Virtual Screening of C. Sativa Constituents for the Identification of Selective Ligands for Cannabinoid Receptor 2



Abstract

1. Introduction
2. Results
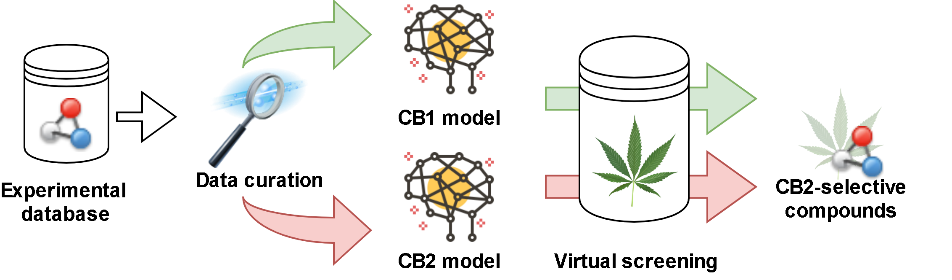
2.1. Study Design
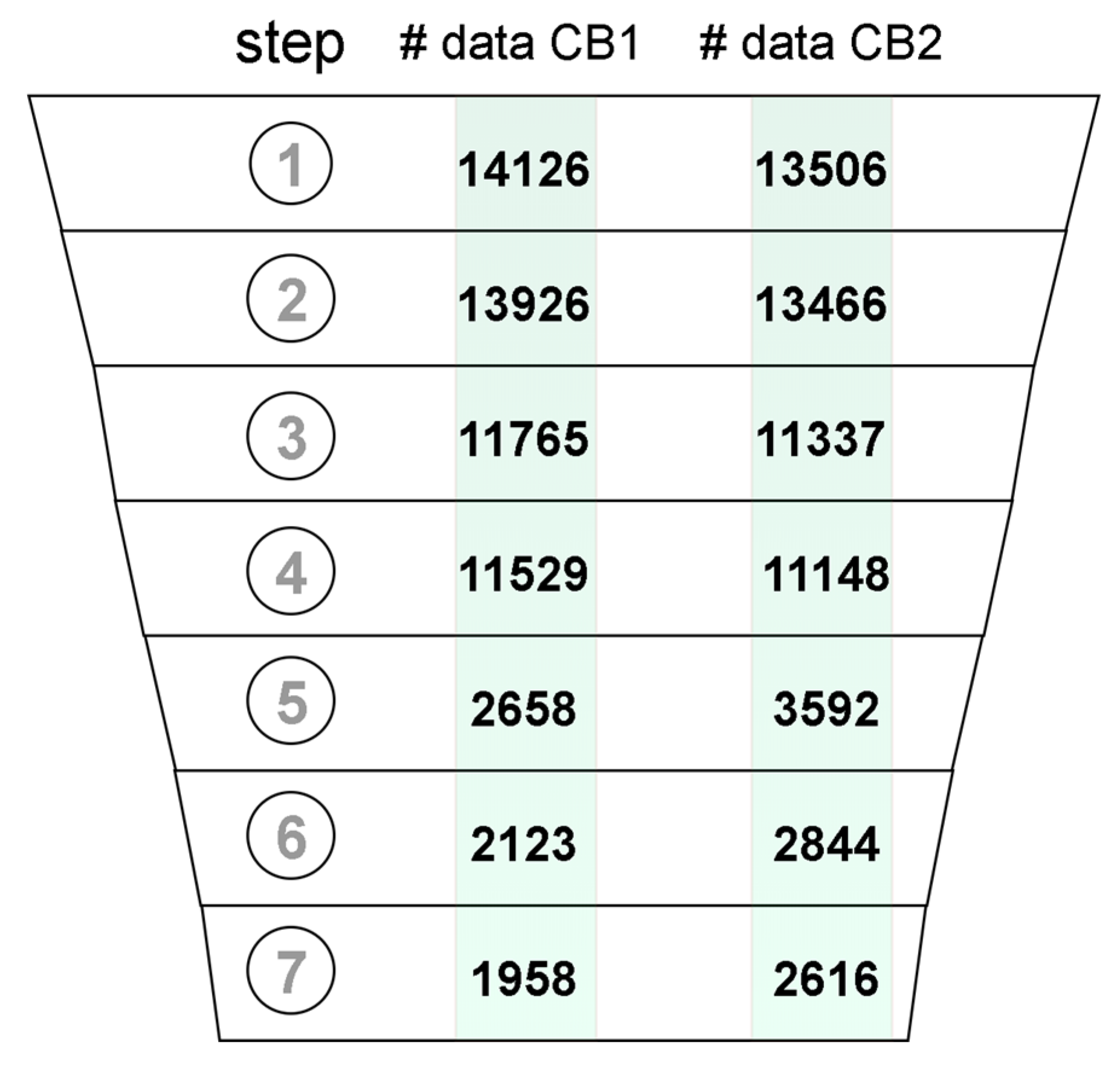
2.2. Data Curation
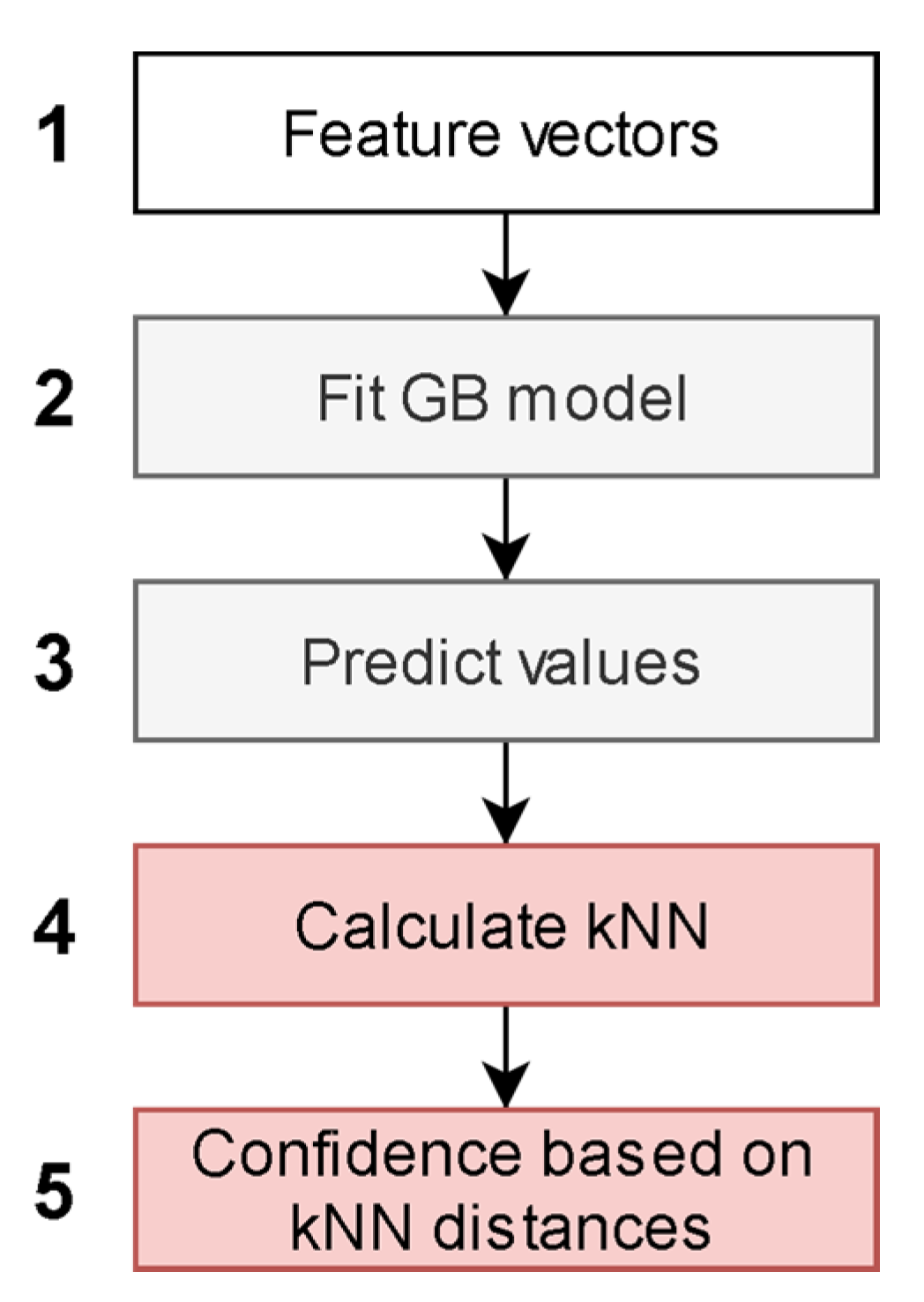
2.3. Model Description
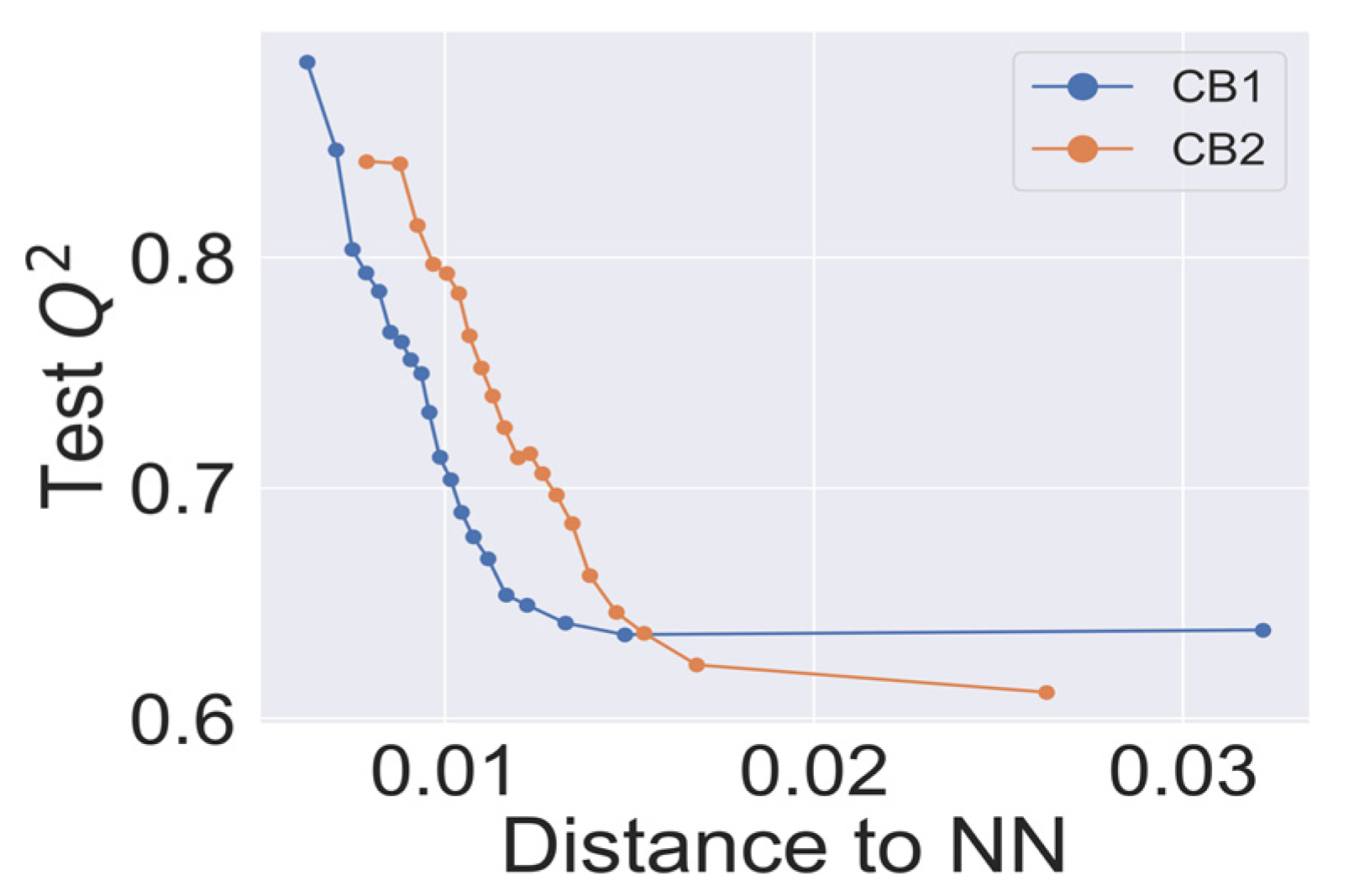
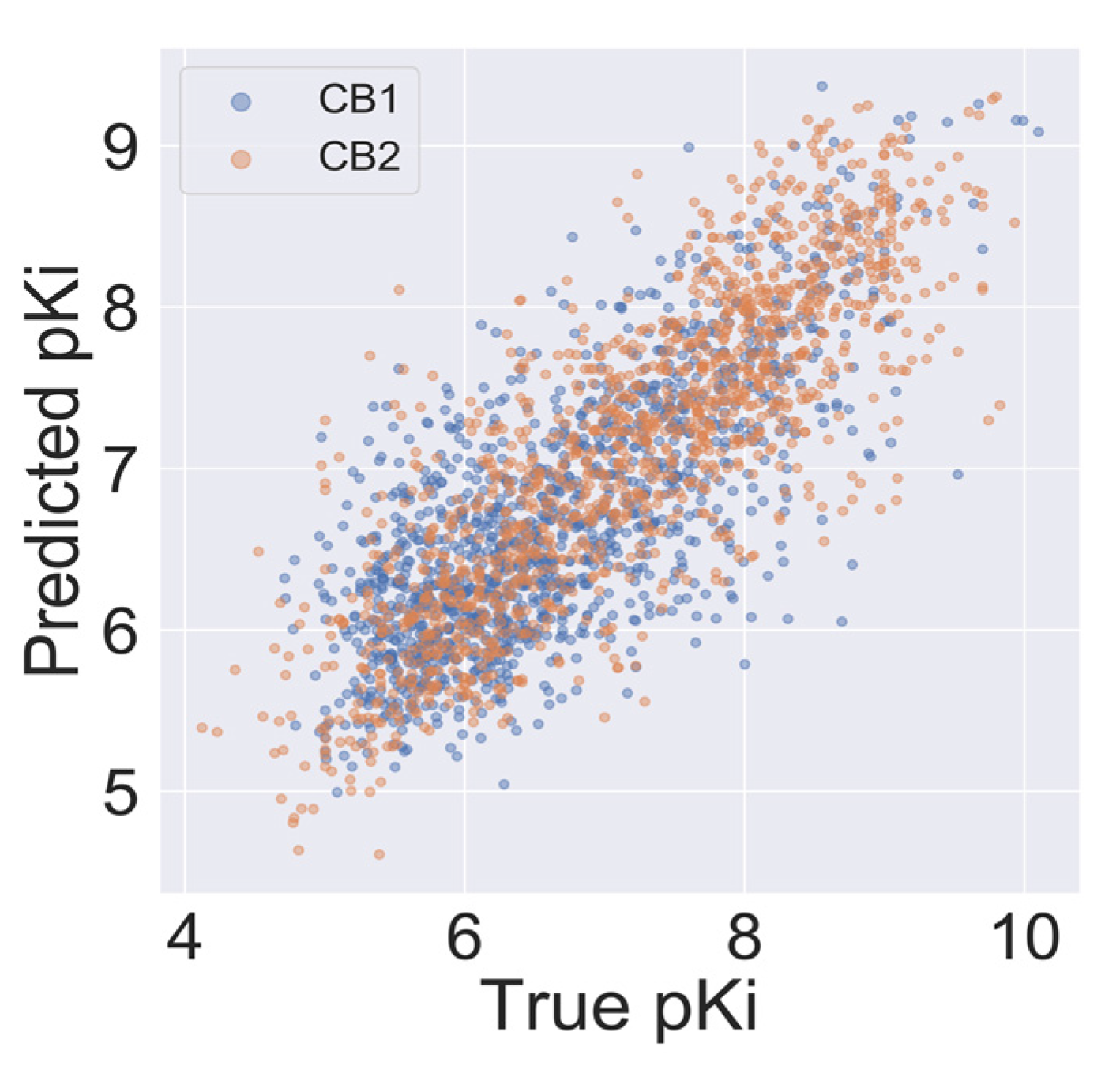
2.4. Model Validation
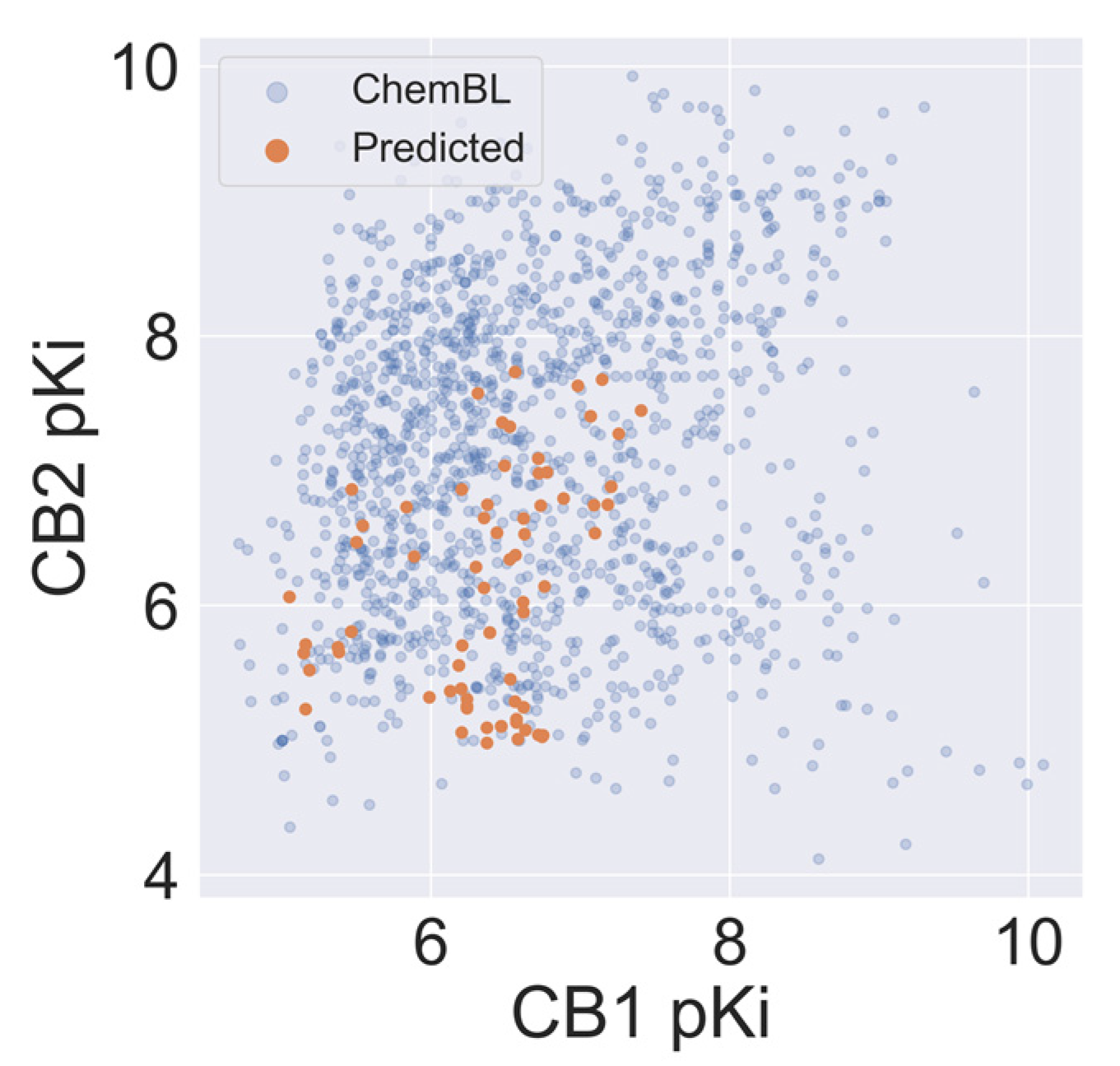
2.5. Virtual Screening of Cannabis Sativa Phytochemicals
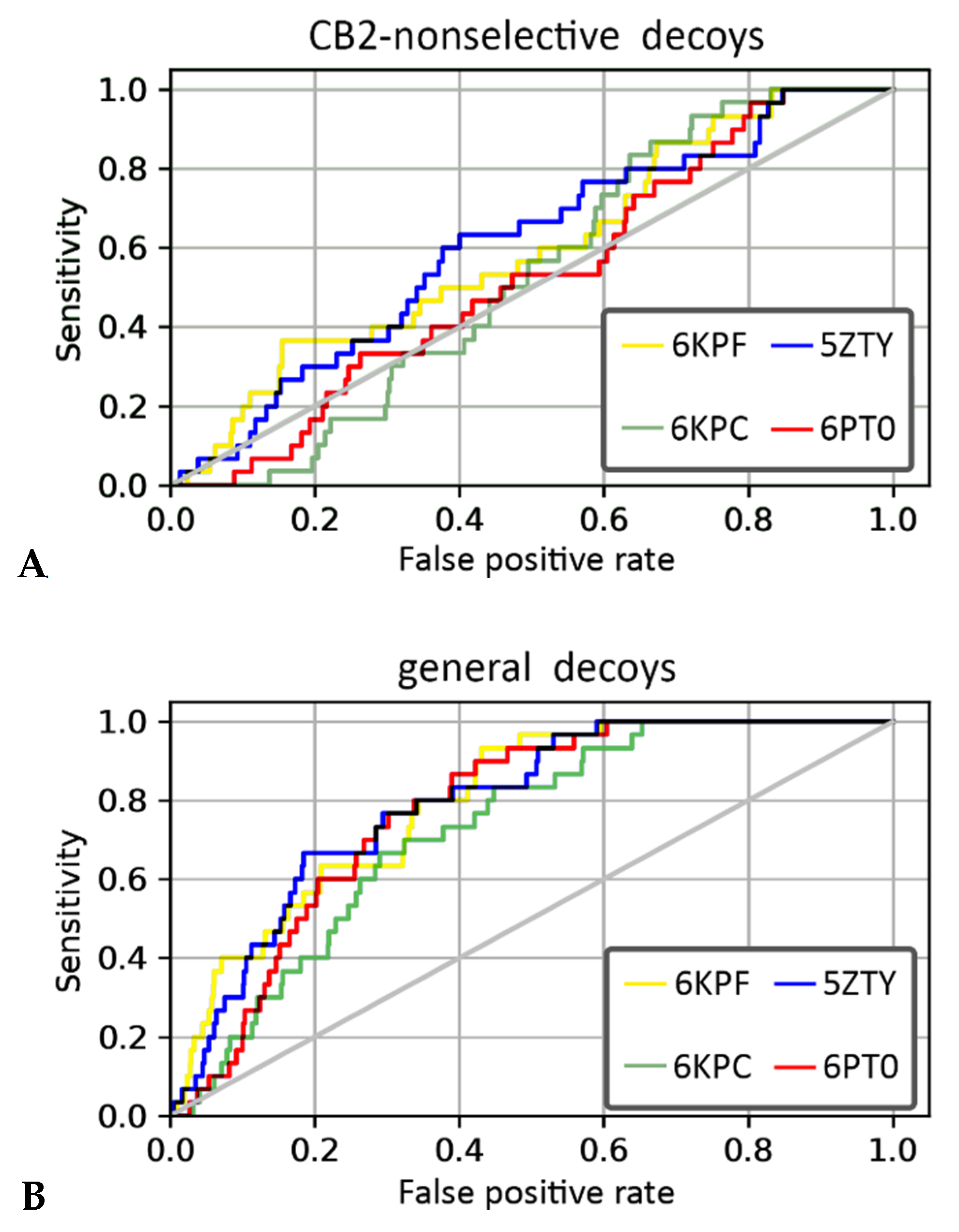
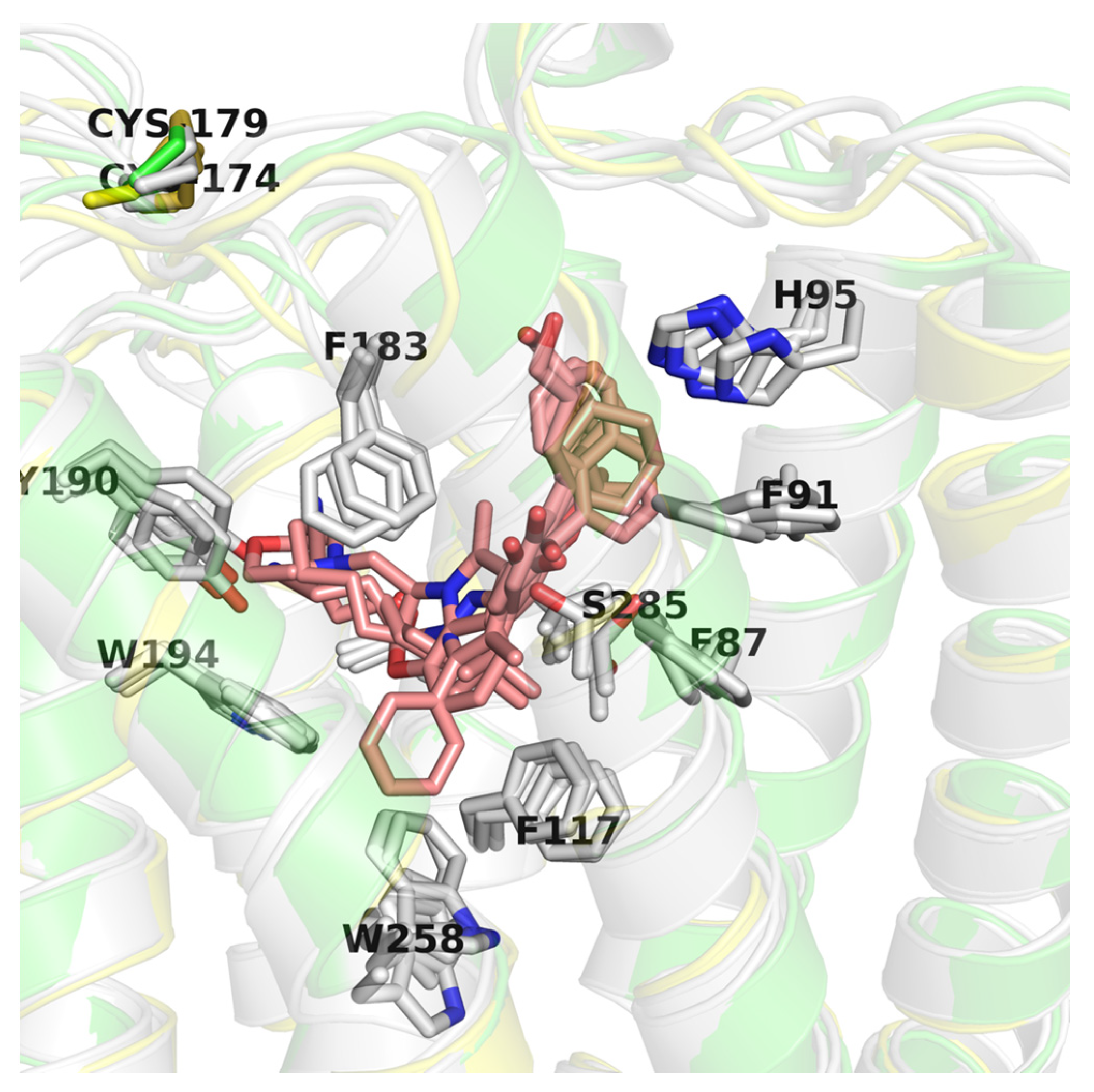
2.6. CB2 Structure-Based Virtual Screening Results
3. Discussion
4. Materials and Methods
4.1. Data Collection
4.2. Data Curation
4.3. Descriptor Calculation
4.4. Machine Learning
4.5. Model Validation
4.6. Prediction of CB2-Selectivity of Cannabis Sativa Ingredients
4.7. Structure-Based Virtual Screening
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cascio, M.G.; Pertwee, R.G.; Marini, P. The pharmacology and therapeutic potential of plant cannabinoids. In Cannabis sativa L.—Botany and Biotechnology; Chandra, S., Lata, H., ElSohly, M.A., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 207–225. ISBN 9783319545646. [Google Scholar]
- ElSohly, M.A.; Radwan, M.M.; Gul, W.; Chandra, S.; Galal, A. Phytochemistry of Cannabis sativa L. Prog. Chem. Org. Nat. Prod. 2017, 103, 1–36. [Google Scholar] [PubMed]
- Andre, C.M.; Hausman, J.-F.; Guerriero, G. Cannabis sativa: The plant of the thousand and one molecules. Front. Plant Sci. 2016, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Aizpurua-Olaizola, O.; Soydaner, U.; Öztürk, E.; Schibano, D.; Simsir, Y.; Navarro, P.; Etxebarria, N.; Usobiaga, A. Evolution of the cannabinoid and terpene content during the growth of Cannabis sativa plants from different chemotypes. J. Nat. Prod. 2016, 79, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Galaj, E.; Bi, G.