Vascular Remodeling in Moyamoya Angiopathy: From Peripheral Blood Mononuclear Cells to Endothelial Cells

 , , , ,
, , , ,  , , ,
, , , 

 add
Show full author list
add
Show full author list
Abstract
1. Introduction
2. Results
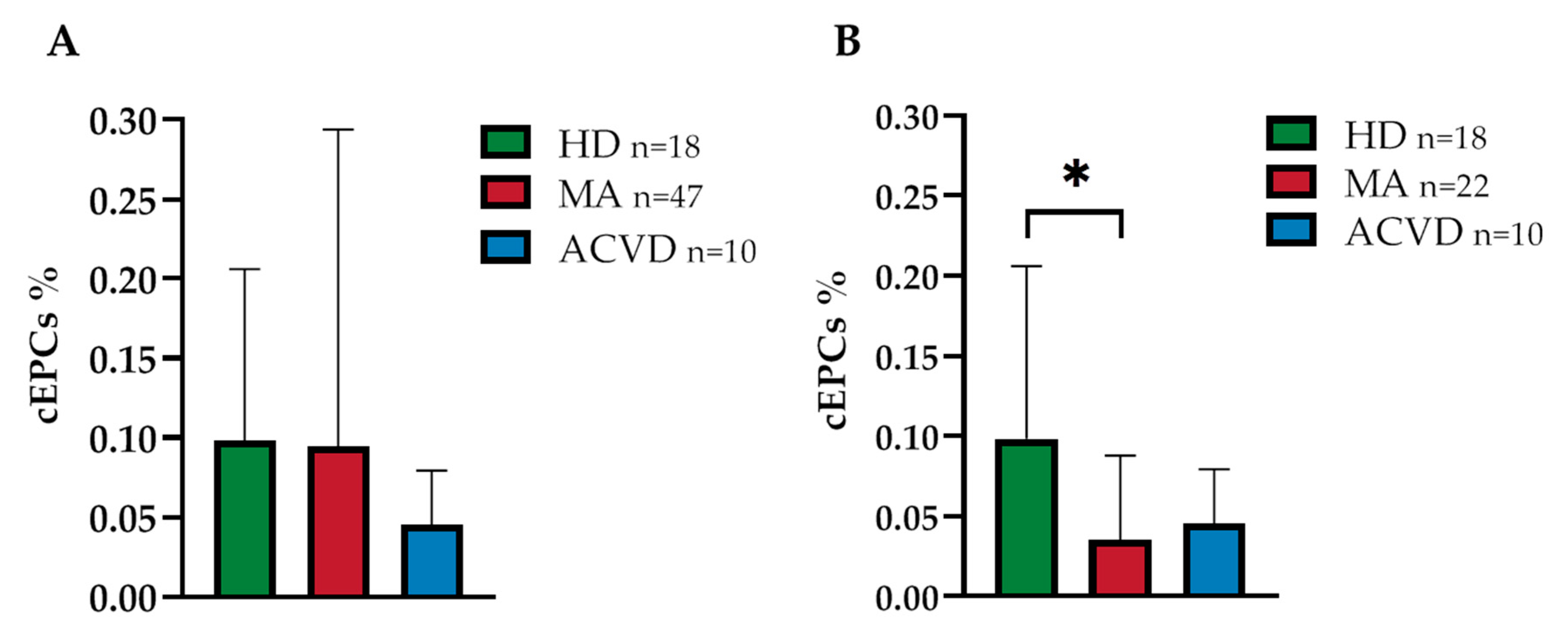
2.1. Reduced EPC Level in Peripheral Blood of a Homogeneous Group of MA Patients
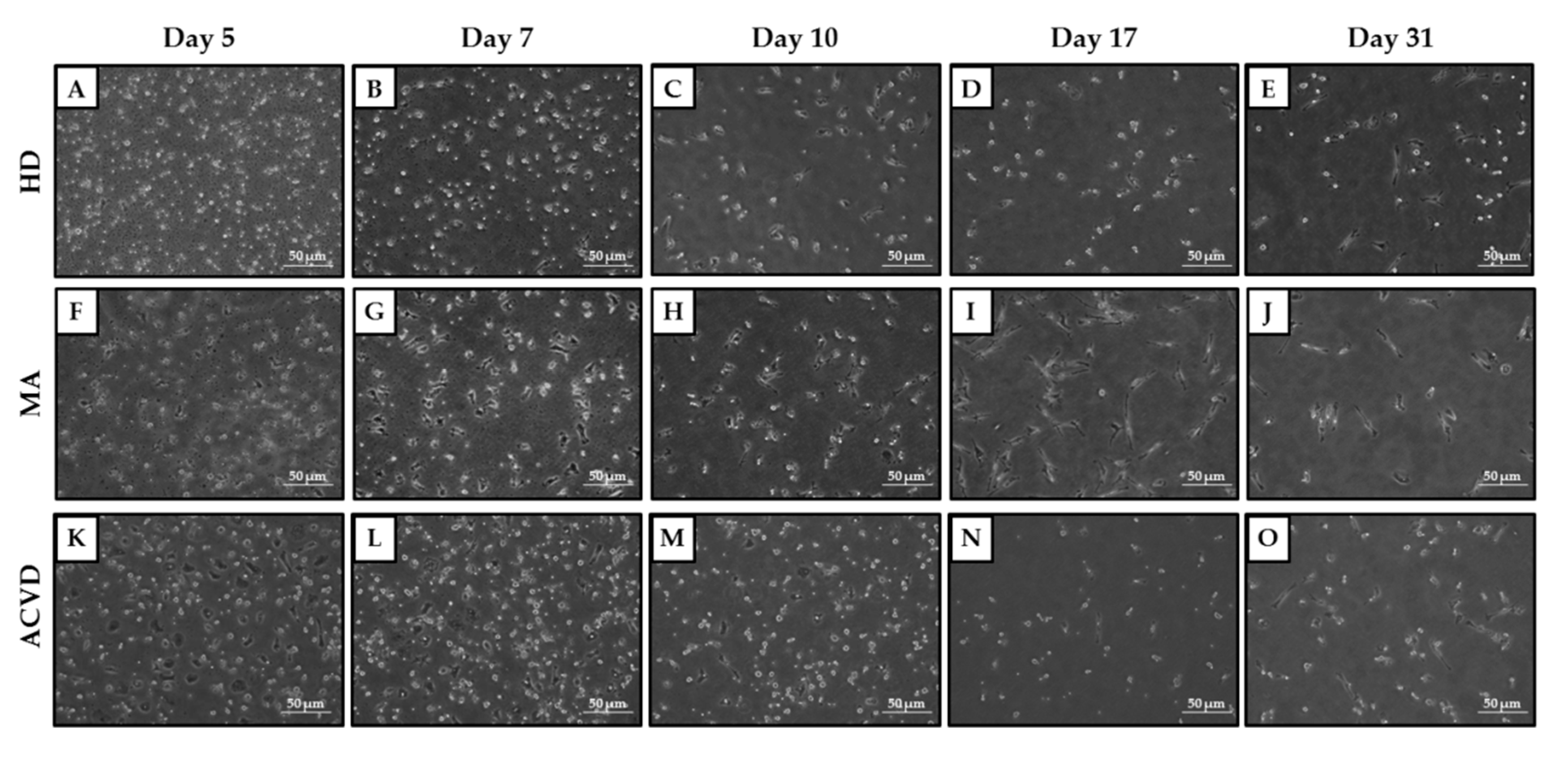
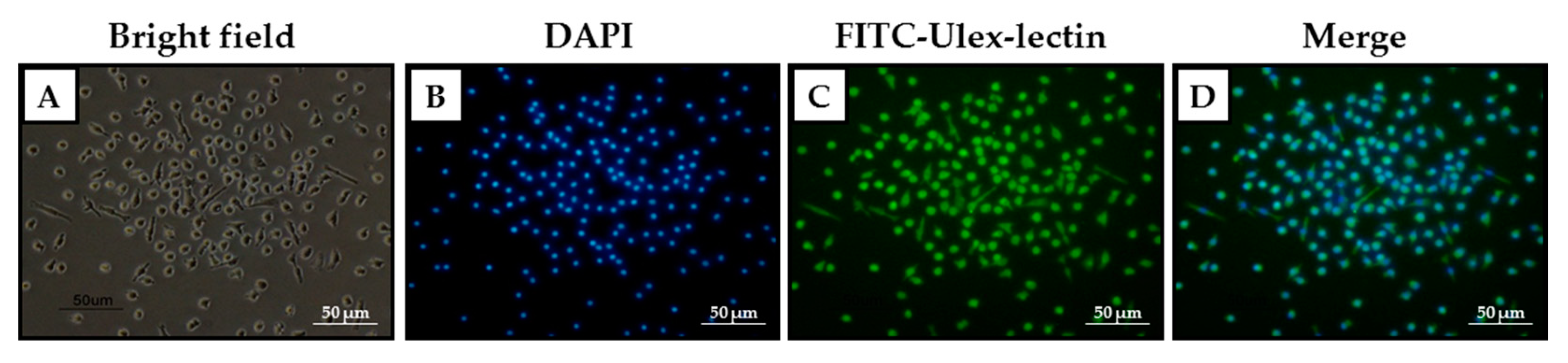
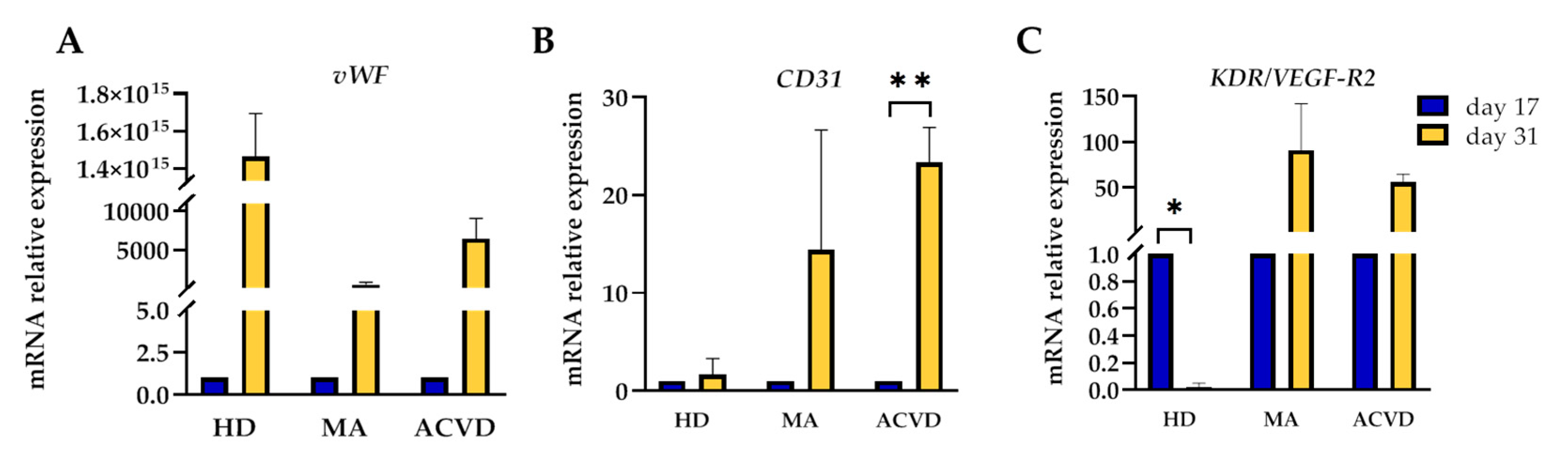
2.2. Identification and Characterization of EPCs in Culture
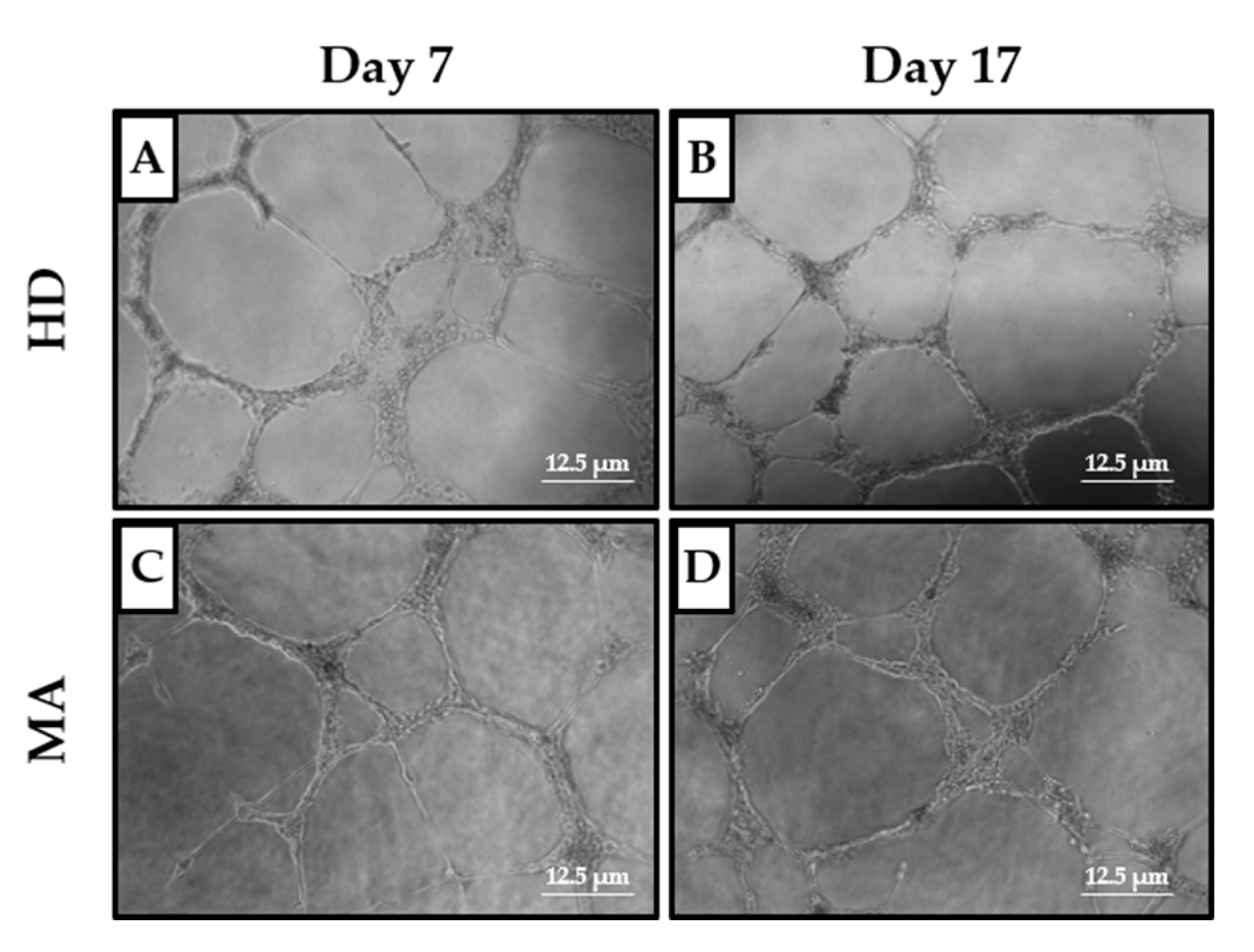
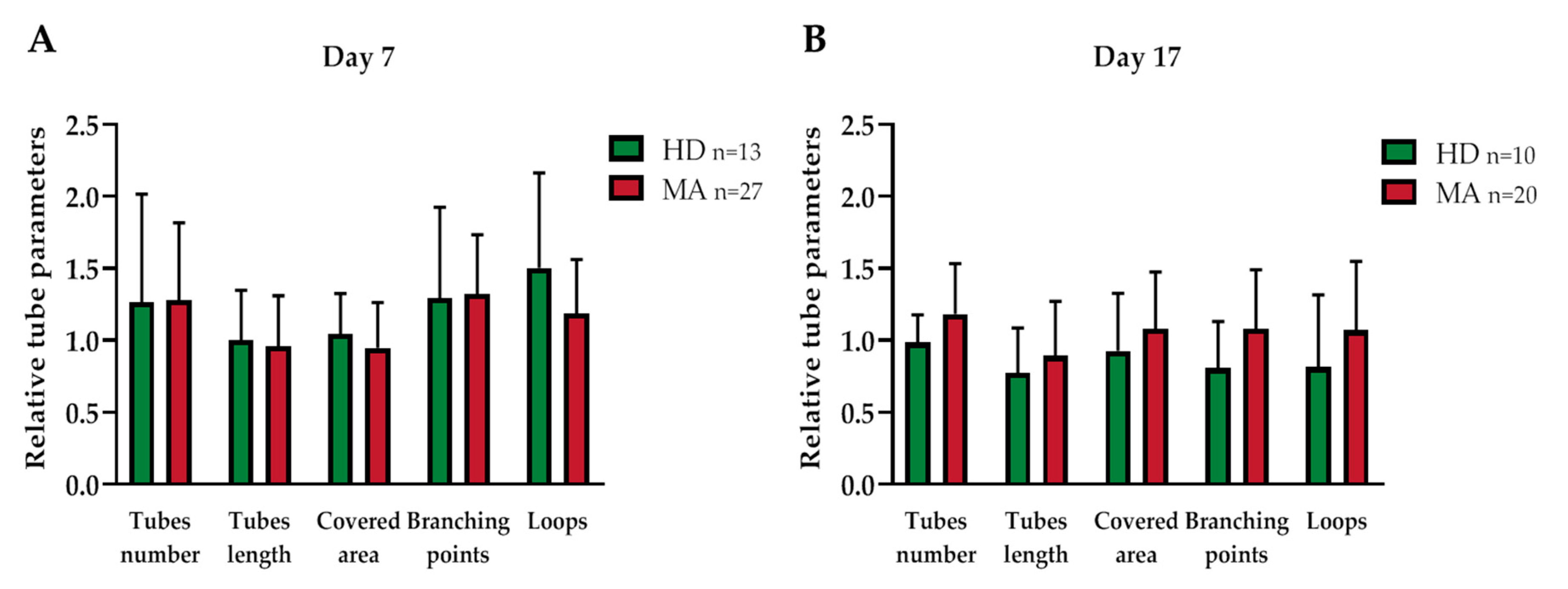
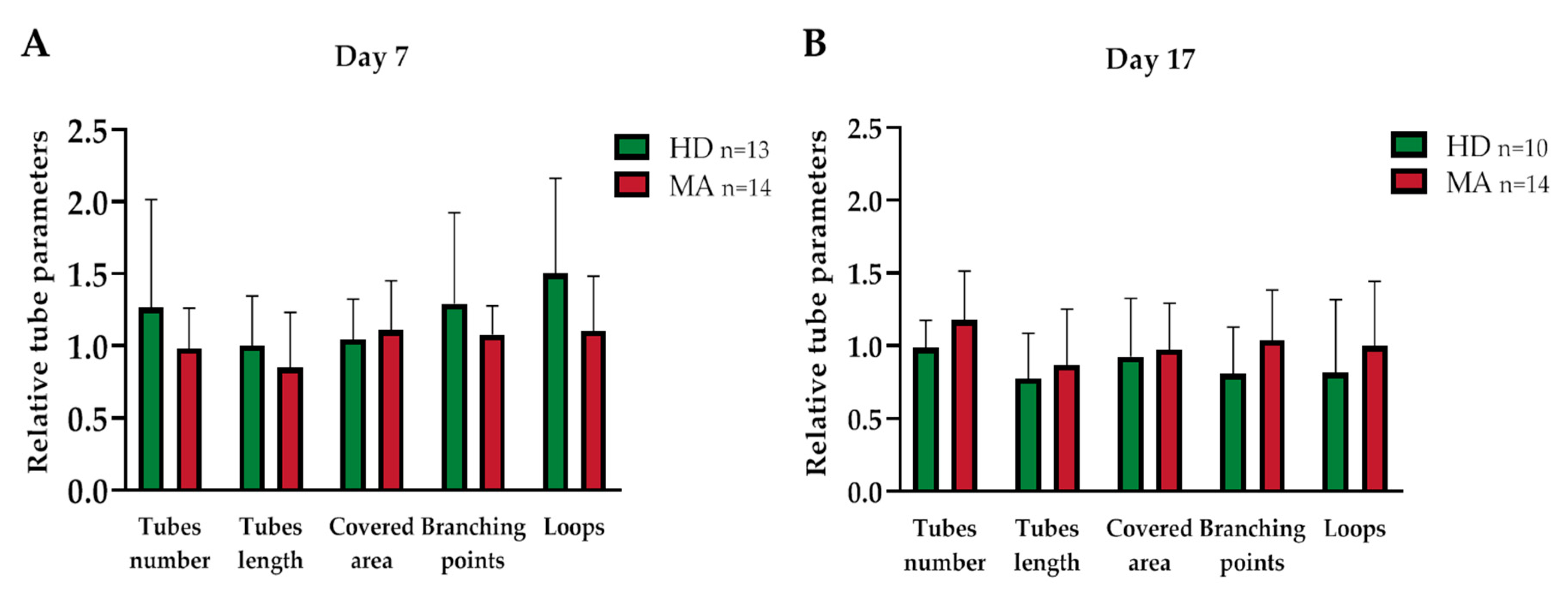
2.3. EPC Paracrine Activity Does Not Influence the Formation of Vessels
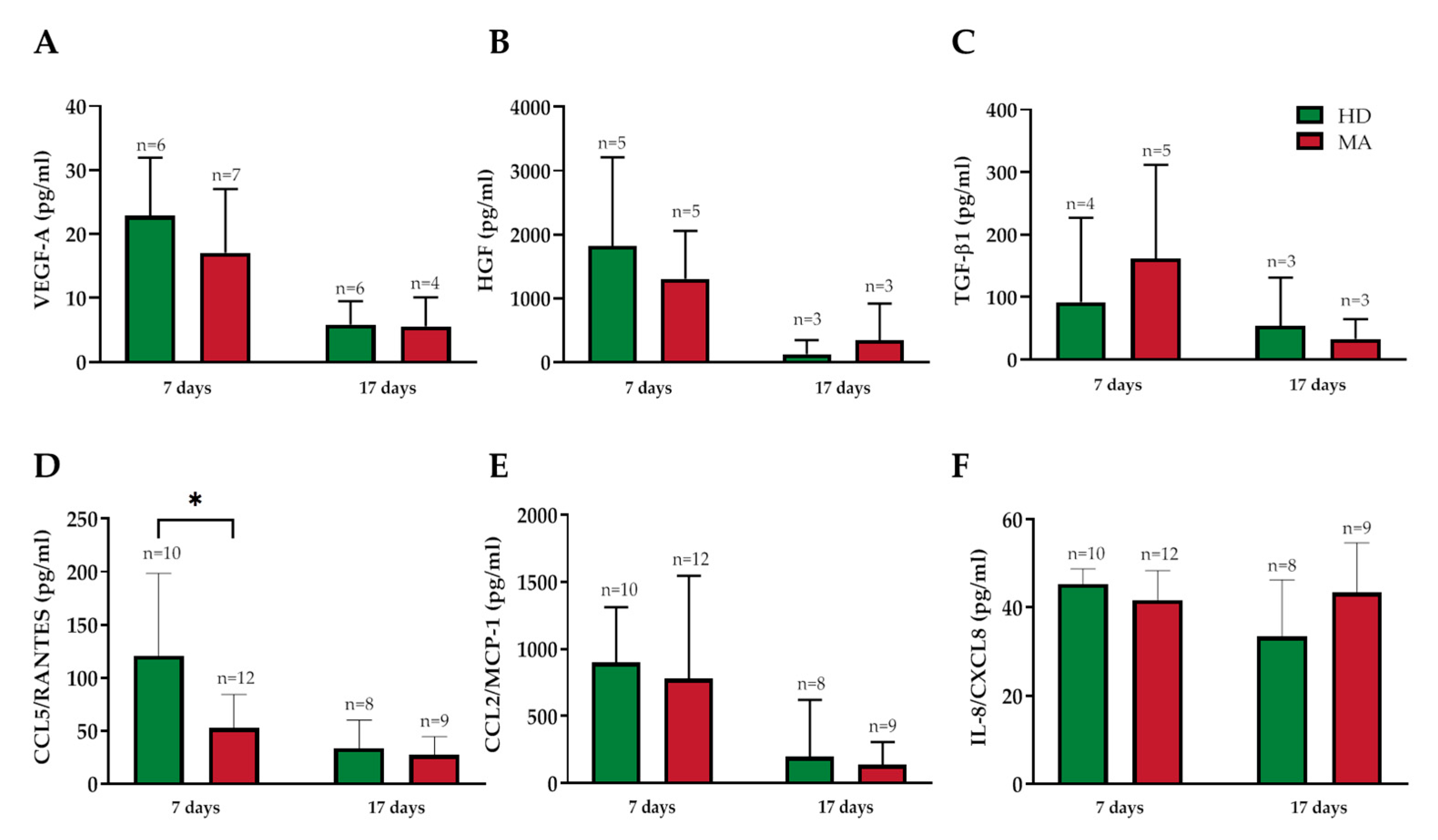
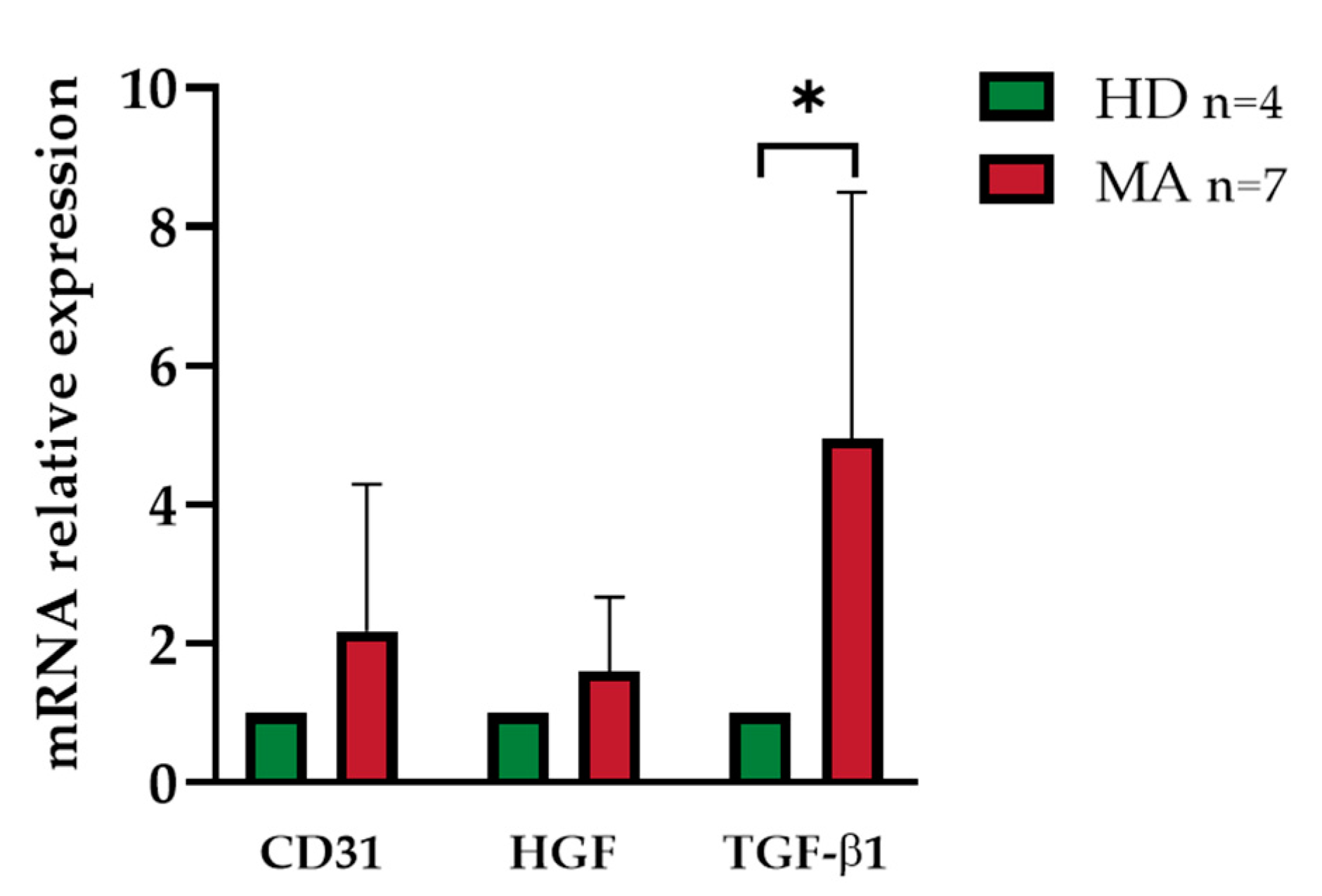
2.4. Release of Endothelial Markers and Inflammatory Cytokines by EPCs from HD and MA Subjects
3. Discussion
4. Materials and Methods
4.1. Moyamoya Patients and Healthy/ACVD Controls: Inclusion Criteria
4.2. Ethical Issues
4.3. Blood and Plasma Samples Collection
4.4. Clinical-Radiological Factors
4.5. Flow Cytometry Analysis
4.6. Isolation and Culture of Endothelial Progenitor Cells
4.7. Fluorescence Microscopy
4.8. Cell Culture
4.9. Tube Formation Assay
4.10. ELISA
4.11. RNA Extraction and Real-Time PCR Analysis
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MA | Moyamoya angiopathy |
| cEPCs | Circulating endothelial progenitor cells |
| HD | Healthy donors |
| ICAs | Internal carotid arteries |
| RNF213 | Ring Finger Protein 213 |
| PBMCs | Peripheral blood mononuclear cells |
| TIA | Transient ischemic attack |
| ACVD | Atherosclerotic cerebrovascular disease |
| WB | White blood |
| SD vWF VEGF-R2 | Standard deviation von Willebrand Factor Vascular endothelial growth factor receptor-2 |
| HUVEC | Human Umbilical Vein Endothelial Cells |
| EGM-MV | Microvascular Endothelial Cell Growth Medium |
| HGF | Hepatocyte growth factor |
| TGF-β1 CCL5 CCL2 | Transforming growth factor-beta 1 Chemokine (C-C motif) ligand 5 Chemokine (C-C motif) ligand 2 |
| β2M | Beta 2-microglobulin |
| EDTA | Ethylenediaminetetraacetic acid |
| EBM | Endothelial Basal Medium |
| n | Number |
| A | Adult |
| P | Pediatric |
| FACS | Fluorescence-activated cell sorting |
| na | Not available |
| ECFCs CECs | Endothelial colony forming cells Circulating endothelial cells |
| AA | Alcohol abuse |
| AD | Antidepressant |
| AE | Antiepileptic |
| AH | Antihypertensive |
| AHHC | Anti-hyperhomocysteinemia |
| AG | Antiaggregants |
| B | Bilateral |
| CCB | Calcium channel blockers |
| CVD | Cerebrovascular disease |
| DL | Dyslipidemia |
| DM | Diabetes mellitus |
| ET | Estroprogestinic therapy |
| f | Female |
| H | Hypertension |
| HHC | Hyperhomocysteinemia |
| HoS | History of smoking |
| HS | Hemorrhagic stroke |
| HT | Head trauma |
| IHH | Ischemic heart disease |
| IS | Ischemic stroke |
| m | Male |
| NIHSS | National Institute of Health Scale |
| PI | Physical inactivity |
| PSY | psychiatric disorder |
| ST | Statins |
| U | Unilateral |
References
- Fukui, M. Guidelines for the diagnosis and treatment of spontaneous occlusion of the circle of Willis (’moyamoya’ disease). Research committee on spontaneous occlusion of the circle of willis (moyamoya disease) of the ministry of health and welfare, Japan. Clin. Neurol. Neurosurg. 1997, 99, S238–S240. [Google Scholar] [CrossRef]
