Gut Microbiota Metabolites in NAFLD Pathogenesis and Therapeutic Implications


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Evidence of the Importance of the Intestinal Microbiota in NAFLD
3. Gut Microbiota Metabolites in the Pathogenesis of NAFLD
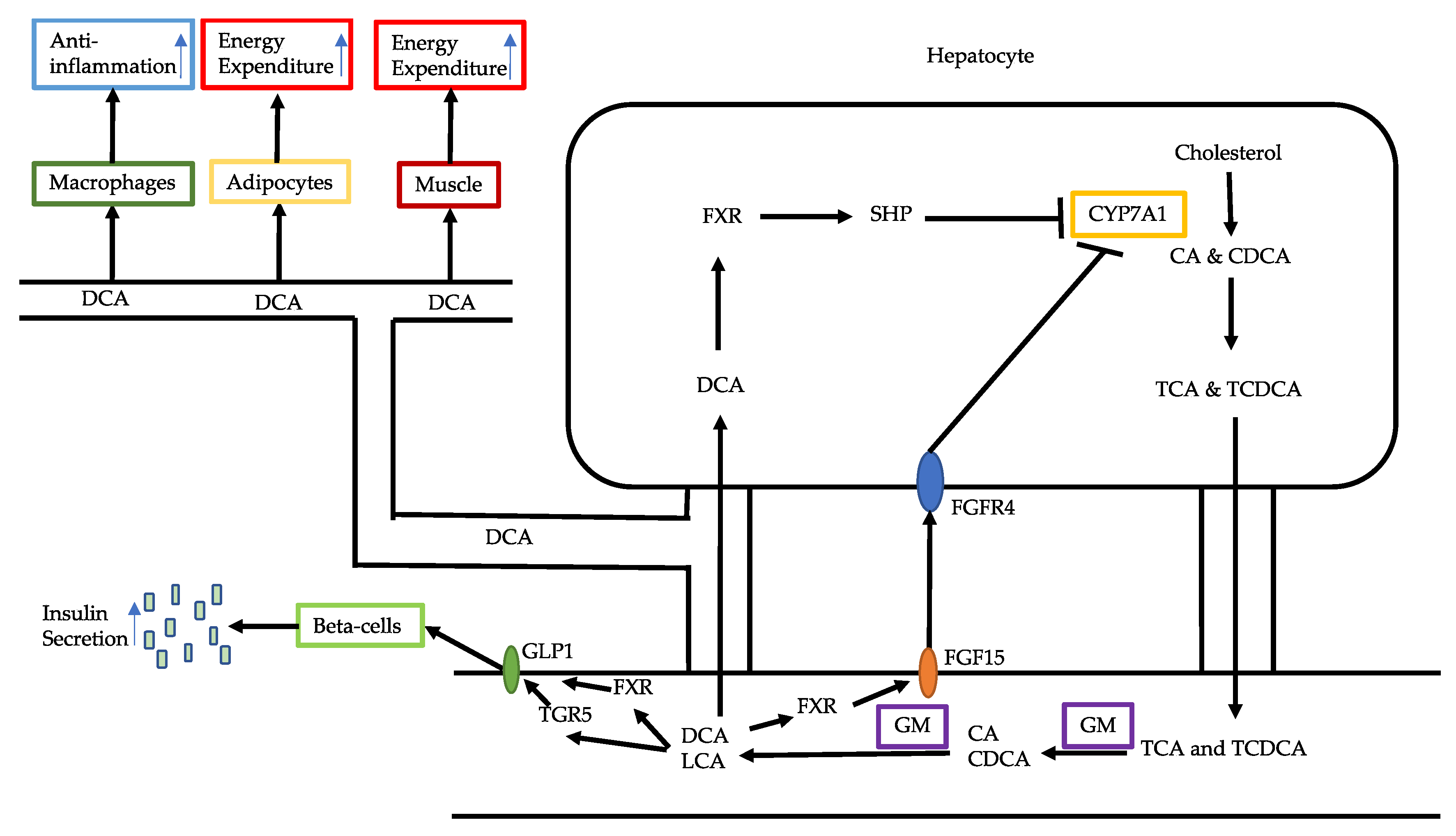
3.1. Bile Acid Alterations
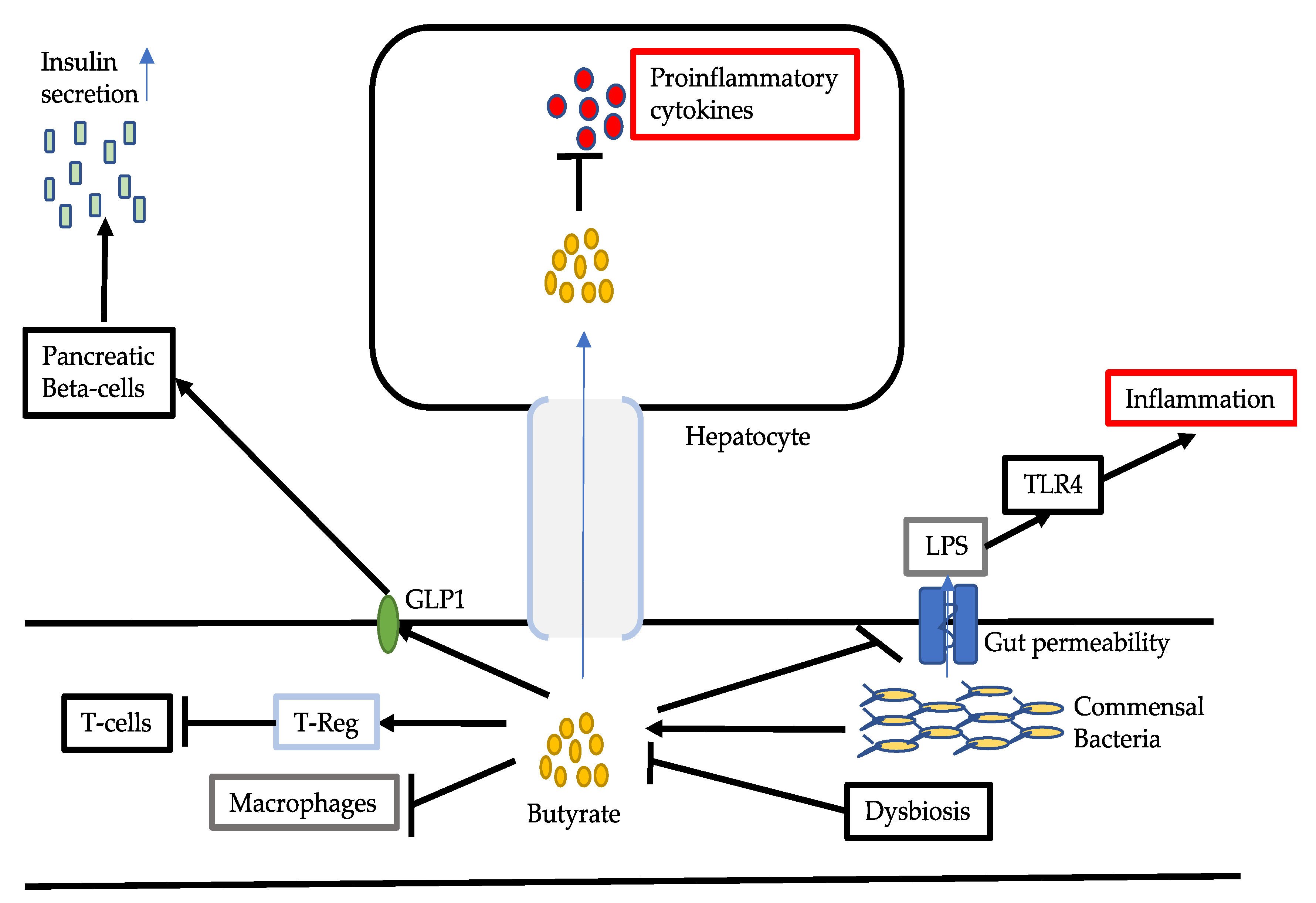
3.2. Reduced Production of Butyrate
3.3. Amino Acids
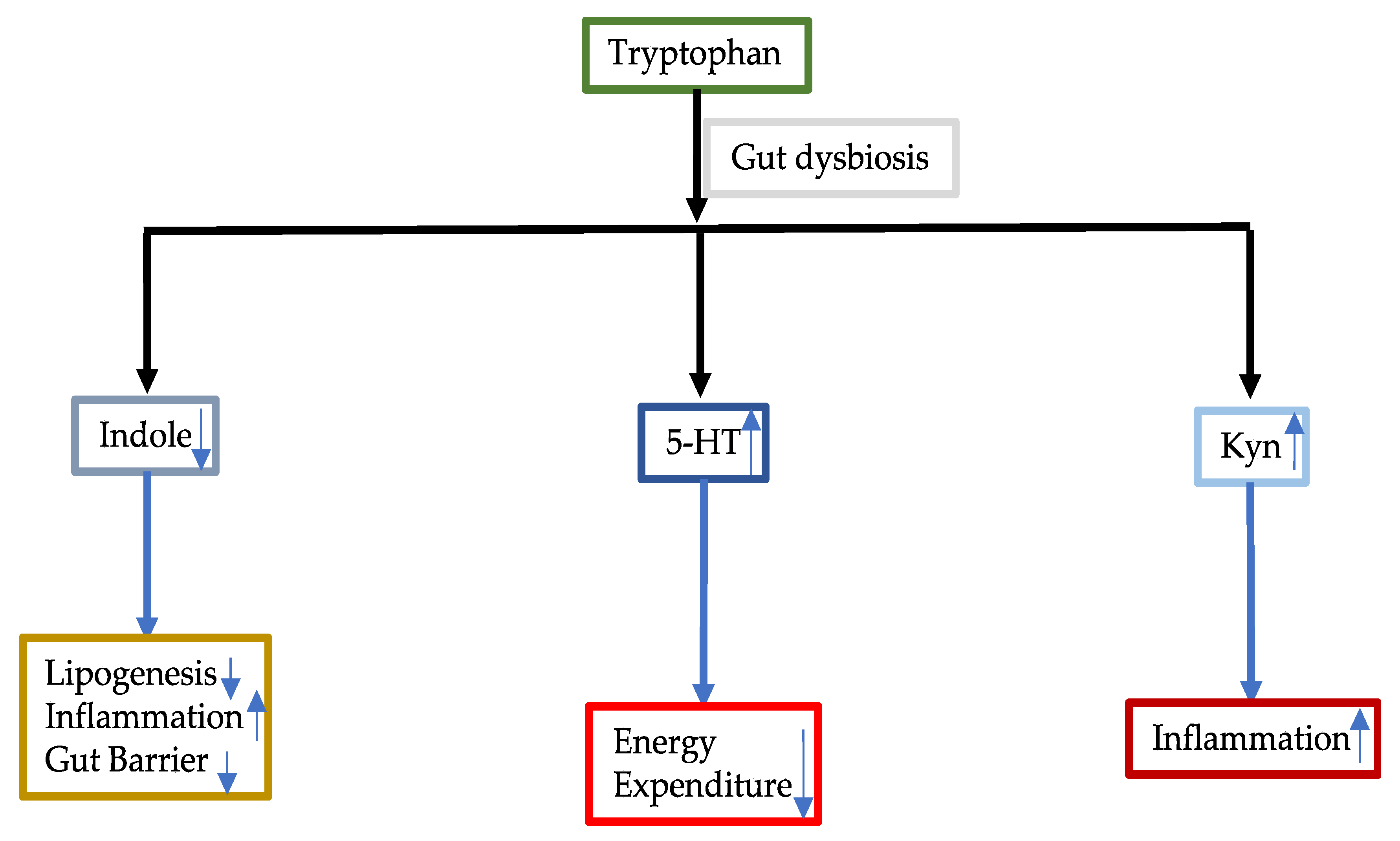
3.3.1. Tryptophan
3.3.2. Phenylalanine
3.3.3. Branched Chain Amino Acids
3.4. Choline and Methionine
3.5. Ethanol
4. Modulation of Gut Microbiota and Its Metabolites
4.1. Probiotics
Lactobacillus
4.2. Prebiotics
4.3. Synbiotics
4.4. FMT
4.5. FMD
5. Conclusions
Funding
Conflicts of Interest
References
- Wong, V.W.; Chitturi, S.; Wong, G.L.; Yu, J.; Chan, H.L.Y.; Farrell, G.C. Pathogenesis and Novel Treatment Options for Non-Alcoholic Steatohepatitis. Lancet Gastroenterol. Hepatol. 2016, 1, 56–67. [Google Scholar] [CrossRef]
- Hazlehurst, J.M.; Woods, C.; Marjot, T.; Cobbold, J.F.; Tomilson, J.W. Non-Alcoholic Fatty Liver Disease and Diabetes. Metabolism 2016, 65, 1096–1108. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Vitetta, L. Bile Acids and Butyrate in the Effects of Probiotics/Synbiotics on Nonalcoholic Fatty Liver Disease. Eur. J. Gastroenterol. Hepatol. 2019, 31, 1475–1476. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Vitetta, L. Mitochondria Could Be a Potential Key Mediator Linking the Intestinal Microbiota to Depression. J. Cell Biochem. 2020, 121, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Aron-Wisnewsky, J.; Vigliotti, C.; Witjes, J.; Le, P.; Holleboom, A.G.; Verheij, J.; Nieuwdorp, M.; Clément, K. Gut Microbiota and Human NAFLD: Disentangling Microbial Signatures From Metabolic Disorders. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 279–297. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Vitetta, L. Letter to the Editors: Could Butyrate Be Incorporated With Farnesoid X Receptor Agonist Cilofexor to Enhance Primary Sclerosing Cholangitis Treatment? Hepatology 2020. in print; Online ahead of print. [Google Scholar] [CrossRef]
- Chen, J.; Vitetta, L. Butyrate in Inflammatory Bowel Disease Therapy. Gastroenterology 2020, 158, 1511. [Google Scholar] [CrossRef]
- Boursier, J.; Mueller, O.; Barret, M.; Machado, M.; Fizanne, L.; Araujo-Perez, F.; Guy, C.D.; Seed, P.C.; Rawls, J.F.; David, L.A.; et al. The Severity of Nonalcoholic Fatty Liver Disease Is Associated With Gut Dysbiosis and Shift in the Metabolic Function of the Gut Microbiota. Hepatology 2016, 63, 764–775. [Google Scholar] [CrossRef]
- Saltzman, E.T.; Palacios, T.; Thomsen, M.; Vitetta, L. Intestinal Microbiome Shifts, Dysbiosis, Inflammation, and Non-Alcoholic Fatty Liver Disease. Front. Microbiol. 2018, 9, 61. [Google Scholar] [CrossRef]
- Schwimmer, J.B.; Johnson, J.S.; Angeles, J.E.; Behling, C.; Belt, P.H.; Borecki, I.; Bross, C.; Durelle, J.; Goyal, N.P.; Hamilton, G.; et al. Microbiome Signatures Associated With Steatohepatitis and Moderate to Severe Fibrosis in Children With Nonalcoholic Fatty Liver Disease. Gastroenterology 2019, 157, 1109–1122. [Google Scholar] [CrossRef]
- Nseir, W.; Artul, S.; Nasrallah, N.; Mahamid, M. The Association between Primary Bacteremia of Presumed Gastrointestinal Origin and Nonalcoholic Fatty Liver Disease. Dig. Liver. Dis. 2016, 48, 343–344. [Google Scholar] [CrossRef] [PubMed]