-H.; Yang, H.-J.; Xi, Z.-X. Cannabidiol attenuates the rewarding effects of cocaine in rats by CB2, 5-HT1A and TRPV1 receptor mechanisms. Neuropharmacology 2020, 167, 107740. [Google Scholar] [CrossRef]
- Aso, E.; Andrés-Benito, P.; Grau-Escolano, J.; Caltana, L.; Brusco, A.; Sanz, P.; Ferrer, I. Cannabidiol-enriched extract reduced the cognitive impairment but not the epileptic seizures in a Lafora disease animal model. In Cannabis and Cannabinoid Res; Mary Ann Liebert, Inc.: New Rochelle, NY, USA, 2019. [Google Scholar]
- Volkow, N.D.; Hampson, A.J.; Baler, R.D. Don’t worry, be happy: Endocannabinoids and cannabis at the intersection of stress and reward. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 285–308. [Google Scholar] [CrossRef]
- Rong, C.; Lee, Y.; Carmona, N.E.; Cha, D.S.; Ragguett, R.-M.; Rosenblat, J.D.; Mansur, R.B.; Ho, R.C.; McIntyre, R.S. Cannabidiol in medical marijuana: Research vistas and potential opportunities. Pharmacol. Res. 2017, 121, 213–218. [Google Scholar] [CrossRef]
- Morales, P.; Hurst, D.P.; Reggio, P.H. Molecular Targets of the Phytocannabinoids: A Complex Picture. Prog. Chem. Org. Nat. Prod. 2017, 103, 103–131. [Google Scholar]
- Devinsky, O.; Cilio, M.R.; Cross, H.; Fernandez-Ruiz, J.; French, J.; Hill, C.; Katz, R.; Di Marzo, V.; Jutras-Aswad, D.; Notcutt, W.G.; et al. Cannabidiol: Pharmacology and potential therapeutic role in epilepsy and other neuropsychiatric disorders. Epilepsia 2014, 55, 791–802. [Google Scholar] [CrossRef]