- Bersano, A.; Guey, S.; Bedini, G.; Nava, S.; Hervé, D.; Vajkoczy, P.; Tatlisumak, T.; Sareela, M.; Van Der Zwan, A.; Klijn, C.J.; et al. Research progresses in understanding the pathophysiology of moyamoya disease. Cerebrovasc. Dis. 2016, 41, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Kossorotoff, M.; Tournier-Lasserve, E.; Herve, D.; Guey, S. Moyamoya disease and syndromes: From genetics to clinical management. Appl. Clin. Genet. 2015, 8, 49–68. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, E.; Saeki, N.; Oishi, H.; Hirai, S.; Yamaura, A. Long-term natural history of hemorrhagic type moyamoya disease in 42 patients. J. Neurosurg. 2000, 93, 976–980. [Google Scholar] [CrossRef]
- Kraemer, M.; Heienbrok, W.; Berlit, P. Moyamoya Disease in Europeans. Stroke 2008, 39, 3193–3200. [Google Scholar] [CrossRef]
- Acker, G.; Goerdes, S.; Schneider, U.C.; Schmiedek, P.; Czabanka, M.; Vajkoczy, P. Distinct clinical and radiographic characteristics of moyamoya disease amongst European Caucasians. Eur. J. Neurol. 2015, 22, 1012–1017. [Google Scholar] [CrossRef]
- Feghali, J.; Xu, R.; Yang, W.; Liew, J.; Tamargo, R.J.; Marsh, E.B.; Huang, J. Racial phenotypes in moyamoya disease: A comparative analysis of clinical presentation and natural history in a single multiethnic cohort of 250 hemispheres. J. Neurosurg. 2019, 1–7. [Google Scholar] [CrossRef]
- Houkin, K.; Ito, M.; Sugiyama, T.; Shichinohe, H.; Nakayama, N.; Kazumata, K.; Kuroda, S. Review of past research and current concepts on the etiology of moyamoya disease. Neurol. Med.-Chir. 2012, 52, 267–277. [Google Scholar] [CrossRef]
- Bedini, G.; Blecharz, K.; Nava, S.; Vajkoczy, P.; Alessandri, G.; Ranieri, M.; Acerbi, F.; Ferroli, P.; Riva, D.; Esposito, S.; et al. Vasculogenic and angiogenic pathways in moyamoya disease. Curr. Med. Chem. 2016, 23, 315–345. [Google Scholar] [CrossRef]
- Kamada, F.; Aoki, Y.; Narisawa, A.; Abe, Y.; Komatsuzaki, S.; Kikuchi, A.; Kanno, J.; Niihori, T.; Ono, M.; Ishii, N.; et al. A genome-wide association study identifies RNF213 as the first Moyamoya disease gene. J. Hum. Genet. 2010, 56, 34–40. [Google Scholar] [CrossRef]
- Guey, S.; Kraemer, M.; Herve, D.; Ludwig, T.E.; Kossorotoff, M.; Bergametti, F.; Schwitalla, J.C.; Choi, S.; Broseus, L.; Callebaut, I.; et al. Rare RNF213 variants in the C-terminal region encompassing the RING-finger domain are associated with moyamoya angiopathy in Caucasians. Eur. J. Hum. Genet. 2017, 25, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Raso, A.; Biassoni, R.; Mascelli, S.; Nozza, P.; Ugolotti, E.; Di Marco, E.; De Marco, P.; Merello, E.; Cama, A.; Pavanello, M.; et al. Moyamoya vasculopathy shows a genetic mutational gradient decreasing from East to West. J. Neurosurg. Sci. 2020, 64, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Fallen, S.; Zhou, Y.; Baxter, D.; Scherler, K.; Kuo, M.F.; Wang, K. The impact of moyamoya disease and RNF213 Mutations on the spectrum of plasma protein and microRNA. J. Clin. Med. 2019, 8, 1648. [Google Scholar] [CrossRef] [PubMed]
- Takeda, M.; Tezuka, T.; Kim, M.; Choi, J.; Oichi, Y.; Kobayashi, H.; Harada, K.H.; Mizushima, T.; Taketani, S.; Koizumi, A.; et al. Moyamoya disease patient mutations in the RING domain of RNF213 reduce its ubiquitin ligase activity and enhance NFκB activation and apoptosis in an AAA+ domain-dependent manner. Biochem. Biophys. Res. Commun. 2020, 525, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Khakoo, A.Y.; Finkel, T. Endothelial progenitor cells. Annu. Rev. Med. 2005, 56, 79–101. [Google Scholar] [CrossRef]
- Asahara, T.; Murohara, T.; Sullivan, A.; Silver, M.; Van Der Zee, R.; Li, T.; Witzenbichler, B.; Schatteman, G.; Isner, J.M. Isolation of putative progenitor endothelial cells for angiogenesis. Science 1997, 275, 964–966. [Google Scholar] [CrossRef]
- Käßmeyer, S.; Plendl, J.; Custodis, P.; Bahramsoltani, M. New insights in vascular development: Vasculogenesis and endothelial progenitor cells. Anat. Histol. Embryol. 2009, 38, 1–11. [Google Scholar] [CrossRef]