- Kolodziejczyk, A.A.; Zheng, D.; Shibolet, O.; Elinav, E. The role of the microbiome in NAFLD and NASH. EMBO Mol. Med. 2019, 11, e9302. [Google Scholar] [CrossRef] [PubMed]
- Roy, T.L.; Llopis, M.; Lepage, P.; Bruenau, A.; Rabot, S.; Bevilacqua, C.; Martin, P.; Philippe, C.; Walker, F.; Bado, A. Intestinal Microbiota Determines Development of Non-Alcoholic Fatty Liver Disease in Mice. Gut 2013, 62, 1787–1794. [Google Scholar] [CrossRef] [PubMed]
- Soderborg, T.K.; Clark, S.E.; Mulligan, C.E.; Janssen, R.C.; Babcock, L.; Ir, D.; Young, B.; Krebs, N.; Lemas, D.J.; Johnson, L.K.; et al. The Gut Microbiota in Infants of Obese Mothers Increases Inflammation and Susceptibility to NAFLD. Nat. Commun. 2018, 9, 4462. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.C.; Ching, Y.H.; Li, Y.P.; Liu, J.Y.; Huang, Y.T.; Huang, Y.W.; Yang, S.S.; Huang, W.C.; Chuang, H.L. Nonalcoholic Fatty Liver Disease Is Exacerbated in High-Fat Diet-Fed Gnotobiotic Mice by Colonization With the Gut Microbiota From Patients With Nonalcoholic Steatohepatitis. Nutrients 2017, 9, 1220. [Google Scholar] [CrossRef] [PubMed]
- Hoyles, L.; Fernandez-Real, J.M.; Federici, M.; Serino, M.; Abbott, J.; Charpentier, J.; Heymes, C.; Luque, J.L.; Anthony, E.; Barton, R.H.; et al. Molecular Phenomics and Metagenomics of Hepatic Steatosis in Non-Diabetic Obese Women. Nat. Med. 2018, 24, 1070–1080. [Google Scholar] [CrossRef]
- Chiang, J.Y.L.; Ferrell, J.M. Bile acid receptors FXR and TGR5 signaling in fatty liver diseases and therapy. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, g554–g573. [Google Scholar] [CrossRef]
- Chen, J.; Thomsen, M.; Vitetta, L. Interaction of Gut Microbiota With Dysregulation of Bile Acids in the Pathogenesis of Nonalcoholic Fatty Liver Disease and Potential Therapeutic Implications of Probiotics. J. Cell Biochem. 2019, 120, 2713–2720. [Google Scholar] [CrossRef]
- Song, Z.; Cai, Y.; Lao, X.; Wang, X.; Lin, X.; Cui, Y.; Kalavagunta, P.K.; Liao, J.; Jin, L.; Shang, J.; et al. Taxonomic Profiling and Populational Patterns of Bacterial Bile Salt Hydrolase (BSH) Genes Based on Worldwide Human Gut Microbiome. Microbiome 2019, 7, 9. [Google Scholar] [CrossRef]
- Chen, J.; Zheng, M.; Liu, J.; Luo, Y.; Yang, W.; Yang, J.; Liu, J.; Zhou, J.; Xu, C.; Zhao, F.; et al. Ratio of Conjugated Chenodeoxycholic to Muricholic Acids Is Associated With Severity of Nonalcoholic Steatohepatitis. Obes. Silver Spring 2019, 27, 2055–2066. [Google Scholar] [CrossRef]
- Miyata, M.; Funaki, A.; Fukuhara, C.; Funaki, A.; Fukuhara, C.; Sumiya, Y.; Sugiura, Y. Taurine Attenuates Hepatic Steatosis in a Genetic Model of Fatty Liver Disease. J. Toxicol. Sci. 2020, 45, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, J.D.; Guo, G.L. Pharmacologic Modulation of Bile Acid-FXR-FGF15/FGF19 Pathway for the Treatment of Nonalcoholic Steatohepatitis. Handb. Exp. Pharmacol. 2019, 256, 325–357. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, E.; Fu, L.; John, L.; Hultgren, B.; Huang, X.; Renz, M.; Stephan, J.P.; Tsai, S.P.; Powell-Braxton, L.; French, D.; et al. Transgenic Mice Expressing Human Fibroblast Growth Factor-19 Display Increased Metabolic Rate and Decreased Adiposity. Endocrinology 2002, 143, 1741–1747. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Sola, G.; Uriarte, I.; Latasa, M.U.; Fernandez-Barrena, M.G.; Urtasun, R.; Elizalde, M.; Barcena-Varela, M.; Jiménez, M.; Chang, H.C.; Barbero, R.; et al. Fibroblast Growth Factor 15/19 (FGF15/19) Protects From Diet-Induced Hepatic Steatosis: Development of an FGF19-based Chimeric Molecule to Promote Fatty Liver Regeneration. Gut 2017, 66, 1818–1828. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Crudele, A.; Panera, N.; Romito, I.; Meroni, M.; De Stefanis, C.; Palma, A.; Comparcola, D.; Fracanzani, A.L.; Miele, L.; et al. β-Klotho Gene Variation Is Associated With Liver Damage in Children With NAFLD. J. Hepatol. 2020, 72, 411–419. [Google Scholar] [CrossRef]