- Russo, E.B.; Burnett, A.; Hall, B.; Parker, K.K. Agonistic properties of cannabidiol at 5-HT1a receptors. Neurochem. Res. 2005, 30, 1037–1043. [Google Scholar] [CrossRef]
- Svíženská, I.; Dubovy, P.; Šulcová, A. Cannabinoid receptors 1 and 2 (CB1 and CB2), their distribution, ligands and functional involvement in nervous system structures—A short review. Pharmacol. Biochem. Behav. 2008, 90, 501–511. [Google Scholar] [CrossRef]
- Marzo, V.D.; Di Marzo, V.; Bifulco, M.; De Petrocellis, L. The endocannabinoid system and its therapeutic exploitation. Nat. Rev. Drug Discov. 2004, 3, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Bátkai, S.; Kunos, G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol. Rev. 2006, 58, 389–462. [Google Scholar] [CrossRef] [PubMed]
- Lyu, J.; Wang, S.; Balius, T.E.; Singh, I.; Levit, A.; Moroz, Y.S.; O’Meara, M.J.; Che, T.; Algaa, E.; Tolmachova, K.; et al. Ultra-large library docking for discovering new chemotypes. Nature 2019, 566, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Jakowiecki, J.; Filipek, S. Hydrophobic ligand entry and exit pathways of the CB1 cannabinoid receptor. J. Chem. Inf. Model. 2016, 56, 2457–2466. [Google Scholar] [CrossRef] [PubMed]
- Latek, D.; Kolinski, M.; Ghoshdastider, U.; Debinski, A.; Bombolewski, R.; Plazinska, A.; Jozwiak, K.; Filipek, S. Modeling of ligand binding to G protein coupled receptors: Cannabinoid CB 1, CB 2 and adrenergic β 2 AR. J. Mol. Model. 2011, 17, 2353–2366. [Google Scholar] [CrossRef] [PubMed]
- Chohan, T.A.; Chen, J.-J.; Qian, H.-Y.; Pan, Y.-L.; Chen, J.-Z. Molecular modeling studies to characterize N-phenylpyrimidin-2-amine selectivity for CDK2 and CDK4 through 3D-QSAR and molecular dynamics simulations. Mol. Biosyst. 2016, 12, 1250–1268. [Google Scholar] [CrossRef]