- Schatteman, G.C.; Dunnwald, M.; Jiao, C. Biology of bone marrow-derived endothelial cell precursors. Am. J. Physiol. Circ. Physiol. 2007, 292, H1–H18. [Google Scholar] [CrossRef]
- Lee, P.S.S.; Poh, K.K. Endothelial progenitor cells in cardiovascular diseases. World J. Stem Cells 2014, 6, 355–366. [Google Scholar] [CrossRef]
- Liu, H.B.; Gong, Y.F.; Yu, C.J.; Sun, Y.Y.; Li, X.Y.; Zhao, D.; Zhang, Z.R. Endothelial progenitor cells in cardiovascular diseases: From biomarker to therapeutic agent. Regen. Med. Res. 2013, 1, 1–9. [Google Scholar] [CrossRef]
- Sugiyama, T.; Kuroda, S.; Nakayama, N.; Tanaka, S.; Houkin, K. Bone marrow-derived endothelial progenitor cells participate in the initiation of moyamoya disease. Neurol. Med.-Chir. 2011, 51, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, T.; Taguchi, A.; Matsuyama, T.; Shimizu, Y.; Kikuchi-Taura, A.; Soma, T.; Stern, D.M.; Yoshikawa, H.; Kasahara, Y.; Moriwaki, H.; et al. Increase in circulating CD34-positive cells in patients with angiographic evidence of moyamoya-like vessels. Br. J. Pharmacol. 2008, 28, 1086–1089. [Google Scholar] [CrossRef] [PubMed]
- Rafat, N.; Beck, G.; Peña-Tapia, P.; Schmiedek, P.; Vajkoczy, P. Increased levels of circulating endothelial progenitor cells in patients with moyamoya disease. Stroke 2009, 40, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Jung, J.H.; Phi, J.H.; Kang, H.S.; Kim, J.E.; Chae, J.H.; Kim, S.J.; Kim, Y.H.; Kim, Y.Y.; Cho, B.K.; et al. Decreased level and defective function of circulating endothelial progenitor cells in children with moyamoya disease. J. Neurosci. Res. 2009, 88, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.H.; Chu, K.; Lee, S.T.; Park, H.K.; Kim, N.H.; Kim, J.; Bahn, J.J.; Song, E.C.; Kim, M.; Lee, S.K.; et al. Circulating endothelial progenitor cells as a pathogenetic marker of moyamoya disease. Br. J. Pharmacol. 2008, 28, 1795–1803. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Du, Q.; Hu, M.; Zhang, J.; Chen, J. Endothelial progenitor cells in moyamoya disease: Current situation and controversial issues. Cell Transplant. 2020, 29, 1–12. [Google Scholar] [CrossRef]
- Bersano, A.; Bedini, G.; Nava, S.; Acerbi, F.; Sebastiano, D.R.; Binelli, S.; Franceschetti, S.; Faragò, G.; Grisoli, M.; Gioppo, A.; et al. GEN-O-MA project: An Italian network studying clinical course and pathogenic pathways of moyamoya disease—Study protocol and preliminary results. Neurol. Sci. 2019, 40, 561–570. [Google Scholar] [CrossRef]
- Corsini, E.; Ciusani, E.; Gaviani, P.; Silvani, A.; Canazza, A.; Bernardi, G.; Calatozzolo, C.; DiMeco, F.; Salmaggi, A. Decrease in circulating endothelial progenitor cells in treated glioma patients. J. Neuro-Oncol. 2012, 108, 123–129. [Google Scholar] [CrossRef]
- George, A.L.; Bangalore-Prakash, P.; Rajoria, S.; Suriano, R.; Shanmugam, A.; Mittelman, A.; Tiwari, R.K. Endothelial progenitor cell biology in disease and tissue regeneration. J. Hematol. Oncol. 2011, 4, 24. [Google Scholar] [CrossRef]
- Ai, J.; Sun, J.; Wan, T.; Ma, J.; Feng, L.; Yao, K. Generation of an anti-angiogenic endothelial progenitor cell line via endostatin gene transfer. Mol. Med. Rep. 2018, 17, 5814–5820. [Google Scholar] [CrossRef]