- Lake, A.D.; Novak, P.; Shipkova, P.; Aranibar, N.; Robertson, D.; Reily, M.D.; Lu, Z.; Lehman-McKeeman, L.D.; Cherrington, N.J. Decreased Hepatotoxic Bile Acid Composition and Altered Synthesis in Progressive Human Nonalcoholic Fatty Liver Disease. Toxicol. Appl. Pharmacol. 2013, 268, 132–140. [Google Scholar] [CrossRef]
- Xi, Y.; Li, H. Role of Farnesoid X Receptor in Hepatic Steatosis in Nonalcoholic Fatty Liver Disease. Biomed. Pharmacother. 2020, 121, 109609. [Google Scholar] [CrossRef]
- Chiang, J.Y.L.; Pathak, P.; Liu, H.; Donepudi, A.; Ferrell, J.; Boehme, S. Intestinal Farnesoid X Receptor and Takeda G Protein Couple Receptor 5 Signaling in Metabolic Regulation. Dig Dis. 2017, 35, 241–245. [Google Scholar] [CrossRef]
- Onoviran, O.F.; Li, D.; Smith, S.T.; Raji, M.A. Effects of Glucagon-Like Peptide 1 Receptor Agonists on Comorbidities in Older Patients With Diabetes Mellitus. Ther. Adv. Chronic Dis. 2019, 10. [Google Scholar] [CrossRef]
- Sun, F.; Wu, S.; Wang, J.; Guo, S.; Chai, S.; Yang, Z.; Li, L.; Zhang, Y.; Ji, L.; Zhan, S. Effect of Glucagon-Like peptide-1 Receptor Agonists on Lipid Profiles Among Type 2 Diabetes: A Systematic Review and Network Meta-Analysis. Clin. Ther. 2015, 37, 225–241. [Google Scholar] [CrossRef]
- Watanabe, M.; Houten, S.M.; Mataki, C.; Christoffolete, M.A.; Kim, B.W.; Sato, H.; Messaddeq, N.; Harney, J.W.; Ezaki, O.; Kodama, T.; et al. Bile Acids Induce Energy Expenditure by Promoting Intracellular Thyroid Hormone Activation. Nature 2006, 439, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Pols, T.W.H.; Nomura, M.; Harach, T.; Lo Sasso, G.; Oosterveer, M.H.; Thomas, C.; Rizzo, G.; Gioiello, A.; Adorini, L.; Pellicciari, R.; et al. TGR5 Activation Inhibits Atherosclerosis by Reducing Macrophage Inflammation and Lipid Loading. Cell Metab. 2011, 14, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, W.; Yu, D.; Forman, B.M.; Huang, W. The G-Protein-Coupled Bile Acid Receptor, Gpbar1 (TGR5), Negatively Regulates Hepatic Inflammatory Response Through Antagonizing Nuclear Factor κ Light-Chain Enhancer of Activated B Cells (NF-κB) in Mice. Hepatology 2011, 54, 1421–1432. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Vitetta, L. Inflammation-Modulating Effect of Butyrate in the Prevention of Colon Cancer by Dietary Fiber. Clin. Colorectal Cancer 2018, 17, e541–e544. [Google Scholar] [CrossRef]
- Chen, J.; Zhao, K.N.; Vitetta, L. Effects of Intestinal Microbial-Elaborated Butyrate on Oncogenic Signaling Pathways. Nutrients 2019, 11, 1026. [Google Scholar] [CrossRef]
- Chen, J.; Vitetta, L. The Role of Butyrate in Attenuating Pathobiont-Induced Hyperinflammation. Immune Netw. 2020, 20, e15. [Google Scholar] [CrossRef]
- Fei, N.; Bruneau, A.; Zhang, X.; Wang, R.; Wang, J.; Rabot, S.; Gérard, P.; Zhao, L. Endotoxin Producers Overgrowing in Human Gut Microbiota as the Causative Agents for Nonalcoholic Fatty Liver Disease. mBio 2020, 11, e03263-19. [Google Scholar] [CrossRef]
- Carpino, G.; Ben, M.D.; Pastori, D.; Carnevale, R.; Baratta, F.; Overi, D.; Francis, H.; Cardinale, V.; Onori, P.; Safarikia, S.; et al. Increased Liver Localization of Lipopolysaccharides in Human and Experimental NAFLD. Hepatology 2019. Online ahead of print. [Google Scholar] [CrossRef]
