef]
- Araneda, O.F.; Tuesta, M. Lung oxidative damage by hypoxia. Oxid. Med. Cell. Longev. 2012, 2012. [Google Scholar] [CrossRef]
- Jiang, C.; Guo, Y.; Yu, H.; Lu, S.; Meng, L. Pleiotropic microRNA-21 in pulmonary remodeling: Novel insights for molecular mechanism and present advancements. Allergy Asthma Clin. Immunol. 2019, 15, 33. [Google Scholar] [CrossRef]
- Sarkar, J.; Gou, D.; Turaka, P.; Viktorova, E.; Ramchandran, R.; Raj, J.U. MicroRNA-21 plays a role in hypoxia-mediated pulmonary artery smooth muscle cell proliferation and migration. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 299, 861–871. [Google Scholar] [CrossRef]
- Kumari, S.; Badana, A.K.; Murali Mohan, G.; Shailender, G.; Malla, R.R. Reactive Oxygen Species: A Key Constituent in Cancer Survival. Biomark. Insights 2018, 13. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, C.; Shan, S.; Liu, X.; Jiang, Z.; Ren, T. TLR4/ROS/miRNA-21 pathway underlies lipopolysaccharide instructed primary tumor outgrowth in lung cancer patients. Oncotarget 2016, 7, 42172–42182. [Google Scholar] [CrossRef]
- Pratheeshkumar, P.; Son, Y.O.; Divya, S.P.; Turcios, L.; Roy, R.V.; Hitron, J.A.; Wang, L.; Kim, D.; Dai, J.; Asha, P.; et al. Hexavalent chromium induces malignant transformation of human lung bronchial epithelial cells via ROS-dependent activation of miR-21-PDCD4 signaling. Oncotarget 2016, 7, 51193–51210. [Google Scholar] [CrossRef] [PubMed]
- Pratheeshkumar, P.; Son, Y.-O.; Divya, S.P.; Wang, L.; Turcios, L.; Roy, R.V.; Hitron, J.A.; Kim, D.; Dai, J.; Asha, P.; et al. Quercetin inhibits Cr(VI)-induced malignant cell transformation by targeting miR-21-PDCD4 signaling pathway. Oncotarget 2017, 8, 52118–52131. [Google Scholar] [CrossRef]
- Jajoo, S.; Mukherjea, D.; Kaur, T.; Sheehan, K.E.; Sheth, S.; Borse, V.; Rybak, L.P.; Ramkumar, V. Essential role of NADPH oxidase-dependent reactive oxygen species generation in regulating MicroRNA-21 expression and function in prostate cancer. Antioxid. Redox Signal. 2013, 19, 1863–1876. [Google Scholar] [CrossRef] [PubMed]
- Ling, M.; Li, Y.; Xu, Y.; Pang, Y.; Shen, L.; Jiang, R.; Zhao, Y.; Yang, X.; Zhang, J.; Zhou, J.; et al. Regulation of miRNA-21 by reactive oxygen species-activated ERK/NF-κB in arsenite-induced cell transformation. Free Radic. Biol. Med. 2012, 52, 1508–1518. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Bowman, L.; Meighan, T.; Pratheeshkumar, P.; Shi, X.; Ding, M. Induction of miR-21-PDCD4 signaling by UVB in JB6 cells involves ROS-mediated MAPK pathways. Exp. Toxicol. Pathol. 2013, 65, 1145–1148. [Google Scholar] [CrossRef] [PubMed]
- Iyer, R.; Lehnert, B.E. Factors underlying the cell growth-related bystander responses to α particles. Cancer Res. 2000, 60, 1290–1298. [Google Scholar] [PubMed]
- Shao, C.; Folkard, M.; Prise, K.M. Role of TGF-Β1 and nitric oxide in the bystander response of irradiated glioma cells. Oncogene 2008, 27, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Gow, M.D.; Seymour, C.B.; Ryan, L.A.; Mothersill, C.E. Induction of Bystander Response in Human Glioma Cells using High-Energy Electrons: A Role for TGF-β1. Radiat. Res. 2010, 173, 769–778. [Google Scholar] [CrossRef]