- Stokes, J.M.; Yang, K.; Swanson, K.; Jin, W.; Cubillos-Ruiz, A.; Donghia, N.M.; MacNair, C.R.; French, S.; Carfrae, L.A.; Bloom-Ackermann, Z.; et al. A Deep Learning Approach to Antibiotic Discovery. Cell 2020, 181, 475–483. [Google Scholar] [CrossRef]
- Kasabe, A.J.; Kulkarni, A.S.; Gaikwad, V.L. QSPR Modeling of biopharmaceutical properties of hydroxypropyl methylcellulose (cellulose ethers) tablets based on its degree of polymerization. AAPS PharmSciTech 2019, 20, 308. [Google Scholar] [CrossRef]
- Mizera, M.; Krause, A.; Zalewski, P.; Skibiński, R.; Cielecka-Piontek, J. Quantitative structure-retention relationship model for the determination of naratriptan hydrochloride and its impurities based on artificial neural networks coupled with genetic algorithm. Talanta 2017, 164, 164–174. [Google Scholar] [CrossRef]
- Mizera, M.; Talaczyńska, A.; Zalewski, P.; Skibiński, R.; Cielecka-Piontek, J. Prediction of HPLC retention times of tebipenem pivoxyl and its degradation products in solid state by applying adaptive artificial neural network with recursive features elimination. Talanta 2015, 137, 174–181. [Google Scholar] [CrossRef]
- Romero-Parra, J.; Chung, H.; Tapia, R.A.; Faúndez, M.; Morales-Verdejo, C.; Lorca, M.; Lagos, C.F.; Di Marzo, V.; David Pessoa-Mahana, C.; Mella, J. Combined CoMFA and CoMSIA 3D-QSAR study of benzimidazole and benzothiophene derivatives with selective affinity for the CB2 cannabinoid receptor. Eur. J. Pharm. Sci. 2017, 101, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lv, Q.; Xu, W.; Lai, X.; Liu, Y.; Tu, G. 4D-QSAR studies of CB 2 cannabinoid receptor inverse agonists: A comparison to 3D-QSAR. Med. Chem. Res. 2019, 28, 498–504. [Google Scholar] [CrossRef]
