Aryl Hydrocarbon Receptor Connects Inflammation to Breast Cancer


Abstract
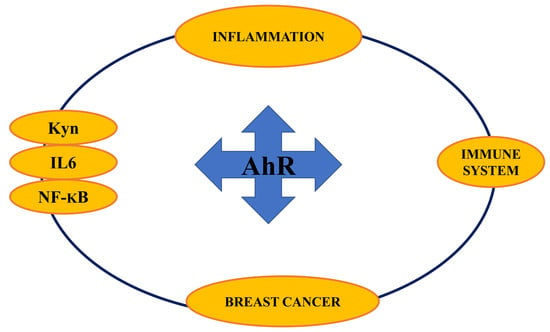
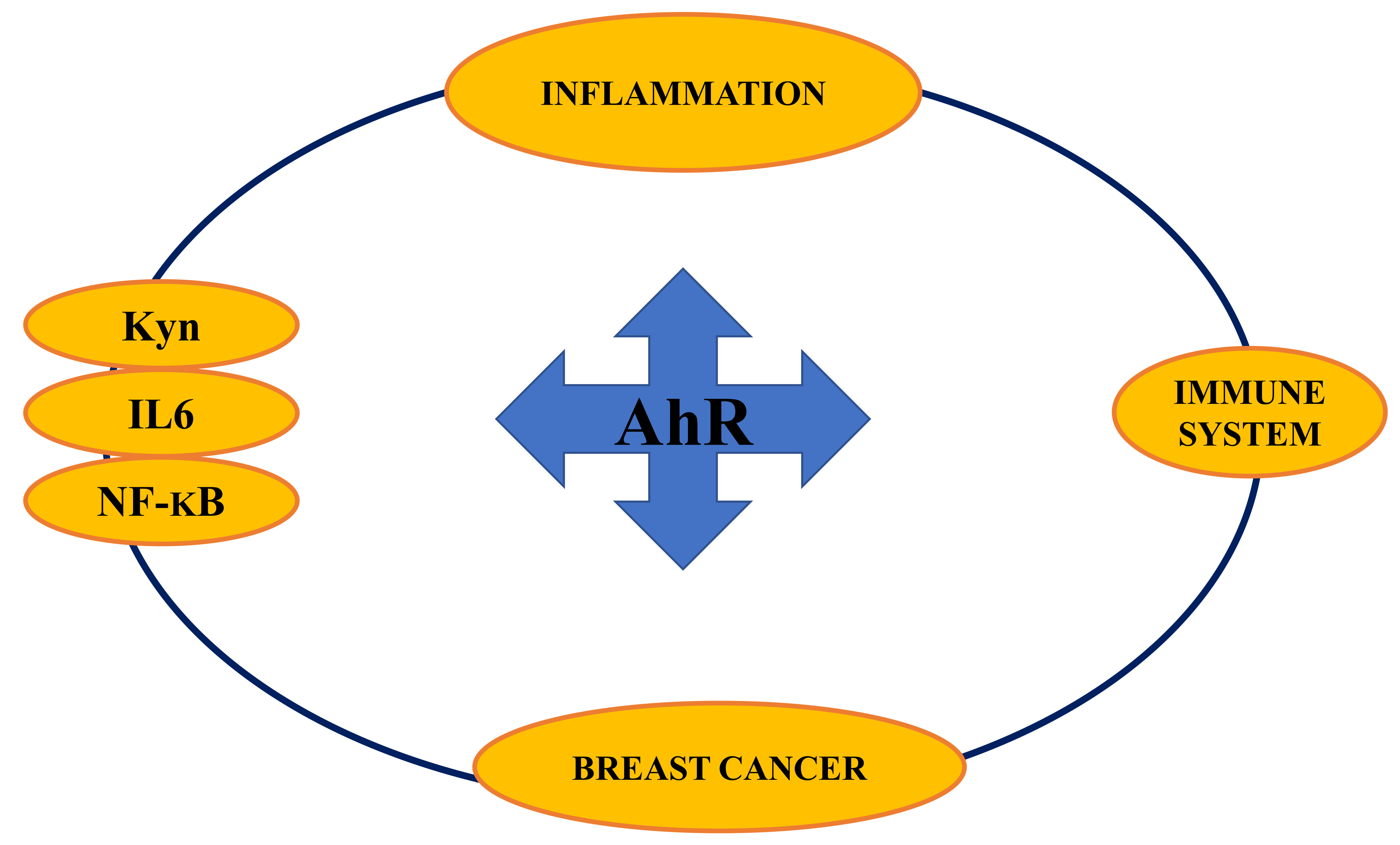{kind=link}
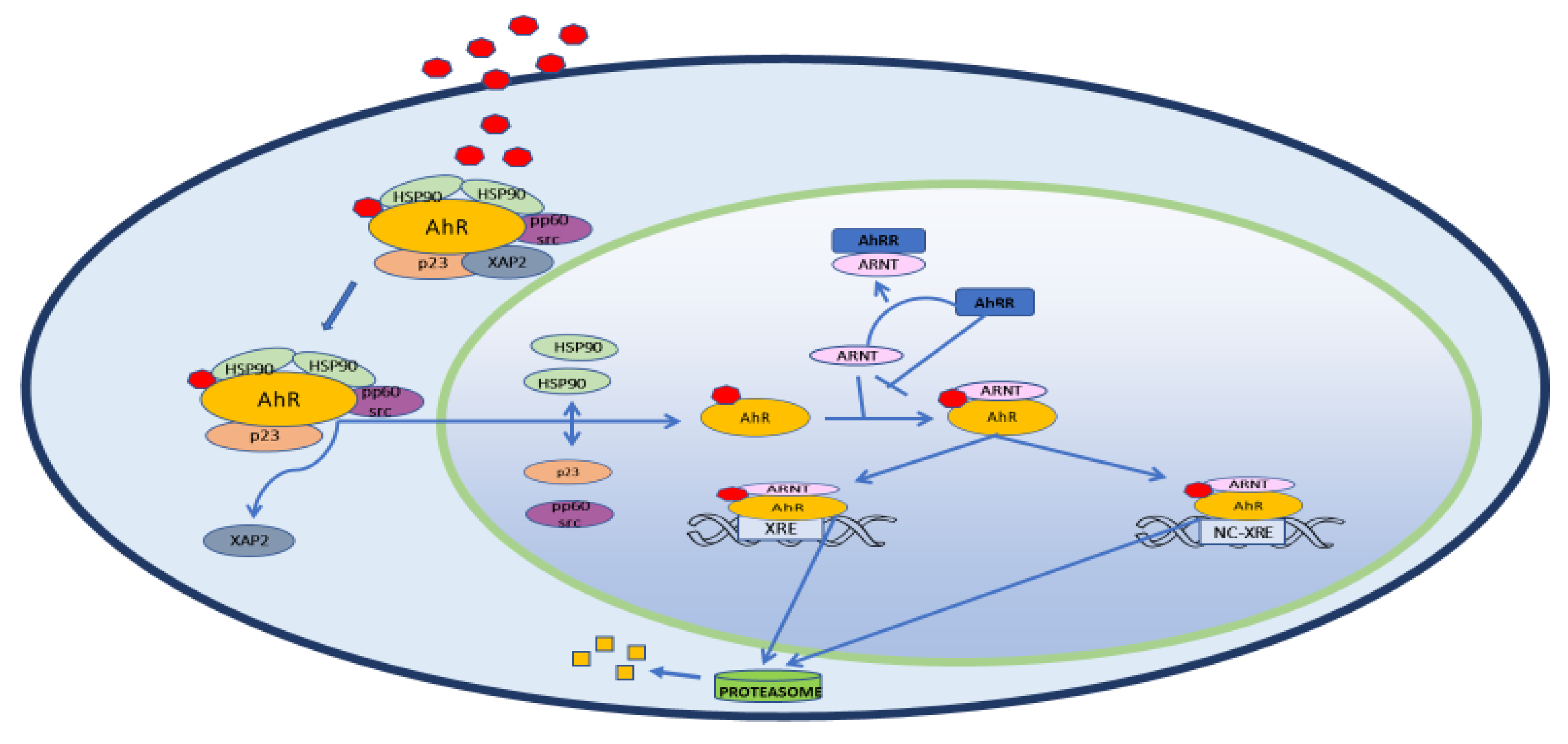{kind=link}
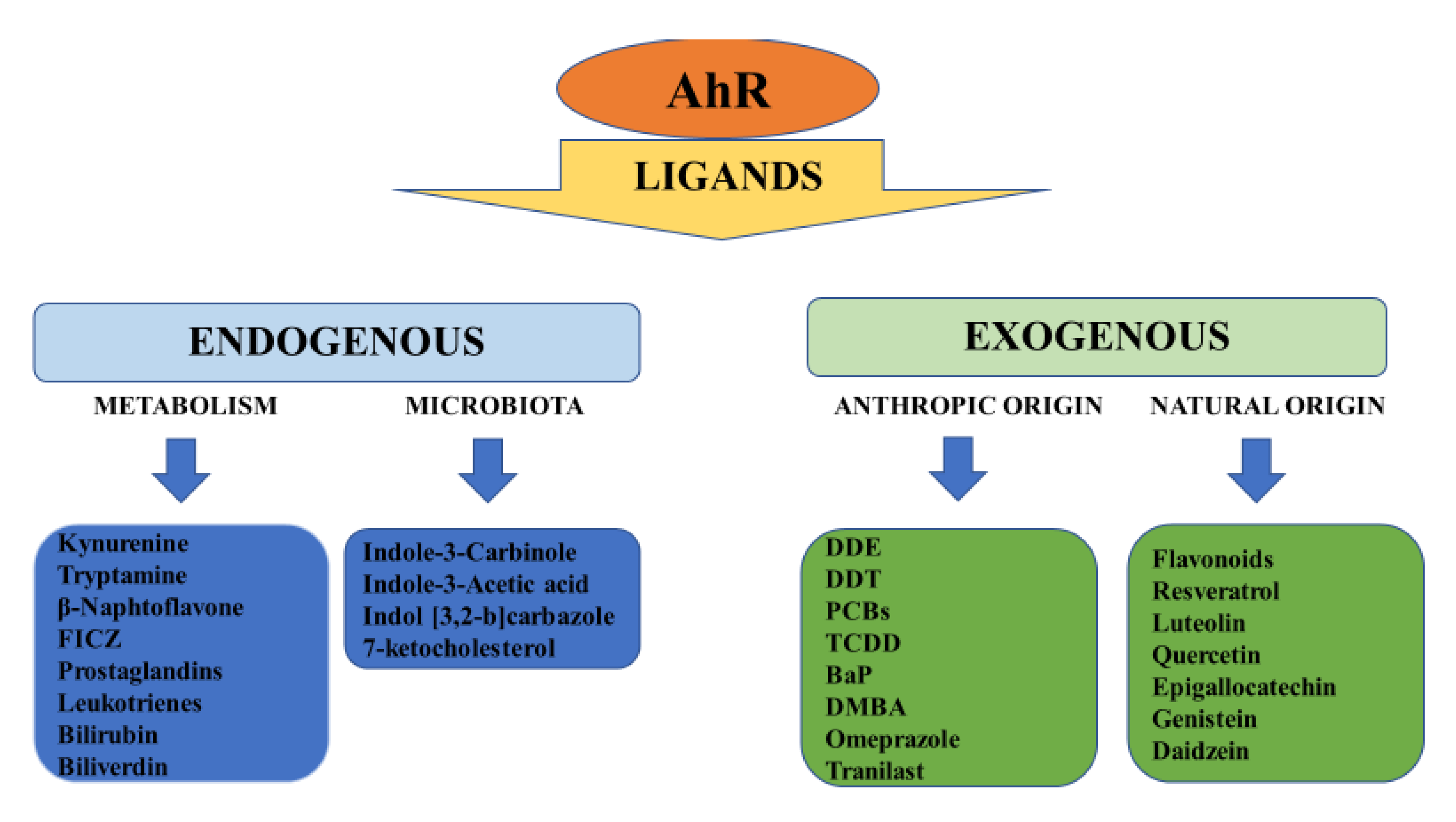{kind=link}
{kind=link}
1. Introduction
2. Structure, Physiology and Target Genes
3. AhR and Inflammation
3.1. AhR in the Immune System
3.2. AhR and NF-ΚB
4. AhR and Cancer
4.1. AhR and Breast Cancer
4.2. AhR and TNBC
5. Conclusions
Funding
Conflicts of Interest
Abbreviations
| AhR | Aryl hydrocarbon receptor |
| AhRR | Aryl hydrocarbon receptor repressor |
| AIP | Immunophilin-like Ah receptor-interacting protein) |
| Aldh3a1 | Aldehyde dehydrogenase family 3, subfamily 1 |
| ARNT | Aryl hydrocarbon nuclear translocator |
| BaP | Benzo[a]pyrene |
| BC | Breast cancer |
| BCRP | Breast cancer resistance proteins |
| BRCA | BReast CAncer gene |
| CDK 4/6 | Cyclin-dependent kinase 4/6 |
| CDKN1A | Cyclin-dependent kinase inhibitor 1A |
| c-Myc | Avian Myelocytomatosis virus oncogene cellular homolog |
| COX2 | Cyclooxygenase-2 |
| CpG | C phosphate G |
| CRM1 | Chromosomal Maintenance 1 |
| CYP1A1 | Cytochrome P450 family 1 subfamily A member 1 |
| CYP1A2 | Cytochrome P450 family 1 subfamily A member 2 |
| CYP1B1 | Cytochrome P450 family 1 subfamily B member 1 |
| DDT | Dichlorodiphenyltrichloroethane |
| DMBA | Dimethyl-benz(a)anthracene |
| E2 | Estradiol |
| ECC-1 | endocervical cancer cells |
| EGFR | Epithelial growth factor receptor |
| EGR1 | Early growth response 1 |
| ESR1 | Estrogen receptor gene |
| ER | Estrogen receptor |
| FICZ | 6-formylindolo[3,2-b] carbazole |
| Gstα1 | Glutathione S-transferase, alpha 1 |
| HAHs | Halogenated aromatic hydrocarbons |
| HepG2 | Hepatoma G2 |
| HER2 | Human epidermal growth factor receptor 2 |
| HIF-1β | Hypoxia-induced factor β |
| HSPs90 | Heat shock proteins 90 |
| IAA | Indole-3-acetic acid |
| ICZ | Indol [3,2-b]carbazole |
| IDO | Indoleamine-2,3-dioxygenase |
| IGF | Insulin growth factor |
| IKKα | Inhibitor kappa B kinase α |
| IL1β | Interleukin 1β |
| IL-6 | Interleukin 6 |
| IL-8 | interleukin-8 |
| iNO | induced nitric oxide oxide |
| KLF6 | Kruppel-like factor 6 |
| Kyn | Kynurenine |
| LPS | Lipopolysaccharide |
| LT-α1β2 | Lymphotoxin-α1β2 |
| MCF 10F | Michigan Cancer Foundation 10 Floating |
| MDA-MB231 | M.D. Anderson - Metastasis Breast 231 |
| MMP | Matrix metalloproteinase |
| mPGE2S | microsomal PGE2 synthase |
| MPR2 | Multidrug resistance protein 2 |
| MPR3 | Multidrug resistance protein 3 |
| mRNA | Messenger ribonucleic acid |
| NC-XREs | Nonconsensus response elements |
| NF-kB | Nuclear Factor kappa-light-chain-enhancer of activated B cells |
| NIK | NF-κB-inducing kinase |
| Nqo1 | NAD(P)H dehydrogenase quinone 1 |