- Lin, Y.; Weisdorf, D.J.; Solovey, A.; Hebbel, R.P. Origins of circulating endothelial cells and endothelial outgrowth from blood. J. Clin. Investig. 2000, 105, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Hur, J.; Yoon, C.H.; Kim, H.; Choi, J.H.; Kang, H.J.; Hwang, K.K.; Oh, B.H.; Lee, M.M.; Park, Y.B. Characterization of two types of endothelial progenitor cells and their different contributions to neovasculogenesis. Arter. Thromb. Vasc. Biol. 2004, 24, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.C.; Chang, S.J.; Chueh, Y.N.; Huang, T.S.; Huang, P.H.; Cheng, S.M.; Tsai, T.N.; Chen, J.W.; Wang, H.W. Distinct angiogenesis roles and surface markers of early and late endothelial progenitor cells revealed by functional group analyses. BMC Genom. 2013, 14, 182. [Google Scholar] [CrossRef]
- Sieveking, D.P.; Buckle, A.; Celermajer, D.S.; Ng, M.K. Strikingly different angiogenic properties of endothelial progenitor cell subpopulations. J. Am. Coll. Cardiol. 2008, 51, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.M.; Zalos, G.; Halcox, J.P.J.; Schenke, W.H.; Waclawiw, M.A.; Quyyumi, A.A.; Finkel, T. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N. Engl. J. Med. 2003, 348, 593–600. [Google Scholar] [CrossRef]
- Vasa, M.; Fichtlscherer, S.; Aicher, A.; Adler, K.; Urbich, C.; Martin, H.; Zeiher, A.M.; Dimmeler, S. Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ. Res. 2001, 89, e1–e7. [Google Scholar] [CrossRef] [PubMed]
- Tepper, O.M.; Galiano, R.D.; Capla, J.M.; Kalka, C.; Gagne, P.J.; Jacobowitz, G.R.; Levine, J.P.; Gurtner, G.C. Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation 2002, 106, 2781–2786. [Google Scholar] [CrossRef]
- Schmidt-Lucke, C.; Rössig, L.; Fichtlscherer, S.; Vasa, M.; Britten, M.; Kämper, U.; Dimmeler, S.; Zeiher, A.M. Reduced number of circulating endothelial progenitor cells predicts future cardiovascular events. Circulation 2005, 111, 2981–2987. [Google Scholar] [CrossRef]
- Ghani, U.; Shuaib, A.; Salam, A.; Nasir, A.; Shuaib, U.; Jeerakathil, T.; Sher, F.; O’Rourke, F.; Nasser, A.M.; Schwindt, B.; et al. Endothelial progenitor cells during cerebrovascular disease. Stroke 2005, 36, 151–153. [Google Scholar] [CrossRef]
- Chu, K.; Jung, K.H.; Lee, S.T.; Park, H.K.; Sinn, D.I.; Kim, J.M.; Kim, N.H.; Kim, J.H.; Kim, S.J.; Song, E.C.; et al. Circulating endothelial progenitor cells as a new marker of endothelial dysfunction or repair in acute stroke* supplemental methods. Stroke 2008, 39, 1441–1447. [Google Scholar] [CrossRef]
- Kang, H.S.; Wang, K.C.; Kim, S.K. Circulating vascular progenitor cells in moyamoya disease. J. Korean Neurosurg. Soc. 2015, 57, 428–431. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Kalka, C.; Masuda, H.; Chen, D.; Silver, M.; Kearney, M.; Magner, M.; Isner, J.M.; Asahara, T. Ischemia- and cytokine-induced mobilization of bone marrow-derived endothelial progenitor cells for neovascularization. Nat. Med. 1999, 5, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Zhang, Z.; Ye, D.; Xing, P.; Zou, Z.; Lei, H.; Duan, L. Gene dysregulation in peripheral blood of moyamoya disease and comparison with other vascular disorders. PLoS ONE 2019, 14, e0221811. [Google Scholar] [CrossRef] [PubMed]
- Geng, C.; Cui, C.; Guo, Y.; Wang, C.; Zhang, J.; Han, W.; Jin, F.; Chen, D.; Jiang, P. Metabolomic profiling revealed potential biomarkers in patients with moyamoya disease. Front. Mol. Neurosci. 2020, 14, 308. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Son, S.M.; Mook-Jung, I.; Moon, Y.J.; Lee, J.Y.; Wang, K.-C.; Kang, H.-S.; Phi, J.H.; Choi, S.A.; Chong, S.; et al. Mitochondrial abnormalities related to the dysfunction of circulating endothelial colony-forming cells in moyamoya disease. J. Neurosurg. 2018, 129, 1151–1159. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.B.; Zhi, H.; Zhang, X.Y.; Liang, J.W.; He, J.; Su, C.; Xia, W.H.; Zhang, G.X.; Tao, J. Mitochondrial dysfunction-mediated decline in angiogenic capacity of endothelial progenitor cells is associated with capillary rarefaction in patients with hypertension via downregulation of CXCR4/JAK2/SIRT5 signaling. Bio. Med. 2019, 42, 64–75. [Google Scholar] [CrossRef]