- Chen, J.-Z.; Han, X.-W.; Liu, Q.; Makriyannis, A.; Wang, J.; Xie, X.-Q. 3D-QSAR studies of arylpyrazole antagonists of cannabinoid receptor subtypes CB1 and CB2. A combined NMR and CoMFA approach. J. Med. Chem. 2006, 49, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Floresta, G.; Apirakkan, O.; Rescifina, A.; Abbate, V. Discovery of high-affinity cannabinoid receptors ligands through a 3D-QSAR ushered by scaffold-hopping analysis. Molecules 2018, 23, 2183. [Google Scholar] [CrossRef]
- Labib, R.M.; Srivedavyasasri, R.; Youssef, F.S.; Ross, S.A. Secondary metabolites isolated from Pinus roxburghii and interpretation of their cannabinoid and opioid binding properties by virtual screening and in vitro studies. Saudi Pharm. J. 2018, 26, 437–444. [Google Scholar] [CrossRef]
- Hua, T.; Vemuri, K.; Nikas, S.P.; Laprairie, R.B.; Wu, Y.; Qu, L.; Pu, M.; Korde, A.; Jiang, S.; Ho, J.-H.; et al. Crystal structures of agonist-bound human cannabinoid receptor CB1. Nature 2017, 547, 468–471. [Google Scholar] [CrossRef]
- Ji, B.; Liu, S.; He, X.; Man, V.H.; Xie, X.-Q.; Wang, J. Prediction of the binding affinities and selectivity for CB1 and CB2 ligands using homology modeling, molecular docking, molecular dynamics simulations, and MM-PBSA binding free energy calculations. ACS Chem. Neurosci. 2020, 11, 1139–1158. [Google Scholar] [CrossRef]
- Hua, T.; Li, X.; Wu, L.; Iliopoulos-Tsoutsouvas, C.; Wang, Y.; Wu, M.; Shen, L.; Johnston, C.A.; Nikas, S.P.; Song, F.; et al. Activation and signaling mechanism revealed by cannabinoid receptor-Gi complex structures. Cell 2020, 180, 655–665.e18. [Google Scholar] [CrossRef]