| OATP | Anionic transport proteins |
| OCTP | Organic cationic transport proteins |
| OSM | OncoStatin M |
| p23 | Proteolytically resistant 23-kDa protein |
| PAHs | Polycyclic aromatic hydrocarbons |
| PAI-1 | plasminogen activator inhibitor-1 |
| PARP | Poly ADP ribose polymerase |
| PCBs | Polychlorinated biphenyls |
| PGE2 | Prostaglandin E2 |
| PLAU | Plasminogen activator urokinase |
| PMA | Phorbol 12-myristate 13-acetate |
| pp60(c-src) | Proto-oncogene tyrosine-protein kinase 60 (Sarcoma) |
| PR | Progesterone receptor |
| RelA | V-Rel reticuloendotheliosis viral oncogene homolog A |
| RelB | V-Rel Reticuloendotheliosis viral oncogene homolog B |
| SAhRMs | Selective AhR modulators |
| STAT3 | Signal transducer and activator of transcription 3 |
| TCDD | 2,3,7,8-Tetrachlorodibenzo-p-dioxin |
| TDO | Tryptophan-2,3-dioxygenase |
| TiPARP | TCDD-inducible poly ADP-ribose polymerase |
| TNBC | Triple negative breast cancer |
| TNFR | Tumor necrosis factor receptor |
| TNFα | Tumor necrosis factor-α |
| Ugt1a6 | UDP glucuronosyltransferase family 1 member A6 |
| Wnt | Wingless/Integrated |
| XA | Xanthurenic acid |
| XAP2 | Hepatitis B virus X-associated protein 2 |
| XPO1 | Exportin 1 |
| XREs | Xenobiotic response elements |
References
- Hahn, M.E.; Karchner, S.I.; Shapiro, M.A.; Perera, S.A. Molecular evolution of two vertebrate aryl hydrocarbon (dioxin) receptors (AHR1 and AHR2) and the PAS family. Proc. Natl. Acad. Sci. USA 1997, 94, 13743–13748. [Google Scholar] [CrossRef] [PubMed]
- Safe, S.H.; Zacharewski, T. Organochlorine exposure and risk for breast cancer. Prog. Clin. Biol. Res. 1997, 396, 133–145. [Google Scholar] [PubMed]
- Poland, A.; Knutson, J.; Glover, E. Studies on the mechanism of action of halogenated aromatic hydrocarbons. Clin. Physiol. Biochem. 1985, 3, 147–154. [Google Scholar] [PubMed]
- Mulero-Navarro, S.; Fernandez-Salguero, P.M. New Trends in Aryl Hydrocarbon Receptor Biology. Front. Cell Dev. Biol. 2016, 4, 45. [Google Scholar] [CrossRef]
- Rothhammer, V.; Quintana, F.J. The aryl hydrocarbon receptor: An environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 2019, 19, 184–197. [Google Scholar] [CrossRef]
- Matthews, J. AHR toxicity and signaling: Role of TIPARP and ADP-ribosylation. Curr. Opin. Toxicol. 2017, 2, 50–57. [Google Scholar] [CrossRef]
- Mimura, J.; Ema, M.; Sogawa, K.; Fujii-Kuriyama, Y. Identification of a novel mechanism of regulation of Ah (dioxin) receptor function. Genes Dev. 1999, 13, 20–25. [Google Scholar] [CrossRef]
- Israel, D.I.; Whitlock, J.P., Jr. Induction of mRNA specific for cytochrome P1-450 in wild type and variant mouse hepatoma cells. J. Biol. Chem. 1983, 258, 10390–10394. [Google Scholar] [PubMed]