- Srinivasan, J.; Britz, G.W.; Newell, D.W. Cerebral revascularization for moyamoya disease in adults. Neurosurg. Clin. N. Am. 2001, 12, 585–594. [Google Scholar] [CrossRef]
- Smith, E.; Scott, R.M. Moyamoya: Epidemiology, presentation, and diagnosis. Neurosurg. Clin. N. Am. 2010, 21, 543–551. [Google Scholar] [CrossRef]
- Tarasów, E.; Kułakowska, A.; Lukasiewicz, A.; Kapica-Topczewska, K.; Korneluk-Sadzyńska, A.; Brzozowska, J.; Drozdowski, W. Moyamoya disease: Diagnostic imaging. Pol. J. Radiol. 2011, 76, 73–79. [Google Scholar]
- Fadini, G.P.; Losordo, U.; Dimmeler, S. Critical reevaluation of endothelial progenitor cell phenotypes for therapeutic and diagnostic use. Circ. Res. 2012, 110, 624–637. [Google Scholar] [CrossRef]
- Ni, G.; Liu, W.; Huang, X.; Zhu, S.; Yue, X.; Chen, Z.; Chen, M.; Liu, X.; Xu, G. Increased levels of circulating SDF-1α and CD34+ CXCR4+ cells in patients with moyamoya disease. Eur. J. Neurol. 2011, 18, 1304–1309. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.S.; Moon, Y.J.; Kim, Y.Y.; Park, W.Y.; Park, A.K.; Wang, K.C.; Kim, J.E.; Phi, J.H.; Lee, J.Y.; Kim, S.K. Smooth-muscle progenitor cells isolated from patients with moyamoya disease: Novel experimental cell model. J. Neurosurg. 2014, 120, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Moon, Y.J.; Lee, H.O.; Park, A.K.; Choi, S.A.; Wang, K.C.; Han, J.W.; Joung, J.G.; Kang, H.S.; Kim, J.E.; et al. Deregulation of retinaldehyde dehydrogenase 2 leads to defective angiogenic function of endothelial colony–Forming cells in pediatric moyamoya disease. Arter. Thromb. Vasc. Biol. 2015, 35, 1670–1677. [Google Scholar] [CrossRef] [PubMed]
- Phi, J.H.; Suzuki, N.; Moon, Y.J.; Park, A.K.; Wang, K.C.; Lee, J.Y.; Choi, S.A.; Chong, S.; Shirane, R.; Kim, S.K. Chemokine ligand 5 (CCL5) derived from endothelial colony-forming cells (ECFCs) mediates recruitment of smooth muscle progenitor Cells (SPCs) toward critical vascular locations in moyamoya disease. PLoS ONE 2017, 12, e0169714. [Google Scholar] [CrossRef]
- Bao, X.Y.; Fan, Y.N.; Liu, Y.; Wang, Q.N.; Zhang, Y.; Zhu, B.; Liu, B.; Duan, L. Circulating endothelial progenitor cells and endothelial cells in moyamoya disease. Brain Behav. 2018, 8, e01035. [Google Scholar] [CrossRef]
- Jangra, A.; Choi, S.A.; Koh, E.J.; Moon, Y.J.; Wang, K.C.; Phi, J.H.; Lee, J.Y.; Kim, S.K. Panobinostat, a histone deacetylase inhibitor, rescues the angiogenic potential of endothelial colony-forming cells in moyamoya disease. Child’s Nerv. Syst. 2019, 35, 823–831. [Google Scholar] [CrossRef]
- Choi, S.A.; Chong, S.; Kwak, P.A.; Moon, Y.J.; Jangra, A.; Phi, J.H.; Lee, J.Y.; Park, S.H.; Kim, S.K. Impaired functional recovery of endothelial colony-forming cells from moyamoya disease in a chronic cerebral hypoperfusion rat model. J. Neurosurgery Pediatr. 2019, 23, 204–213. [Google Scholar] [CrossRef]
- Nagata, E.; Masuda, H.; Nakayama, T.; Netsu, S.; Yuzawa, H.; Fujii, N.; Kohara, S.; Sorimachi, T.; Osada, T.; Imazeki, R.; et al. Author correction: Insufficient production of IL-10 from M2 macrophages impairs in vitro endothelial progenitor cell differentiation in patients with Moyamoya disease. Sci. Rep. 2020, 10, 1. [Google Scholar] [CrossRef]
- Hojo, M.; Hoshimaru, M.; Miyamoto, S.; Taki, W.; Nagata, I.; Asahi, M.; Matsuura, N.; Ishizaki, R.; Kikuchi, H.; Hashimoto, N. Role of transforming growth factor—β1 in the pathogenesis of moyamoya disease. J. Neurosurg. 1998, 89, 623–629. [Google Scholar] [CrossRef]
- Urbich, C.; Dimmeler, S. Endothelial progenitor cells. Circ. Res. 2004, 95, 343–353. [Google Scholar] [CrossRef]