- Xing, C.; Zhuang, Y.; Xu, T.-H.; Feng, Z.; Zhou, X.E.; Chen, M.; Wang, L.; Meng, X.; Xue, Y.; Wang, J.; et al. Cryo-EM structure of the human cannabinoid receptor CB2-Gi signaling complex. Cell 2020, 180, 645–654.e13. [Google Scholar] [CrossRef]
- Li, X.; Hua, T.; Vemuri, K.; Ho, J.-H.; Wu, Y.; Wu, L.; Popov, P.; Benchama, O.; Zvonok, N.; Locke, K.; et al. Crystal structure of the human cannabinoid receptor CB2. Cell 2019, 176, 459–467.e13. [Google Scholar] [CrossRef]
- Tropsha, A. Best practices for QSAR model development, validation, and exploitation. Mol. Inform. 2010, 29, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Latek, D.; Rutkowska, E.; Niewieczerzal, S.; Cielecka-Piontek, J. Drug-induced diabetes type 2: In silico study involving class B GPCRs. PLoS ONE 2019, 14, e0208892. [Google Scholar] [CrossRef] [PubMed]
- Pasznik, P.; Rutkowska, E.; Niewieczerzal, S.; Cielecka-Piontek, J.; Latek, D. Potential off-target effects of beta-blockers on gut hormone receptors: In silico study including GUT-DOCK—A web service for small-molecule docking. PLoS ONE 2019, 14, e0210705. [Google Scholar] [CrossRef]
- Ancuceanu, R.; Dinu, M.; Neaga, I.; Laszlo, F.G.; Boda, D. Development of QSAR machine learning-based models to forecast the effect of substances on malignant melanoma cells. Oncol. Lett. 2019, 17, 4188–4196. [Google Scholar] [CrossRef]
- Kwon, S.; Bae, H.; Jo, J.; Yoon, S. Comprehensive ensemble in QSAR prediction for drug discovery. BMC Bioinform. 2019, 20, 521. [Google Scholar] [CrossRef]
- Shoyama, Y.; Nishioka, I. Cannabis. XIII. Two new spiro-compounds, cannabispirol and acetyl cannabispirol. Chem. Pharm. Bull. 1978, 26, 3641–3646. [Google Scholar] [CrossRef]