- Nebert, D.W.; Roe, A.L.; Dieter, M.Z.; Solis, W.A.; Yang, Y.; Dalton, T.P. Role of the aromatic hydrocarbon receptor and [Ah] gene battery in the oxidative stress response, cell cycle control, and apoptosis. Biochem. Pharmacol. 2000, 59, 65–85. [Google Scholar] [CrossRef]
- Tijet, N.; Boutros, P.C.; Moffat, I.D.; Okey, A.B.; Tuomisto, J.; Pohjanvirta, R. Aryl hydrocarbon receptor regulates distinct dioxin-dependent and dioxin-independent gene batteries. Mol. Pharmacol. 2006, 69, 140–153. [Google Scholar] [CrossRef]
- Bock, K.W. From TCDD-mediated toxicity to searches of physiologic AHR functions. Biochem. Pharmacol. 2018, 155, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Marlowe, J.L.; Puga, A. Aryl hydrocarbon receptor, cell cycle regulation, toxicity, and tumorigenesis. J. Cell Biochem. 2005, 96, 1174–1184. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Cao, Z.; Wang, X. Role of aryl hydrocarbon receptor in cancer. Biochim. Biophys. Acta 2013, 1836, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Morales-Hernández, A.; González-Rico, F.J.; Román, A.C.; Rico-Leo, E.; Alvarez-Barrientos, A.; Sánchez, L.; Macia, Á.; Heras, S.R.; García-Pérez, J.L.; Merino, J.M.; et al. Alu retrotransposons promote differentiation of human carcinoma cells through the aryl hydrocarbon receptor. Nucleic Acids Res. 2016, 44, 4665–4683. [Google Scholar] [CrossRef]
- Pohjanvirta, R.; Viluksela, M. Novel Aspects of Toxicity Mechanisms of Dioxins and Related Compounds. Int. J. Mol. Sci. 2020, 21, 2342. [Google Scholar] [CrossRef]
- Wright, E.J.; De Castro, K.P.; Joshi, A.D.; Elferink, C.J. Canonical and non-canonical aryl hydrocarbon receptor signaling pathways. Curr. Opin. Toxicol. 2017, 2, 87–92. [Google Scholar] [CrossRef]
- Qian, X.; Hulit, J.; Suyama, K.; Eugenin, E.A.; Belbin, T.J.; Loudig, O.; Smirnova, T.; Zhou, Z.N.; Segall, J.; Locker, J.; et al. p21CIP1 mediates reciprocal switching between proliferation and invasion during metastasis. Oncogene 2013, 32, 2292–2303. [Google Scholar] [CrossRef]
- Esser, C.; Rannug, A. The aryl hydrocarbon receptor in barrier organ physiology, immunology, and toxicology. Pharmacol. Rev. 2015, 67, 259–279. [Google Scholar] [CrossRef]
- Neavin, D.R.; Liu, D.; Ray, B.; Weinshilboum, R.M. The Role of the Aryl Hydrocarbon Receptor (AHR) in Immune and Inflammatory Diseases. Int. J. Mol. Sci. 2018, 19, 3851. [Google Scholar] [CrossRef]
- Hubbard, T.D.; Murray, I.A.; Perdew, G.H. Indole and Tryptophan Metabolism: Endogenous and Dietary Routes to Ah Receptor Activation. Drug Metab. Dispos. 2015, 43, 1522–1535. [Google Scholar] [CrossRef]