- Sharifnia, T.; Antoun, J.; Verriere, T.G.C.; Suarez, G.; Wattacheril, J.; Wilson, K.T.; Peek, R.M., Jr.; Abumrad, N.N.; Flynn, C.R. Hepatic TLR4 Signaling in Obese NAFLD. Am. J. Physiol Gastrointest. Liver Physiol. 2015, 309, g270–g278. [Google Scholar] [CrossRef]
- Zhou, D.; Pan, Q.; Xin, F.Z.; Zhang, R.N.; He, C.X.; Chen, G.Y.; Liu, C.; Chen, Y.W.; Fan, J.G. Sodium Butyrate Attenuates High-Fat Diet-Induced Steatohepatitis in Mice by Improving Gut Microbiota and Gastrointestinal Barrier. World J. Gastroenterol. 2017, 23, 60–75. [Google Scholar] [CrossRef]
- Zhou, D.; Chen, Y.W.; Zhao, Z.H.; Yang, R.X.; Xin, F.Z.; Liu, X.L.; Pan, Q.; Zhou, H.; Fan, J.G. Sodium Butyrate Reduces High-Fat Diet-Induced Non-Alcoholic Steatohepatitis Through Upregulation of Hepatic GLP-1R Expression. Exp. Mol. Med. 2018, 50, 1–12. [Google Scholar] [CrossRef]
- Kaji, I.; Karaki, S.I.; Kuwahara, A. Short-Chain Fatty Acid Receptor and Its Contribution to Glucagon-Like Peptide-1 Release. Digestion 2014, 89, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Baumann, A.; Jin, C.J.; Brandt, A.; Sellmann, C.; Nier, A.; Burkard, M.; Venturelli, S.; Bergheim, I. Oral Supplementation of Sodium Butyrate Attenuates the Progression of Non-Alcoholic Steatohepatitis. Nutrients 2020, 12, 951. [Google Scholar] [CrossRef]
- Leung, C.; Rivera, L.; Furness, J.B.; Angus, P.W. The Role of the Gut Microbiota in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Agus, A.; Planchais, J.; Sokol, H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Ritze, Y.; Bardos, G.; Hubert, A.; Böhle, M.; Bischoff, S.C. Effect of Tryptophan Supplementation on Diet-Induced Non-Alcoholic Fatty Liver Disease in Mice. Br. J. Nutr. 2014, 112, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Shimada, Y.; Kinoshita, M.; Harada, K.; Mizutani, M.; Masahata, K.; Kayama, H.; Takeda, K. Commensal Bacteria-Dependent Indole Production Enhances Epithelial Barrier Function in the Colon. PLoS ONE 2013, 8, e80604. [Google Scholar] [CrossRef]
- Ma, L.; Li, H.; Hu, J.; Zheng, J.; Zhou, J.; Botchlett, R.; Matthews, D.; Zeng, T.; Chen, L.; Xiao, X.; et al. Indole Alleviates Diet-Induced Hepatic Steatosis and Inflammation in a ManneInvolving Myeloid Cell PFKFB3. Hepatology 2020. Online ahead of print. [Google Scholar] [CrossRef]
- Ji, Y.; Gao, Y.; Chen, H.; Yin, Y.; Zhang, W. Indole-3-Acetic Acid Alleviates Nonalcoholic Fatty Liver Disease in Mice via Attenuation of Hepatic Lipogenesis, and Oxidative and Inflammatory Stress. Nutrients 2019, 11, 2062. [Google Scholar] [CrossRef]
- Crane, J.D.; Palanivel, R.; Mottillo, E.P.; Bujak, A.L.; Wang, H.; Ford, R.J.; Collins, A.; Blümer, R.M.; Fullerton, M.D.; Yabut, J.M.; et al. Inhibiting Peripheral Serotonin Synthesis Reduces Obesity and Metabolic Dysfunction by Promoting Brown Adipose Tissue Thermogenesis. Nat. Med. 2015, 21, 166–172. [Google Scholar] [CrossRef]
- Choi, W.; Namkung, J.; Hwang, I.; Kim, H.; Lim, A.; Park, H.J.; Lee, H.W.; Han, K.H.; Park, S.; Jeong, J.S.; et al. Serotonin Signals Through a Gut-Liver Axis to Regulate Hepatic Steatosis. Nat. Commun. 2018, 9, 4824. [Google Scholar] [CrossRef] [PubMed]
- Wolowczuk, I.; Hennart, B.; Leloire, A.; Bessede, A.; Soichot, M.; Taront, S.; Caiazzo, R.; Raverdy, V.; Pigeyre, M.; Guillemin, G.J.; et al. Tryptophan Metabolism Activation by Indoleamine 2,3-dioxygenase in Adipose Tissue of Obese Women: An Attempt to Maintain Immune Homeostasis and Vascular Tone. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 303, r135–r143. [Google Scholar] [CrossRef] [PubMed]