- Molnar, J.; Csiszar, K.; Nishioka, I.; Shoyama, Y. The effects of cannabispiro compounds and tetrahydrocannabidiolic acid on the plasmid transfer and maintenance in Escherichia coli. Acta Microbiol. Hung. 1986, 33, 221–231. [Google Scholar]
- Molnár, J.; Szabó, D.; Pusztai, R.; Mucsi, I.; Berek, L.; Ocsovszki, I.; Kawata, E.; Shoyama, Y. Membrane associated antitumor effects of crocine-, ginsenoside- and cannabinoid derivates. Anticancer Res. 2000, 20, 861–867. [Google Scholar]
- León, L.G.; Donadel, O.J.; Tonn, C.E.; Padrón, J.M. Tessaric acid derivatives induce G2/M cell cycle arrest in human solid tumor cell lines. Bioorg. Med. Chem. 2009, 17, 6251–6256. [Google Scholar] [CrossRef]
- Crombie, L.; Crombie, W.M.L. Cannabinoid acids and esters: Miniaturized synthesis and chromatographic study. Phytochemistry 1977, 16, 1413–1420. [Google Scholar] [CrossRef]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Zhang, P.; Wang, Y.; Qin, C.; Chen, S.; He, W.; Tao, L.; Tan, Y.; Gao, D.; Wang, B.; et al. CMAUP: A database of collective molecular activities of useful plants. Nucleic Acids Res. 2019, 47, D1118–D1127. [Google Scholar] [CrossRef]
- Fourches, D.; Muratov, E.; Tropsha, A. Trust, but verify: On the importance of chemical structure curation in cheminformatics and QSAR modeling research. J. Chem. Inf. Model. 2010, 50, 1189–1204. [Google Scholar] [CrossRef] [PubMed]
- Fourches, D.; Muratov, E.; Tropsha, A. Trust, but verify II: A Practical guide to chemogenomics data curation. J. Chem. Inf. Model. 2016, 56, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Landrum, G. Rdkit documentation. Release 2013, 1, 1–79. [Google Scholar]
- Resources. Available online: http://db-gpcr.chem.uw.edu.pl/ (accessed on 28 April 2020).