- Safe, S.; Han, H.; Goldsby, J.; Mohankumar, K.; Chapkin, R.S. Aryl Hydrocarbon Receptor (AhR) Ligands as Selective AhR Modulators: Genomic Studies. Curr. Opin. Toxicol. 2018, 11–12, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Salguero, P.M.; Hilbert, D.M.; Rudikoff, S.; Ward, J.M.; Gonzalez, F.J. Aryl-hydrocarbon receptor-deficient mice are resistant to 2,3,7,8 tetrachlorodibenzo -p-dioxin-induced toxicity. Toxicol. Appl. Pharmacol. 1996, 140, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, F.J.; Fernandez-Salguero, P. The aryl hydrocarbon receptor: Studies using the AHR-null mice. Drug Metab Dispos. 1998, 26, 1194–1198. [Google Scholar]
- Jaeger, C.; Khazaal, A.Q.; Xu, C.; Sun, M.; Krager, S.L.; Tischkau, S.A. Aryl Hydrocarbon Receptor Deficiency Alters Circadian and Metabolic Rhythmicity. J. Biol. Rhythms. 2017, 32, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Hennig, B.; Meerarani, P.; Slim, R.; Toborek, M.; Daugherty, A.; Silverstone, A.E.; Robertson, L.W. Proinflammatory properties of coplanar PCBs: In vitro and in vivo evidence. Toxicol. Appl. Pharmacol. 2002, 181, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Rabson, A.B.; Gallo, M.A. Ah receptor and NF-kappaB interactions: Mechanisms and physiological implications. Chem. Biol. Interact. 2002, 141, 97–115. [Google Scholar] [CrossRef]
- Dalton, T.P.; Puga, A.; Shertzer, H.G. Induction of cellular oxidative stress by aryl hydrocarbon receptor activation. Chem. Biol. Interact. 2002, 141, 77–95. [Google Scholar] [CrossRef]
- Vogel, C.F.; Sciullo, E.; Matsumura, F. Activation of inflammatory mediators and potential role of ah-receptor ligands in foam cell formation. Cardiovasc. Toxicol. 2004, 4, 363–373. [Google Scholar] [CrossRef]
- Martinez, J.M.; Baek, S.J.; Mays, D.M.; Tithof, P.K.; Eling, T.E.; Walker, N.J. EGR1 is a novel target for AhR agonists in human lung epithelial cells. Toxicol. Sci. 2004, 82, 429–435. [Google Scholar] [CrossRef]
- Martey, C.A.; Baglole, C.J.; Gasiewicz, T.A.; Sime, P.J.; Phipps, R.P. The aryl hydrocarbon receptor is a regulator of cigarette smoke induction of the cyclooxygenase and prostaglandin pathways in human lung fibroblasts. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, L391–L399. [Google Scholar] [CrossRef]
- Thatcher, T.H.; Maggirwar, S.B.; Baglole, C.J.; Lakatos, H.F.; Gasiewicz, T.A.; Phipps, R.P.; Sime, P.J. Aryl hydrocarbon receptor-deficient mice develop heightened inflammatory responses to cigarette smoke and endotoxin associated with rapid loss of the nuclear factor-kappaB component RelB. Am. J. Pathol. 2007, 170, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.H.; Chang, H.; Chang, J.T.; Lin, P. Aryl hydrocarbon receptor in association with RelA modulates IL-6 expression in non-smoking lung cancer. Oncogene 2012, 31, 2555–2565. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-Acosta, O.; Vega, L.; Estrada-Muñiz, E.; Rodríguez, M.S.; Gonzalez, F.J.; Elizondo, G. Activation of aryl hydrocarbon receptor regulates the LPS/IFNγ-induced inflammatory response by inducing ubiquitin-proteosomal and lysosomal degradation of RelA/p65. Biochem. Pharmacol. 2018, 155, 141–149. [Google Scholar] [CrossRef]