- Currier, A.R.; Ziegler, M.H.; Riley, M.M.; Babcock, T.A.; Telbis, V.P.; Carlin, J.M. Tumor Necrosis Factor-Alpha and Lipopolysaccharide Enhance Interferon-Induced Antichlamydial Indoleamine Dioxygenase Activity Independently. J. Interferon Cytokine Res. 2000, 20, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Hissong, B.D.; Carlin, J.M. Potentiation of Interferon-Induced Indoleamine 2,3-dioxygenase mRNA in Human Mononuclear Phagocytes by Lipopolysaccharide and interleukin-1. J. Interferon Cytokine Res. 1997, 17, 387–393. [Google Scholar] [CrossRef]
- Piirsalu, M.; Taalberg, E.; Lillevali, K.; Tian, L.; Zilmer, M.; Vasar, E. Treatment With Lipopolysaccharide Induces Distinct Changes in Metabolite Profile and Body Weight in 129Sv and Bl6 Mouse Strains. Front. Pharmacol. 2020, 11, 371. [Google Scholar] [CrossRef]
- Haroon, E.; Welle, J.R.; Woolwine, B.J.; Goldsmith, D.R.; Baer, W.; Patel, T.; Felger, J.C.; Miller, A.H. Associations Among Peripheral and Central Kynurenine Pathway Metabolites and Inflammation in Depression. Neuropsychopharmacology 2020, 45, 998–1007. [Google Scholar] [CrossRef]
- Badawy, A.A.; Guillemin, G. The Plasma [Kynurenine]/[Tryptophan] Ratio and Indoleamine 2,3-Dioxygenase: Time for Appraisal. Int. J. Tryptophan Res. 2019, 12. [Google Scholar] [CrossRef]
- Cussotto, S.; Delgado, I.; Anesi, A.; Dexpert, S.; Aubert, A.; Beau, C.; Forestier, D.; Ledaguenel, P.; Magne, E.; Mattivi, F.; et al. Tryptophan metabolic pathways are altered in obesity and are associated with systemic inflammation. Front Immunol. 2020, 11, 557. [Google Scholar] [CrossRef]
- Sunny, N.E.; Kalavalapalli, S.; Bril, F.; Garrett, T.J.; Nautiyal, M.; Mathew, J.T.; Williams, C.M.; Cusi, K. Cross-Talk Between Branched-Chain Amino Acids and Hepatic Mitochondria Is Compromised in Nonalcoholic Fatty Liver Disease. Am. J. Physiol. Endocrinol. Metab. 2015, 309, e311–e319. [Google Scholar] [CrossRef]
- Gaggini, M.; Carli, F.; Rosso, C.; Buzzigoli, E.; Marietti, M.; Della Latta, V.; Ciociaro, D.; Abate, M.L.; Gambino, R.; Cassader, M.; et al. Altered Amino Acid Concentrations in NAFLD: Impact of Obesity and Insulin Resistance. Hepatology 2018, 67, 145–158. [Google Scholar] [CrossRef]
- Lynch, C.J.; Adams, S.H. Branched-Chain Amino Acids in Metabolic Signalling and Insulin Resistance. Nat. Rev. Endocrinol. 2014, 10, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Adeva, M.M.; Calvino, J.; Souto, G.; Donapetry, C. Insulin Resistance and the Metabolism of Branched-Chain Amino Acids in Humans. Amino Acids 2012, 43, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, H.K.; Gudmundsdottir, V.; Nielsen, H.B.; Hyotylainen, T.; Nielsen, T.; Jensen, B.A.; Forslund, K.; Hildebrand, F.; Prifti, E.; Falony, G.; et al. Human Gut Microbes Impact Host Serum Metabolome and Insulin Sensitivity. Nature 2016, 535, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Arao, Y.; Kawai, H.; Kamimura, K.; Kobayashi, T.; Nakano, O.; Hayatsu, M.; Ushiki, T.; Terai, S. Effect of Methionine/Choline-Deficient Diet and High-Fat Diet-Induced Steatohepatitis on Mitochondrial Homeostasis in Mice. Biochem. Biophys. Res. Commun. 2020, 527, 365–371. [Google Scholar] [CrossRef]