- Moriwaki, H.; Tian, Y.-S.; Kawashita, N.; Takagi, T. Mordred: A molecular descriptor calculator. J. Cheminform. 2018, 10, 4. [Google Scholar] [CrossRef]
- Ke, G.; Meng, Q.; Finley, T.; Wang, T.; Chen, W.; Ma, W.; Ye, Q.; Liu, T.-Y. LightGBM: A highly efficient gradient boosting decision tree. In Advances in Neural Information Processing Systems 30; Guyon, I., Luxburg, U.V., Bengio, S., Wallach, H., Fergus, R., Vishwanathan, S., Garnett, R., Eds.; Curran Associates Inc.: New York, NY, USA, 2017; pp. 3146–3154. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Shao, Z.; Yan, W.; Chapman, K.; Ramesh, K.; Ferrell, A.J.; Yin, J.; Wang, X.; Xu, Q.; Rosenbaum, D.M. Structure of an allosteric modulator bound to the CB1 cannabinoid receptor. Nat. Chem. Biol. 2019, 15, 1199–1205. [Google Scholar] [CrossRef]
- Release, S. Others 4: Schrödinger Suite 2017-4 Protein Preparation Wizard; Epik, Schrödinger, LLC: New York, NY, USA, 2017. [Google Scholar]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Release, S. 2: LigPrep; Schrödinger, LLC: New York, NY, USA, 2017. [Google Scholar]
- Paulke, A.; Proschak, E.; Sommer, K.; Achenbach, J.; Wunder, C.; Toennes, S.W. Synthetic cannabinoids: In silico prediction of the cannabinoid receptor 1 affinity by a quantitative structure-activity relationship model. Toxicol. Lett. 2016, 245, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Kumar, A. CORAL: QSAR models of CB1 cannabinoid receptor inhibitors based on local and global SMILES attributes with the index of ideality of correlation and the correlation contradiction index. Chemom. Intell. Lab. Syst. 2020, 200, 103982. [Google Scholar] [CrossRef]
- Ghasemi, F.; Mehridehnavi, A.; Fassihi, A.; Pérez-Sánchez, H. Deep neural network in QSAR studies using deep belief network. Appl. Soft. Comput. 2018, 62, 251–258. [Google Scholar] [CrossRef]
- Grayson, M. Cannabis. Nature 2015, 525, S1. [Google Scholar] [CrossRef] [PubMed]
- Mangoato, I.M.; Mahadevappa, C.P.; Matsabisa, M.G. Cannabis sativa L. Extracts can reverse drug resistance in colorectal carcinoma cells in vitro. Synergy 2019, 9, 100056. [Google Scholar] [CrossRef]
- Martinenghi, L.D.; Jønsson, R.; Lund, T.; Jenssen, H. Isolation, purification, and antimicrobial characterization of cannabidiolic acid and cannabidiol from Cannabis sativa L. Biomolecules 2020, 10, 900. [Google Scholar] [CrossRef] [PubMed]
- Stonehouse, G.C.; McCarron, B.J.; Guignardi, Z.S.; El Mehdawi, A.F.; Lima, L.W.; Fakra, S.C.; Pilon-Smits, E.A.H. Selenium metabolism in hemp (Cannabis sativa L.)—Potential for Phytoremediation and biofortification. Environ. Sci. Technol. 2020, 54, 4221–4230. [Google Scholar] [CrossRef]
- Frassinetti, S.; Gabriele, M.; Moccia, E.; Longo, V.; Di Gioia, D. Antimicrobial and antibiofilm activity of Cannabis sativa L. seeds extract against Staphylococcus aureus and growth effects on probiotic Lactobacillus spp. LWT 2020, 124, 109149. [Google Scholar] [CrossRef]
- Sangiovanni, E.; Fumagalli, M.; Pacchetti, B.; Piazza, S.; Magnavacca, A.; Khalilpour, S.; Melzi, G.; Martinelli, G.; Dell’Agli, M. Cannabis sativa L. extract and cannabidiol inhibit in vitro mediators of skin inflammation and wound injury. Phytother. Res. 2019, 33, 2083–2093. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Predicted Value | Similar Molecule for ChEMBL | Tanimoto Coefficient | |||
|---|---|---|---|---|---|---|
| CB1 pKi | CB2 pKi | Structure | CB1 pKi | CB2 pKi | ||
C1 (4-hydroxy-6-methoxyspiro[1,2-dihydroindene-3,4’-cyclohexane]-1’-yl) acetate | 5.46 | 6.87 |  CHEMBL1201151 | 4.64 | N/A | 0.28 |
C2 2-[(2R,4aR,8R,8aR)-8-hydroxy-4a,8-dimethyl-1,2,3,4,5,6,7,8a-octahydronaphthalen-2-yl]prop-2-enoic acid | 5.57 | 6.59 |  CHEMBL256753 | 6.95 | 8.03 | 0.18 |
C3 6-carboxy-2-methyl-2-(4-methylpent-3-enyl)-7-pentylchromen-5-olate | 6.41 | 7.29 |  | 7.33 | 6.04 | 0.41 |
| Metric | CB2-Non-Selective Decoys | General Decoys | ||||||
|---|---|---|---|---|---|---|---|---|
| 6KPF | 6KPC | 6PT0 | 5ZTY | 6KPF | 6KPC | 6PT0 | 5ZTY | |
| EF 2% | 0 | 0 | 0 | 1.7 | 3.3 | 0 | 0 | 3.3 |
| EF 5% | 0.67 | 0 | 0 | 1.3 | 4.7 | 1.3 | 1.3 | 3.3 |
| EF 10% | 1.7 | 0 | 0.33 | 1 | 4.0 | 2 | 1.7 | 3 |
| ROC AUC | 0.6 | 0.53 | 0.53 | 0.6 | 0.8 | 0.73 | 0.77 | 0.79 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mizera, M.; Latek, D.; Cielecka-Piontek, J. Virtual Screening of C. Sativa Constituents for the Identification of Selective Ligands for Cannabinoid Receptor 2. Int. J. Mol. Sci. 2020, 21, 5308. https://doi.org/10.3390/ijms21155308
Mizera M, Latek D, Cielecka-Piontek J. Virtual Screening of C. Sativa Constituents for the Identification of Selective Ligands for Cannabinoid Receptor 2. International Journal of Molecular Sciences. 2020; 21(15):5308. https://doi.org/10.3390/ijms21155308
Chicago/Turabian StyleMizera, Mikołaj, Dorota Latek, and Judyta Cielecka-Piontek. 2020. "Virtual Screening of C. Sativa Constituents for the Identification of Selective Ligands for Cannabinoid Receptor 2" International Journal of Molecular Sciences 21, no. 15: 5308. https://doi.org/10.3390/ijms21155308
APA StyleMizera, M., Latek, D., & Cielecka-Piontek, J. (2020). Virtual Screening of C. Sativa Constituents for the Identification of Selective Ligands for Cannabinoid Receptor 2. International Journal of Molecular Sciences, 21(15), 5308. https://doi.org/10.3390/ijms21155308