- Gharavi, N.; El-Kadi, A.O. Role of nitric oxide in downregulation of cytochrome P450 1a1 and NADPH: Quinone oxidoreductase 1 by tumor necrosis factor-alpha and lipopolysaccharide which triggers tumoral induction and promotion. J. Pharm. Sci. 2007, 96, 2795–2807. [Google Scholar] [CrossRef] [PubMed]
- Podechard, N.; Lecureur, V.; Le Ferrec, E.; Guenon, I.; Sparfel, L.; Gilot, D.; Gordon, J.R.; Lagente, V.; Fardel, O. Interleukin-8 induction by the environmental contaminant benzo(a)pyrene is aryl hydrocarbon receptor-dependent and leads to lung inflammation. Toxicol. Lett. 2008, 177, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Stockinger, B.; Hirota, K.; Duarte, J.; Veldhoen, M. External influences on the immune system via activation of the aryl hydrocarbon receptor. Semin. Immunol. 2011, 23, 99–105. [Google Scholar] [CrossRef]
- Poland, A.; Knutson, J.C. 2,3,7,8-tetrachlorodibenzo-p-dioxin and related halogenated aromatic hydrocarbons: Examination of the mechanism of toxicity. Annu Rev. Pharmacol. Toxicol. 1982, 22, 517–554. [Google Scholar] [CrossRef] [PubMed]
- Holsapple, M.P.; Morris, D.L.; Wood, S.C.; Snyder, N.K. 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced changes in immunocompetence: Possible mechanisms. Annu. Rev. Pharmacol. Toxicol. 1991, 31, 73–100. [Google Scholar] [CrossRef]
- Sulentic, C.E.; Holsapple, M.P.; Kaminski, N.E. Aryl hydrocarbon receptor-dependent suppression by 2,3,7,8-tetrachlorodibenzo-p-dioxin of IgM secretion in activated B cells. Mol. Pharmacol. 1998, 53, 623–629. [Google Scholar] [CrossRef]
- Doi, H.; Baba, T.; Tohyama, C.; Nohara, K. Functional activation of aryl hydrocarbon receptor (AhR) in primary T cells by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Chemosphere 2003, 52, 655–662. [Google Scholar] [CrossRef]
- Wei, P.; Hu, G.H.; Kang, H.Y.; Yao, H.B.; Kou, W.; Zhang, C.; Hong, S.L. Role of the aryl hydrocarbon receptor in the pathogenesis of chronic rhinosinusitis with nasal polyps. Inflammation 2014, 37, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Quintana, F.J.; Basso, A.S.; Iglesias, A.H.; Korn, T.; Farez, M.F.; Bettelli, E.; Caccamo, M.; Oukka, M.; Weiner, H.L. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008, 453, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Salguero, P.M.; Ward, J.M.; Sundberg, J.P.; Gonzalez, F.J. Lesions of aryl-hydrocarbon receptor-deficient mice. Vet. Pathol. 1997, 34, 605–614. [Google Scholar] [CrossRef] [PubMed]
- NF-kB Transcription Factors—Inducers Physical Stress. Available online: http://www.bu.edu/nf-kb/physiological-mediators/inducers/ (accessed on 19 May 2020).