- Hernandez, G.V.; Smith, V.A.; Melnyk, M.; Burd, M.A.; Sprayberry, K.A.; Edwards, M.S.; Peterson, D.G.; Bennet, D.C.; Fanter, R.K.; Columbus, D.A.; et al. Dysregulated FXR-FGF19 Signaling and Choline Metabolism Are Associated With Gut Dysbiosis and Hyperplasia in a Novel Pig Model of Pediatric NASH. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, g582–g609. [Google Scholar] [CrossRef]
- Cope, K.; Risby, T.; Diehl, A.M. Increased Gastrointestinal Ethanol Production in Obese Mice: Implications for Fatty Liver Disease Pathogenesis. Gastroenterology 2000, 119, 1340–1347. [Google Scholar] [CrossRef]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of Gut Microbiomes in Nonalcoholic Steatohepatitis (NASH) Patients: A Connection between Endogenous Alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef]
- Yuan, J.; Chen, C.; Cui, J.; Lu, J.; Yan, C.; Wei, X.; Zhao, X.; Li, N.; Li, S.; Xue, G.; et al. Fatty Liver Disease Caused by High-Alcohol-Producing Klebsiella Pneumoniae. Cell Metab. 2019, 30, 675–688. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, Z.; Li, H.; Zhao, J.; Wei, X.; Lin, W.; Zhao, X.; Jiang, A.; Yuan, J. Endogenous Ethanol Produced by Intestinal Bacteria Induces Mitochondrial Dysfunction in Non-Alcoholic Fatty Liver Disease. J. Gastroenterol. Hepatol. 2020. Online ahead of print. [Google Scholar] [CrossRef]
- Engstler, A.J.; Aumiller, T.; Degen, C.; Dürr, M.; Weiss, E.; Maier, I.B.; Schattenberg, J.M.; Jin, C.J.; Sellmann, C.; Bergheim, I. Insulin Resistance Alters Hepatic Ethanol Metabolism: Studies in Mice and Children With Non-Alcoholic Fatty Liver Disease. Gut 2016, 65, 1564–1571. [Google Scholar] [CrossRef]
- Briskey, D.; Heritage, M.; Jaskowski, L.; Peake, J.; Gobe, G.; Subramaniam, V.N.; Crawford, D.; Campbell, C.; Vitetta, L. Probiotics Modify Tight-Junction Proteins in an Animal Model of Nonalcoholic Fatty Liver Disease. Therap. Adv. Gastroenterol. 2016, 9, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Okubo, H.; Sakoda, H.; Kushiyama, A.; Fujishiro, M.; Nakatsu, Y.; Fukushima, T.; Matsunaga, Y.; Kamata, H.; Asahara, T.; Yoshida, Y.; et al. Lactobacillus Casei Strain Shirota Protects Against Nonalcoholic Steatohepatitis Development in a Rodent Model. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, g911–g918. [Google Scholar] [CrossRef] [PubMed]
- Naito, E.; Yoshida, Y.; Makino, K.; Kounoshi, Y.; Kunihiro, S.; Takahashi, R.; Matsuzaki, T.; Miyazaki, K.; Ishikawa, F. Beneficial Effect of Oral Administration of Lactobacillus Casei Strain Shirota on Insulin Resistance in Diet-Induced Obesity Mice. J. Appl. Microbiol. 2011, 110, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Wagnerberger, S.; Spruss, A.; Kanuri, G.; Stahl, C.; Schröder, M.; Vetter, W.; Bischoff, S.C.; Bergheim, I. Lactobacillus Casei Shirota Protects From Fructose-Induced Liver Steatosis: A Mouse Model. J. Nutr. Biochem. 2013, 24, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Chen, L.; Zhao, Y.; Wang, C.; Duan, C.; Yang, G.; Niu, C.; Li, S. Lactobacillus Plantarum NA136 Ameliorates Nonalcoholic Fatty Liver Disease by Modulating Gut Microbiota, Improving Intestinal Barrier Integrity, and Attenuating Inflammation. Appl. Microbiol. Biotechnol. 2020, 104, 5273–5282. [Google Scholar] [CrossRef]
- Nguyen, T.D.T.; Kang, J.H.; Lee, M.S. Characterization of Lactobacillus Plantarum PH04, a Potential Probiotic Bacterium With Cholesterol-Lowering Effects. Int. J. Food Microbiol. 2007, 113, 358–361. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, N.; Xi, A.; Ahmed, Z.; Zhang, B.; Bai, X. Effects of Lactobacillus Plantarum MA2 Isolated From Tibet Kefir on Lipid Metabolism and Intestinal Microflora of Rats Fed on High-Cholesterol Diet. Appl. Microbiol. Biotechnol. 2009, 84, 341–347. [Google Scholar] [CrossRef]