- Palii, C.G.; Vulesevic, B.; Fraineau, S.; Pranckeviciene, E.; Griffith, A.J.; Chu, A.; Faralli, H.; Li, Y.; McNeill, B.; Sun, J.; et al. Trichostatin A enhances vascular repair by injected human endothelial progenitors through increasing the expression of TAL1-dependent genes. Cell Stem Cell 2014, 14, 644–657. [Google Scholar] [CrossRef] [PubMed]
- Prasain, N.; Lee, M.R.; Vemula, S.; Meador, J.L.; Yoshimoto, M.; Ferkowicz, M.J.; Fett, A.; Gupta, M.; Rapp, B.M.; Saadatzadeh, M.R.; et al. Differentiation of human pluripotent stem cells to cells similar to cord-blood endothelial colony–forming cells. Nat. Biotechnol. 2014, 32, 1151–1157. [Google Scholar] [CrossRef]
- Fraineau, S.; Palii, C.G.; McNeill, B.; Ritso, M.; Shelley, W.C.; Prasain, N.; Chu, A.; Vion, E.; Rieck, K.; Nilufar, S.; et al. Epigenetic activation of pro-angiogenic signaling pathways in human endothelial progenitors increases vasculogenesis. Stem Cell Rep. 2017, 9, 1573–1587. [Google Scholar] [CrossRef] [PubMed]
- Au, P.; Daheron, L.M.; Duda, D.G.; Cohen, K.S.; Tyrrell, J.A.; Lanning, R.M.; Fukumura, D.; Scadden, D.T.; Jain, R.K. Differential in vivo potential of endothelial progenitor cells from human umbilical cord blood and adult peripheral blood to form functional long-lasting vessels. Blood 2008, 111, 1302–1305. [Google Scholar] [CrossRef]
- Melero-Martin, J.M.; Khan, Z.A.; Picard, A.; Wu, X.; Paruchuri, S.; Bischoff, J. In vivo vasculogenic potential of human blood-derived endothelial progenitor cells. Blood 2007, 109, 4761–4768. [Google Scholar] [CrossRef]
- Reinisch, A.; Hofmann, N.A.; Obenauf, A.C.; Kashofer, K.; Rohde, E.; Schallmoser, K.; Flicker, K.; Lanzer, G.; Linkesch, W.; Speicher, M.R.; et al. Humanized large-scale expanded endothelial colony–forming cells function in vitro and in vivo. Blood 2009, 113, 6716–6725. [Google Scholar] [CrossRef] [PubMed]
- Yoder, M.C.; Mead, L.E.; Prater, D.; Krier, T.R.; Mroueh, K.N.; Li, F.; Krasich, R.; Temm, C.J.; Prchal, J.T.; Ingram, D.A. Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood 2006, 109, 1801–1809. [Google Scholar] [CrossRef]
- Cooke, J.P.; Losordo, D.W. Modulating the vascular response to limb ischemia. Circ. Res. 2015, 116, 1561–1578. [Google Scholar] [CrossRef]
- Fraineau, S.; Palii, C.G.; Allan, D.S.; Brand, M. Epigenetic regulation of endothelial-cell-mediated vascular repair. FEBS J. 2015, 282, 1605–1629. [Google Scholar] [CrossRef]
- Park, K.M.; Gerecht, S. Harnessing developmental processes for vascular engineering and regeneration. Development 2014, 141, 2760–2769. [Google Scholar] [CrossRef]
- Shantsila, E.; Watson, T.; Lip, G.Y.H. Endothelial progenitor cells in cardiovascular disorders. J. Am. Coll. Cardiol. 2007, 49, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Alphonse, R.S.; Vadivel, A.; Fung, M.; Shelley, W.C.; Critser, P.J.; Ionescu, L.; O’Reilly, M.; Ohls, R.K.; McConaghy, S.; Eaton, F.; et al. Existence, functional impairment, and lung repair potential of endothelial colony-forming cells in oxygen-induced arrested alveolar growth. Circulation 2014, 129, 2144–2157. [Google Scholar] [CrossRef] [PubMed]
- Bouvard, C.; Gafsou, B.; Dizier, B.; Galy-Fauroux, I.; Lokajczyk, A.; Boisson-Vidal, C.; Fischer, A.M.; Helley, D. α6-integrin subunit plays a major role in the proangiogenic properties of endothelial progenitor cells. Arter. Thromb. Vasc. Biol. 2010, 30, 1569–1575. [Google Scholar] [CrossRef] [PubMed]