- Tian, Y.; Ke, S.; Denison, M.S.; Rabson, A.B.; Gallo, M.A. Ah receptor and NF-kappaB interactions, a potential mechanism for dioxin toxicity. J. Biol. Chem. 1999, 274, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Sulentic, C.E.; Kang, J.S.; Na, Y.J.; Kaminski, N.E. Interactions at a dioxin responsive element (DRE) and an overlapping kappaB site within the hs4 domain of the 3′ alpha immunoglobulin heavy chain enhancer. Toxicology 2004, 200, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Salisbury, R.L.; Sulentic, C.E. The AhR and NF-κB/Rel Proteins Mediate the Inhibitory Effect of 2,3,7,8-Tetrachlorodibenzo-p-Dioxin on the 3’ Immunoglobulin Heavy Chain Regulatory Region. Toxicol. Sci. 2015, 148, 443–459. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.V.; Boverhof, D.R.; Burgoon, L.D.; Fielden, M.R.; Zacharewski, T.R. Comparative analysis of dioxin response elements in human, mouse and rat genomic sequences. Nucleic Acids Res. 2004, 32, 4512–4523. [Google Scholar] [CrossRef]
- Hanieh, H. Toward understanding the role of aryl hydrocarbon receptor in the immune system: Current progress and future trends. Biomed. Res. Int. 2014, 2014, 520763. [Google Scholar] [CrossRef]
- Vogel, C.F.; Matsumura, F. A new cross-talk between the aryl hydrocarbon receptor and RelB, a member of the NF-kappaB family. Biochem. Pharmacol. 2009, 77, 734–745. [Google Scholar] [CrossRef]
- Vogel, C.F.; Khan, E.M.; Leung, P.S.; Gershwin, M.E.; Chang, W.L.; Wu, D.; Haarmann-Stemmann, T.; Hoffmann, A.; Denison, M.S. Cross-talk between aryl hydrocarbon receptor and the inflammatory response: A role for nuclear factor-κB. J. Biol. Chem. 2014, 289, 1866–1875. [Google Scholar] [CrossRef]
- D’Amato, N.C.; Rogers, T.J.; Gordon, M.A.; Greene, L.I.; Cochrane, D.R.; Spoelstra, N.S.; Nemkov, T.G.; D’Alessandro, A.; Hansen, K.C.; Richer, J.K. A TDO2-AhR signaling axis facilitates anoikis resistance and metastasis in triple-negative breast cancer. Cancer Res. 2015, 75, 4651–4664. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Guarnieri, T. Non-Steroidal Anti Inflammatory Drugs As Gatekeepers Of Colon Carcinoma Highlight New Scenarios Beyond Cyclooxygenases Inhibition. Curr. Cancer Drug Targets 2016, 16, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.A.; Khatami, M.; Baglole, C.J.; Sun, J.; Harris, S.A.; Moon, E.Y.; Al-Mulla, F.; Al-Temaimi, R.; Brown, D.G.; Colacci, A.; et al. Environmental immune disruptors, inflammation and cancer risk. Carcinogenesis 2015, 36 (Suppl. S1), S232–S253. [Google Scholar] [CrossRef]
- Jensen, B.A.; Leeman, R.J.; Schlezinger, J.J.; Sherr, D.H. Aryl hydrocarbon receptor (AhR) agonists suppress interleukin-6 expression by bone marrow stromal cells: An immunotoxicology study. Environ. Health 2003, 2, 16. [Google Scholar] [CrossRef]
- Stobbe-Maicherski, N.; Wolff, S.; Wolff, C.; Abel, J.; Sydlik, U.; Frauenstein, K.; Haarmann-Stemmann, T. The interleukin-6-type cytokine oncostatin M induces aryl hydrocarbon receptor expression in a STAT3-dependent manner in human HepG2 hepatoma cells. FEBS J. 2013, 280, 6681–6690. [Google Scholar] [CrossRef]
- Guarnieri, T.; Abruzzo, P.M.; Bolotta, A. More than a cell biosensor: Aryl hydrocarbon receptor at the intersection of physiology and inflammation. Am. J. Physiol. Cell Physiol. 2020, 318, C1078–C1082. [Google Scholar] [CrossRef]
- Vogel, C.F.A.; Van Winkle, L.S.; Esser, C.; Haarmann-Stemmann, T. The aryl hydrocarbon receptor as a target of environmental stressors—Implications for pollution mediated stress and inflammatory responses. Redox Biol. 2020, 101530. [Google Scholar] [CrossRef]
- Sansone, P.; Storci, G.; Tavolari, S.; Guarnieri, T.; Giovannini, C.; Taffurelli, M.; Ceccarelli, C.; Santini, D.; Paterini, P.; Marcu, K.B.; et al. IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J. Clin. Investig. 2007, 117, 3988–4002. [Google Scholar] [CrossRef]
- Esquivel-Velázquez, M.; Ostoa-Saloma, P.; Palacios-Arreola, M.I.; Nava-Castro, K.E.; Castro, J.I.; Morales-Montor, J. The role of cytokines in breast cancer development and progression. J. Interferon Cytokine Res. 2015, 35, 1–16. [Google Scholar] [CrossRef]
- Baek, H.J.; Kim, S.E.; Choi, E.K.; Kim, J.K.; Shin, D.H.; Park, E.J.; Kim, T.H.; Kim, J.Y.; Kim, K.G.; Deng, C.X.; et al. Inhibition of Estrogen Signaling Reduces the Incidence of BRCA1-associated Mammary Tumor Formation. Int. J. Biol. Sci. 2018, 14, 1755–1768. [Google Scholar] [CrossRef] [PubMed]
- Romagnolo, D.F.; Papoutsis, A.J.; Laukaitis, C.; Selmin, O.I. Constitutive expression of AhR and BRCA-1 promoter CpG hypermethylation as biomarkers of ERα-negative breast tumorigenesis. BMC Cancer 2015, 15, 1026. [Google Scholar] [CrossRef] [PubMed]
- American Cancer Society. Cancer Facts & Figures 2016; American Cancer Society: Atlanta, GA, USA, 2016. [Google Scholar]