- Park, E.J.; Lee, Y.S.; Kim, S.M.; Park, G.S.; Lee, Y.H.; Jeong, D.Y.; Kang, J.; Lee, H.J. Beneficial Effects of Lactobacillus plantarum Strains on Non-Alcoholic Fatty Liver Disease in High Fat/High Fructose Diet-Fed Rats. Nutrients 2020, 12, 542. [Google Scholar] [CrossRef]
- Ritze, Y.; Bardos, G.; Claus, A.; Ehrmann, V.; Bergheim, I.; Schwiertz, A.; Bischoff, S.C. Lactobacillus Rhamnosus GG Protects Against Non-Alcoholic Fatty Liver Disease in Mice. PLoS ONE 2014, 9, e80169. [Google Scholar] [CrossRef]
- Kim, B.; Park, K.; Ji, Y.; Park, S.; Holzapfel, W.; Hyun, C.K. Protective Effects of Lactobacillus Rhamnosus GG Against Dyslipidemia in High-Fat Diet-Induced Obese Mice. Biochem. Biophys. Res. Commun. 2016, 473, 530–536. [Google Scholar] [CrossRef]
- Liu, Q.; Liu, Y.; Li, F.; Gu, Z.; Liu, M.; Shao, T.; Zhang, L.; Zhou, G.; Pan, C.; He, L.; et al. Probiotic Culture Supernatant Improves Metabolic Function Through FGF21-Adiponectin Pathway in Mice. J. Nutr. Biochem. 2020, 75, 108256. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.W.; Gilliland, S.E. Effect of Fermented Milk (Yogurt) Containing Lactobacillus Acidophilus L1 on Serum Cholesterol in Hypercholesterolemic Humans. J. Am. Coll. Nutr. 1999, 18, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Sohn, W.; Jun, D.W.; Lee, K.N.; Lee, H.L.; Lee, O.Y.; Choi, H.S.; Yoon, B.C. Lactobacillus Paracasei Induces M2-Dominant Kupffer Cell Polarization in a Mouse Model of Nonalcoholic Steatohepatitis. Dig. Dis. Sci. 2015, 60, 3340–3350. [Google Scholar] [CrossRef] [PubMed]
- Xin, J.; Zeng, D.; Wang, H.; Ni, X.; Yi, D.; Pan, K.; Jing, B. Preventing Non-Alcoholic Fatty Liver Disease Through Lactobacillus Johnsonii BS15 by Attenuating Inflammation and Mitochondrial Injury and Improving Gut Environment in Obese Mice. Appl. Microbiol. Biotechnol. 2014, 98, 6817–6829. [Google Scholar] [CrossRef]
- Hsieh, F.; Lee, C.; Chai, C.; Chen, W.T.; Lu, Y.C.; Wu, C.S. Oral Administration of Lactobacillus Reuteri GMNL-263 Improves Insulin Resistance and Ameliorates Hepatic Steatosis in High Fructose-Fed Rats. Nutr. Metab. Lond. 2013, 10, 35. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Yun, S.; Park, M.; Park, J.H.; Jeong, S.Y.; Park, H.O. Anti-Obesity Effect of Lactobacillus Gasseri BNR17 in High-Sucrose Diet-Induced Obese Mice. PLoS ONE 2013, 8, e54617. [Google Scholar] [CrossRef]
- Naudin, C.R.; Maner-Smith, K.; Owens, J.A.; Wynn, G.M.; Robinson, B.S.; Matthews, J.D.; Reedy, A.R.; Luo, L.; Wolfarth, A.A.; Darby, T.M.; et al. Lactococcus Lactis Subsp. Cremoris Elicits Protection Against Metabolic Changes Induced by a Western-style Diet. Gastroenterology 2020. Online ahead of print. [Google Scholar] [CrossRef]
- Xiao, M.; Lin, S.; Shen, Z.; Luo, W.; Wang, X. Systematic Review With Meta-Analysis: The Effects of Probiotics in Nonalcoholic Fatty Liver Disease. Gastroenterol. Res. Pract. 2019, 2019, 1484598. [Google Scholar] [CrossRef]
- Jena, P.K.; Sheng, L.; Li, Y.; Wan, Y.Y. Probiotics VSL#3 Are Effective in Reversing Non-Alcoholic Steatohepatitis in a Mouse Model. Hepatobiliary Surg. Nutr. 2020, 9, 170–182. [Google Scholar] [CrossRef]
- Alisi, A.; Bedogni, G.; Baviera, G.; Giorgio, V.; Porro, E.; Paris, C.; Giammaria, P.; Reali, L.; Anania, F.; Nobili, V. Randomised Clinical Trial: The Beneficial Effects of VSL#3 in Obese Children With Non-Alcoholic Steatohepatitis. Aliment. Pharmacol. Ther. 2014, 39, 1276–1285. [Google Scholar]