- Saif, J.; Schwarz, T.M.; Chau, D.Y.S.; Henstock, J.; Sami, P.; Leicht, S.F.; Hermann, P.C.; Alcala, S.; Mulero, F.; Shakesheff, K.M.; et al. Combination of injectable multiple growth factor-releasing scaffolds and cell therapy as an advanced modality to enhance tissue neovascularization. Arter. Thromb. Vasc. Biol. 2010, 30, 1897–1904. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, T.M.; Leicht, S.F.; Radic, T.; Rodriguez-Arabaolaza, I.; Hermann, P.C.; Berger, F.; Saif, J.; Böcker, W.; Ellwart, J.W.; Aicher, A.; et al. Vascular incorporation of endothelial colony-forming cells is essential for functional recovery of murine ischemic tissue following cell therapy. Arter. Thromb. Vasc. Biol. 2011, 32, e13–e21. [Google Scholar] [CrossRef]
- Delorme, B.; Basire, A.; Gentile, C.; Sabatier, F.; Monsonis, F.; Desouches, C.; Blot-Chabaud, M.; Uzan, G.; Sampol, J.; Dignat-George, F. Presence of endothelial progenitor cells, distinct from mature endothelial cells, within human CD146+ blood cells. Thromb. Haemost. 2005, 94, 1270–1279. [Google Scholar] [CrossRef]
- Budzyń, M.; Gryszczyńka, B.; Boruczkowski, M.; Kaczmarek, M.; Begier-Krasińska, B.; Osińska, A.; Bukowska, A.; Iskra, M.; Kasprzak, M.P. The potential role of circulating endothelial cells and endothelial progenitor cells in the prediction of left ventricular hypertrophy in hypertensive patients. Front. Physiol. 2019, 10, 1005. [Google Scholar] [CrossRef]
- Chavakis, E.; Urbich, C.; Dimmeler, S. Homing and engraftment of progenitor cells: A prerequisite for cell therapy. J. Mol. Cell. Cardiol. 2008, 45, 514–522. [Google Scholar] [CrossRef]
- Zhao, L.; Sun, W.; Liang, H.; Gao, T.; Liu, Y.; Sun, Y.; Zhang, S.; Li, C. Therapeutic effect of autologous bone marrow stem cell mobilization combined with anti-infective therapy on moyamoya disease. Saudi J. Biol. Sci. 2020, 27, 676–681. [Google Scholar] [CrossRef]
- Research Committee on the Pathology and Treatment of Spontaneous Occlusion of the Circle of Willis. Health labour sciences research grant for research on measures for intractable diseases guidelines for diagnosis and treatment of moyamoya disease (spontaneous occlusion of the circle of willis). Neurol. Med.-Chir. 2012, 52, 245–266. [Google Scholar] [CrossRef]
- Lee, C.W.; Huang, P.H.; Huang, S.S.; Leu, H.-B.; Huang, C.C.; Wu, T.C.; Chen, J.W.; Lin, S.J. Decreased circulating endothelial progenitor cell levels and function in essential hypertensive patients with electrocardiographic left ventricular hypertrophy. Hypertens. Res. 2011, 34, 999–1003. [Google Scholar] [CrossRef] [PubMed]
- Rossi, F.; Bertone, C.; Montanile, F.; Miglietta, F.; Lubrano, C.; Gandini, L.; Santiemma, V. HDL cholesterol is a strong determinant of endothelial progenitor cells in hypercholesterolemic subjects. Microvasc. Res. 2010, 80, 274–279. [Google Scholar] [CrossRef]
- Fadini, G.P.; Miorin, M.; Facco, M.; Bonamico, S.; Baesso, I.; Grego, F.; Menegolo, M.; De Kreutzenberg, S.V.; Tiengo, A.; Agostini, C.; et al. Circulating endothelial progenitor cells are reduced in peripheral vascular complications of type 2 diabetes mellitus. J. Am. Coll. Cardiol. 2005, 45, 1449–1457. [Google Scholar] [CrossRef] [PubMed]
- Fadini, G.P.; Pucci, L.; Vanacore, R.; Baesso, I.; Penno, G.; Balbarini, A.; Di Stefano, R.; Miccoli, R.; De Kreutzenberg, S.; Coracina, A.; et al. Glucose tolerance is negatively associated with circulating progenitor cell levels. Diabetologia 2007, 50, 2156–2163. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, J.; Takaku, A. Cerebrovascular “moyamoya” disease. Arch. Neurol. 1969, 20, 288–299. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subjects | 7 days | p Value | 17 days | p Value | 31 days | p Value |
|---|---|---|---|---|---|---|
| HD | 26.75 ± 16.97 | 9.25 ± 2.28 | 6.75 ± 2.28 | |||
| MA | 28.89 ± 30.33 | 0.647 | 10.78 ± 13.46 | 0.681 | 5.5 ± 8.07 | 0.603 |
| ACVD | 19.5 ± 4.5 | 0.187 | 11 ± 5 | 0.787 | 5.5 ± 5.5 | 0.858 |