- Sun, Y.S.; Zhao, Z.; Yang, Z.N.; Xu, F.; Lu, H.J.; Zhu, Z.Y.; Shi, W.; Jiang, J.; Yao, P.P.; Zhu, H.P. Risk Factors and Preventions of Breast Cancer. Int. J. Biol. Sci. 2017, 13, 1387–1397. [Google Scholar] [CrossRef] [PubMed]
- Sasser, A.K.; Sullivan, N.J.; Studebaker, A.W.; Hendey, L.F.; Axel, A.E.; Hall, B.M. Interleukin-6 is a potent growth factor for ER-alpha-positive human breast cancer. FASEB J. 2007, 21, 3763–3770. [Google Scholar] [CrossRef] [PubMed]
- Baumgarten, S.C.; Frasor, J. Minireview: Inflammation: An instigator of more aggressive estrogen receptor (ER) positive breast cancers. Mol. Endocrinol. 2012, 26, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Wolff, M.S.; Toniolo, P.G.; Lee, E.W.; Rivera, M.; Dubin, N. Blood levels of organochlorine residues and risk of breast cancer. J. Natl. Cancer Inst. 1993, 85, 648–652. [Google Scholar] [CrossRef]
- Charlier, C.; Albert, A.; Herman, P.; Hamoir, E.; Gaspard, U.; Meurisse, M.; Plomteux, G. Breast cancer and serum organochlorine residues. Occup. Environ. Med. 2003, 60, 348–351. [Google Scholar] [CrossRef] [PubMed]
- DiNatale, B.C.; Schroeder, J.C.; Perdew, G.H. Ah receptor antagonism inhibits constitutive and cytokine inducible IL6 production in head and neck tumor cell lines. Mol. Carcinog. 2011, 50, 173–183. [Google Scholar] [CrossRef]
- Sovak, M.A.; Bellas, R.E.; Kim, D.W.; Zanieski, G.J.; Rogers, A.E.; Traish, A.M.; Sonenshein, G.E. Aberrant nuclear factor-kappaB/Rel expression and the pathogenesis of breast cancer. J. Clin. Investig. 1997, 100, 2952–2960. [Google Scholar] [CrossRef]
- Kim, D.W.; Sovak, M.A.; Zanieski, G.; Nonet, G.; Romieu-Mourez, R.; Lau, A.W.; Hafer, L.J.; Yaswen, P.; Stampfer, M.; Rogers, A.E.; et al. Activation of NF-kappaB/Rel occurs early during neoplastic transformation of mammary cells. Carcinogenesis 2000, 21, 871–879. [Google Scholar] [CrossRef]
- Kim, D.W.; Gazourian, L.; Quadri, S.A.; Romieu-Mourez, R.; Sherr, D.H.; Sonenshein, G.E. The RelA NF-kappaB subunit and the aryl hydrocarbon receptor (AhR) cooperate to transactivate the c-myc promoter in mammary cells. Oncogene 2000, 19, 5498–5506. [Google Scholar] [CrossRef] [PubMed]
- Currier, N.; Solomon, S.E.; Demicco, E.G.; Chang, D.L.; Farago, M.; Ying, H.; Dominguez, I.; Sonenshein, G.E.; Cardiff, R.D.; Xiao, Z.X.; et al. Oncogenic signaling pathways activated in DMBA-induced mouse mammary tumors. Toxicol. Pathol. 2005, 33, 726–737. [Google Scholar] [CrossRef] [PubMed]
- Vyas, D.; Lopez-Hisijos, N.; Shah, P.; Deshpande, K.S.; Basson, M.D.; Vyas, A.; Chaturvedi, L.S. A Second-Generation Proteasome Inhibitor and Doxorubicin Modulates IL-6, pSTAT-3 and NF-kB Activity in MDA-MB-231 Breast Cancer Cells. J. Nanosci. Nanotechnol. 2017, 17, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S. Interleukin-6 Family Cytokines. Cold Spring Harb. Perspect. Biol. 2018, 10, a028415. [Google Scholar] [CrossRef]
- Sakamoto, K.; Wehde, B.L.; Yoo, K.H.; Kim, T.; Rajbhandari, N.; Shin, H.Y.; Triplett, A.A.; Rädler, P.D.; Schuler, F.; Villunger, A.; et al. Janus Kinase 1 Is Essential for Inflammatory Cytokine Signaling and Mammary Gland Remodeling. Mol. Cell Biol. 2016, 36, 1673–1690. [Google Scholar] [CrossRef]
- Dijsselbloem, N.; Vanden Berghe, W.; De Naeyer, A.; Haegeman, G. Soy isoflavone phyto-pharmaceuticals in interleukin-6 affections. Multi-purpose nutraceuticals at the crossroad of hormone replacement, anti-cancer and anti-inflammatory therapy. Biochem. Pharmacol. 2004, 68, 1171–1185. [Google Scholar] [CrossRef]
- Shimura, T.; Shibata, M.; Gonda, K.; Murakami, Y.; Noda, M.; Tachibana, K.; Abe, N.; Ohtake, T. Prognostic impact of interleukin-6 and C-reactive protein on patients with breast cancer. Oncol. Lett. 2019, 17, 5139–5146. [Google Scholar] [CrossRef]
- Jin, K.; Pandey, N.B.; Popel, A.S. Simultaneous blockade of IL-6 and CCL5 signaling for synergistic inhibition of triple-negative breast cancer growth and metastasis. Breast Cancer Res. 2018, 20, 54. [Google Scholar] [CrossRef]
- Hollingshead, B.D.; Beischlag, T.V.; Dinatale, B.C.; Ramadoss, P.; Perdew, G.H. Inflammatory signaling and aryl hydrocarbon receptor mediate synergistic induction of interleukin 6 in MCF-7 cells. Cancer Res. 2008, 68, 3609–3617. [Google Scholar] [CrossRef]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef]
- Novikov, O.; Wang, Z.; Stanford, E.A.; Parks, A.J.; Ramirez-Cardenas, A.; Landesman, E.; Laklouk, I.; Sarita-Reyes, C.; Gusenleitner, D.; Li, A.; et al. An Aryl Hydrocarbon Receptor-Mediated Amplification Loop That Enforces Cell Migration in ER-/PR-/Her2- Human Breast Cancer Cells. Mol. Pharmacol. 2016, 90, 674–688. [Google Scholar] [CrossRef]
- Dietrich, C.; Kaina, B. The aryl hydrocarbon receptor (AhR) in the regulation of cell-cell contact and tumor growth. Carcinogenesis 2010, 31, 1319–1328. [Google Scholar] [CrossRef] [PubMed]