- Temme, J.; Bauer, G. Low-Dose Gamma Irradiation Enhances Superoxide Anion Production by Nonirradiated Cells Through TGF-β1-Dependent Bystander Signaling. Radiat. Res. 2013, 179, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Chen, X.; Tian, W.; Yin, X.; Wang, J.; Yang, H. The role of TGF-β1-miR-21-ROS pathway in bystander responses induced by irradiated non-small-cell lung cancer cells. Br. J. Cancer 2014, 111, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ng, W.-L.; Wang, P.; Tian, L.; Werner, E.; Wang, H.; Doetsch, P.; Wang, Y. MicroRNA-21 Modulates the Levels of Reactive Oxygen Species Levels by Targeting SOD3 and TNFα. Cancer Res. 2012, 72, 4707–4713. [Google Scholar] [CrossRef]
- Lin, C.Y.; Lee, H.C.; Fu, C.Y.; Ding, Y.Y.; Chen, J.S.; Lee, M.H.; Huang, W.J.; Tsai, H.J. MiR-1 and miR-206 target different genes to have opposing roles during angiogenesis in zebrafish embryos. Nat. Commun. 2013, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yuan, Y.; Li, J.; Ren, H.; Cai, Q.; Chen, X.; Liang, H.; Shan, H.; Fu, Z.D.; Gao, X.; et al. MicroRNA-1 aggravates cardiac oxidative stress by post-transcriptional modification of the antioxidant network. Cell Stress Chaperones 2015, 20, 411–420. [Google Scholar] [CrossRef]
- Wang, L.; Xu, J.; Liu, H.; Li, J.; Hao, H. PM2.5 inhibits SOD1 expression by up-regulating microRNA-206 and promotes ROS accumulation and disease progression in asthmatic mice. Int. Immunopharmacol. 2019, 76, 105871. [Google Scholar] [CrossRef]
- He, Q.; Pu, J.; Yuan, A.; Yao, T.; Ying, X.; Zhao, Y.; Xu, L.; Tong, H.; He, B. Liver X receptor agonist treatment attenuates cardiac dysfunction in type 2 diabetic db/db mice. Cardiovasc. Diabetol. 2014, 13, 149. [Google Scholar] [CrossRef]
- Yu, X.Y.; Song, Y.H.; Geng, Y.J.; Lin, Q.X.; Shan, Z.X.; Lin, S.G.; Li, Y. Glucose induces apoptosis of cardiomyocytes via microRNA-1 and IGF-1. Biochem. Biophys. Res. Commun. 2008, 376, 548–552. [Google Scholar] [CrossRef]
- Singh, A.; Happel, C.; Manna, S.K.; Acquaah-Mensah, G.; Carrerero, J.; Kumar, S.; Nasipuri, P.; Krausz, K.W.; Wakabayashi, N.; Dewi, R.; et al. Transcription factor NRF2 regulates miR-1 and miR-206 to drive tumorigenesis. J. Clin. Investig. 2013, 123, 2921–2934. [Google Scholar] [CrossRef]
- Lee, S.; Lim, S.; Ham, O.; Lee, S.Y.; Lee, C.Y.; Park, J.H.; Lee, J.; Seo, H.H.; Yun, I.; Han, S.M.; et al. ROS-mediated bidirectional regulation of miRNA results in distinct pathologic heart conditions. Biochem. Biophys. Res. Commun. 2015, 465, 349–355. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Climent, M.; Viggiani, G.; Chen, Y.-W.; Coulis, G.; Castaldi, A. MicroRNA and ROS Crosstalk in Cardiac and Pulmonary Diseases. Int. J. Mol. Sci. 2020, 21, 4370. https://doi.org/10.3390/ijms21124370
Climent M, Viggiani G, Chen Y-W, Coulis G, Castaldi A. MicroRNA and ROS Crosstalk in Cardiac and Pulmonary Diseases. International Journal of Molecular Sciences. 2020; 21(12):4370. https://doi.org/10.3390/ijms21124370
Chicago/Turabian StyleCliment, Montserrat, Giacomo Viggiani, Ya-Wen Chen, Gerald Coulis, and Alessandra Castaldi. 2020. "MicroRNA and ROS Crosstalk in Cardiac and Pulmonary Diseases" International Journal of Molecular Sciences 21, no. 12: 4370. https://doi.org/10.3390/ijms21124370
APA StyleCliment, M., Viggiani, G., Chen, Y.-W., Coulis, G., & Castaldi, A. (2020). MicroRNA and ROS Crosstalk in Cardiac and Pulmonary Diseases. International Journal of Molecular Sciences, 21(12), 4370. https://doi.org/10.3390/ijms21124370