| Day 7 | Day 17 | |||||
|---|---|---|---|---|---|---|
| Parameters | HD | MA | p Value | HD | MA | p Value |
| n | 13 | 27 | 10 | 20 | ||
| Covered area | 1.045 ± 0.268 | 0.947 ± 0.309 | 0.329 | 0.926 ± 0.380 | 1.080 ± 0.383 | 0.330 |
| Branching points | 1.293 ± 0.606 | 1.323 ± 0.403 | 0.881 | 0.808 ± 0.306 | 1.081 ± 0.399 | 0.059 |
| Total loops | 1.502 ± 0.636 | 1.187 ± 0.368 | 0.13 | 0.813 ± 0.477 | 1.07 ± 0.467 | 0.198 |
| Total tubes | 1.265 ± 0.722 | 1.278 ± 0.528 | 0.956 | 0.985 ± 0.181 | 1.182 ± 0.342 | 0.056 |
| Total tube length | 1.004 ± 0.331 | 0.957 ± 0.346 | 0.691 | 0.774 ± 0.296 | 0.892 ± 0.37 | 0.375 |
| Day 7 | Day 17 | |||||
|---|---|---|---|---|---|---|
| Parameters | HD | MA | p Value | HD | MA | p Value |
| n | 13 | 14 | 10 | 14 | ||
| Covered area | 1.045 ± 0.268 | 1.108 ± 0.331 | 0.602 | 0.926 ± 0.380 | 0.974 ± 0.308 | 0.757 |
| Branching points | 1.293 ± 0.606 | 1.079 ± 0.192 | 0.259 | 0.808 ± 0.306 | 1.037 ± 0.335 | 0.113 |
| Total loops | 1.502 ± 0.636 | 1.103 ± 0.367 | 0.073 | 0.813 ± 0.477 | 0.998 ± 0.429 | 0.364 |
| Total tubes | 1.265 ± 0.722 | 0.983 ± 0.271 | 0.222 | 0.985 ± 0.181 | 1.175 ± 0.325 | 0.094 |
| Total tube length | 1.004 ± 0.331 | 0.849 ± 0.369 | 0.279 | 0.774 ± 0.296 | 0.868 ± 0.37 | 0.516 |
| Reference | n | Age | Female (%) | Ethnicity | Prior Surgical Treatment (%) | EPC Identification | EPC Level |
|---|---|---|---|---|---|---|---|
| [25] | 24 | A | 66.67% | na | 87.5% | Colony counting | ↓ |
| [22] | 4 | A | 50% | na | na | FACS | ↑ |
| [23] | 20 | A/P | 70% | na | 100% | FACS | ↑ |
| [24] | 28 | P | 50% | na | 100% | FACS; colony counting | ↓; ↓ |
| [51] | 18 | A | 50% | na | 100% | FACS | ↑ |
| [52] | 17 | A/P | 68% | na | 100% | FACS | = |
| [53] | 6 | A/P | 50% | na | na | FACS; colony counting | =; ↓ |
| [54] | 12 | A/P | 75% | na | na | FACS | = |
| [55] | 66 | A | 43.94% | na | 100% | FACS | = |
| [56] | 5 | P | 40% | na | na | FACS | = |
| [57] | 4 | P | 50% | na | na | FACS | = |
| [58] | 23 7 | A A | 52.17% 71.42% | na na | 100% 0% | Colony counting Colony counting | ↓ ↓ |
| Present study | 22 | A | 86.36% | Caucasian | 100% | FACS | ↓ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tinelli, F.; Nava, S.; Arioli, F.; Bedini, G.; Scelzo, E.; Lisini, D.; Faragò, G.; Gioppo, A.; Ciceri, E.F.; Acerbi, F.; et al. Vascular Remodeling in Moyamoya Angiopathy: From Peripheral Blood Mononuclear Cells to Endothelial Cells. Int. J. Mol. Sci. 2020, 21, 5763. https://doi.org/10.3390/ijms21165763
Tinelli F, Nava S, Arioli F, Bedini G, Scelzo E, Lisini D, Faragò G, Gioppo A, Ciceri EF, Acerbi F, et al. Vascular Remodeling in Moyamoya Angiopathy: From Peripheral Blood Mononuclear Cells to Endothelial Cells. International Journal of Molecular Sciences. 2020; 21(16):5763. https://doi.org/10.3390/ijms21165763
Chicago/Turabian StyleTinelli, Francesca, Sara Nava, Francesco Arioli, Gloria Bedini, Emma Scelzo, Daniela Lisini, Giuseppe Faragò, Andrea Gioppo, Elisa F. Ciceri, Francesco Acerbi, and et al. 2020. "Vascular Remodeling in Moyamoya Angiopathy: From Peripheral Blood Mononuclear Cells to Endothelial Cells" International Journal of Molecular Sciences 21, no. 16: 5763. https://doi.org/10.3390/ijms21165763
APA StyleTinelli, F., Nava, S., Arioli, F., Bedini, G., Scelzo, E., Lisini, D., Faragò, G., Gioppo, A., Ciceri, E. F., Acerbi, F., Ferroli, P., Vetrano, I. G., Esposito, S., Saletti, V., Pantaleoni, C., Zibordi, F., Nardocci, N., Zedde, M. L., Pezzini, A., ... Gatti, L. (2020). Vascular Remodeling in Moyamoya Angiopathy: From Peripheral Blood Mononuclear Cells to Endothelial Cells. International Journal of Molecular Sciences, 21(16), 5763. https://doi.org/10.3390/ijms21165763