- Opitz, C.A.; Somarribas Patterson, L.F.; Mohapatra, S.R.; Dewi, D.L.; Sadik, A.; Platten, M.; Trump, S. The therapeutic potential of targeting tryptophan catabolism in cancer. Br. J. Cancer 2020, 122, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Litzenburger, U.M.; Opitz, C.A.; Sahm, F.; Rauschenbach, K.J.; Trump, S.; Winter, M.; Ott, M.; Ochs, K.; Lutz, C.; Liu, X.; et al. Constitutive IDO expression in human cancer is sustained by an autocrine signaling loop involving IL-6, STAT3 and the AHR. Oncotarget 2014, 5, 1038–1051. [Google Scholar] [CrossRef] [PubMed]
- Vacher, S.; Castagnet, P.; Chemlali, W.; Lallemand, F.; Meseure, D.; Pocard, M.; Bieche, I.; Perrot-Applanat, M. High AHR expression in breast tumors correlates with expression of genes from several signaling pathways namely inflammation and endogenous tryptophan metabolism. PLoS ONE 2018, 13, e0190619. [Google Scholar] [CrossRef]
- Goode, G.D.; Ballard, B.R.; Manning, H.C.; Freeman, M.L.; Kang, Y.; Eltom, S.E. Knockdown of aberrantly upregulated aryl hydrocarbon receptor reduces tumor growth and metastasis of MDA-MB-231 human breast cancer cell line. Int. J. Cancer 2013, 133, 2769–2780. [Google Scholar] [CrossRef]
- Donovan, M.G.; Selmin, O.I.; Doetschman, T.C.; Romagnolo, D.F. Epigenetic Activation of BRCA1 by Genistein In Vivo and Triple Negative Breast Cancer Cells Linked to Antagonism toward Aryl Hydrocarbon Receptor. Nutrients 2019, 11, 2559. [Google Scholar] [CrossRef]
- Choi, M.J.; Lee, E.J.; Park, J.S.; Kim, S.N.; Park, E.M.; Kim, H.S. Anti-inflammatory mechanism of galangin in lipopolysaccharide-stimulated microglia: Critical role of PPAR-γ signaling pathway. Biochem. Pharmacol. 2017, 144, 120–131. [Google Scholar] [CrossRef]
- Romagnolo, D.F.; Daniels, K.D.; Grunwald, J.T.; Ramos, S.A.; Propper, C.R.; Selmin, O.I. Epigenetics of breast cancer: Modifying role of environmental and bioactive food compounds. Mol. Nutr. Food Res. 2016, 60, 1310–1329. [Google Scholar] [CrossRef]
- Kamińska, M.; Ciszewski, T.; Łopacka-Szatan, K.; Miotła, P.; Starosławska, E. Breast cancer risk factors. Prz. Menopauzalny 2015, 14, 196–202. [Google Scholar] [CrossRef]
- Todoric, J.; Antonucci, L.; Karin, M. Targeting Inflammation in Cancer Prevention and Therapy. Cancer Prev. Res. 2016, 9, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.; Li, C.F.; Kuo, C.C.; Tsai, K.K.; Hou, M.F.; Hung, W.C. Cancer/stroma interplay via cyclooxygenase-2 and indoleamine 2,3-dioxygenase promotes breast cancer progression. Breast Cancer Res. 2014, 16, 410. [Google Scholar] [CrossRef] [PubMed]
- Papoutsis, A.J.; Selmin, O.I.; Borg, J.L.; Romagnolo, D.F. Gestational exposure to the AhR agonist 2,3,7,8-tetrachlorodibenzo-p-dioxin induces BRCA-1 promoter hypermethylation and reduces BRCA-1 expression in mammary tissue of rat offspring: Preventive effects of resveratrol. Mol. Carcinog. 2015, 54, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Saito, R.; Miki, Y.; Hata, S.; Ishida, T.; Suzuki, T.; Ohuchi, N.; Sasano, H. Aryl hydrocarbon receptor induced intratumoral aromatase in breast cancer. Breast Cancer Res. Treat. 2017, 161, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, F.; Takeyama, K.; Matsumoto, T.; Kitagawa, H.; Yamamoto, Y.; Nohara, K.; Tohyama, C.; Krust, A.; Mimura, J.; Chambon, P.; et al. Modulation of oestrogen receptor signalling by association with the activated dioxin receptor. Nature 2003, 423, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Wormke, M.; Stoner, M.; Saville, B.; Walker, K.; Abdelrahim, M.; Burghardt, R.; Safe, S. The aryl hydrocarbon receptor mediates degradation of estrogen receptor alpha through activation of proteasomes. Mol. Cell Biol. 2003, 23, 1843–1855. [Google Scholar] [CrossRef]
- Safe, S.; Cheng, Y.; Jin, U.H. The Aryl Hydrocarbon Receptor (AhR) as a Drug Target for Cancer Chemotherapy. Curr. Opin. Toxicol. 2017, 2, 24–29. [Google Scholar] [CrossRef]



© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guarnieri, T. Aryl Hydrocarbon Receptor Connects Inflammation to Breast Cancer. Int. J. Mol. Sci. 2020, 21, 5264. https://doi.org/10.3390/ijms21155264
Guarnieri T. Aryl Hydrocarbon Receptor Connects Inflammation to Breast Cancer. International Journal of Molecular Sciences. 2020; 21(15):5264. https://doi.org/10.3390/ijms21155264
Chicago/Turabian StyleGuarnieri, Tiziana. 2020. "Aryl Hydrocarbon Receptor Connects Inflammation to Breast Cancer" International Journal of Molecular Sciences 21, no. 15: 5264. https://doi.org/10.3390/ijms21155264
APA StyleGuarnieri, T. (2020). Aryl Hydrocarbon Receptor Connects Inflammation to Breast Cancer. International Journal of Molecular Sciences, 21(15), 5264. https://doi.org/10.3390/ijms21155264