- Mei, L.; Tang, Y.; Li, M.; Yang, P.; Liu, Z.; Yuan, J.; Zheng, P. Co-Administration of Cholesterol-Lowering Probiotics and Anthraquinone From Cassia Obtusifolia, L. Ameliorate Non-Alcoholic Fatty Liver. PLoS ONE 2015, 10, e0138078. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; He, J.; Gao, N.; Lu, X.; Li, M.; Wu, X.; Liu, Z.; Jin, Y.; Liu, J.; Xu, J.; et al. Probiotics May Delay the Progression of Nonalcoholic Fatty Liver Disease by Restoring the Gut Microbiota Structure and Improving Intestinal Endotoxemia. Sci. Rep. 2017, 7, 45176. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kim, H.; Jeong, D.; Kang, I.B.; Chon, J.W.; Kim, H.S.; Song, K.Y.; Seo, K.H. Kefir Alleviates Obesity and Hepatic Steatosis in High-Fat Diet-Fed Mice by Modulation of Gut Microbiota and Mycobiota: Targeted and Untargeted Community Analysis With Correlation of Biomarkers. J. Nutr. Biochem. 2017, 44, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Takai, A.; Kikuchi, K.; Ichimura, M.; Tsuneyama, K.; Moritoki, Y.; Matsumoto, K.; Tsunashima, H.; Onda, T.; Kuniyoshi, N.; Nariyama, T.; et al. Fructo-Oligosaccharides Ameliorate Steatohepatitis, Visceral Adiposity, and Associated Chronic Inflammation via Increased Production of Short-Chain Fatty Acids in a Mouse Model of Non-Alcoholic Steatohepatitis. BMC Gastroenterol. 2020, 20, 46. [Google Scholar] [CrossRef] [PubMed]
- Kok, N.; Roberfroid, M.; Delzenne, N. Dietary Oligofructose Modifies the Impact of Fructose on Hepatic Triacylglycerol Metabolism. Metabolism 1996, 45, 1547–1550. [Google Scholar] [CrossRef]
- Delzenne, N.M.; Kok, N. Effects of Fructans-Type Prebiotics on Lipid Metabolism. Am. J. Clin. Nutr. 2001, 73 (Suppl. 2), 456s–458s. [Google Scholar] [CrossRef]
- Matsumoto, K.; Ichimura, M.; Tsuneyama, K.; Moritoki, Y.; Tsunashima, H.; Omagari, K.; Hara, M.; Yasuda, I.; Miyakawa, H.; Kikuchi, K. Fructo-Oligosaccharides and Intestinal Barrier Function in a Methionine-Choline-Deficient Mouse Model of Nonalcoholic Steatohepatitis. PLoS ONE 2017, 12, e0175406. [Google Scholar] [CrossRef]
- Borges Haubert, N.J.; Marchini, J.S.; Carvalho Cunha, S.F.; Suen, V.M.; Padovan, G.J.; Jordao, A.A.J.; Marchini Alves, C.M.; Marchini, J.F.; Vannucchi, H. Choline and Fructooligosaccharide: Non-Alcoholic Fatty Liver Disease, Cardiac Fat Deposition, and Oxidative Stress Markers. Nutr. Metab. Insights 2015, 8, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.Y.L.; Orr, D.; Plank, L.D.; Vatanen, T.; O’Sullivan, J.M.; Murphy, R. Randomised Double-Blind Placebo-Controlled Trial of Inulin With Metronidazole in Non-Alcoholic Fatty Liver Disease (NAFLD). Nutrients 2020, 12, 937. [Google Scholar] [CrossRef]
- Weitkunat, K.; Schumann, S.; Petzke, K.J.; Blaut, M.; Loh, G.; Klaus, S. Effects of Dietary Inulin on Bacterial Growth, Short-Chain Fatty Acid Production and Hepatic Lipid Metabolism in Gnotobiotic Mice. J. Nutr. Biochem. 2015, 26, 929–937. [Google Scholar] [CrossRef]
- Liu, L.; Li, P.; Liu, Y.; Zhang, Y. Efficacy of Probiotics and Synbiotics in Patients With Nonalcoholic Fatty Liver Disease: A Meta-Analysis. Dig. Dis. Sci. 2019, 64, 3402–3412. [Google Scholar] [CrossRef]
- Aguayo, G.A.; Donneau, A.; Vaillant, M.T.; Schritz, A.; Franco, O.H.; Stranges, S.; Malisoux, L.; Guillaume, M.; Witte, D.R. Agreement Between 35 Published Frailty Scores in the General Population. Am. J. Epidemiol. 2017, 186, 420–434. [Google Scholar] [CrossRef] [PubMed]
- Yao, F.; Jia, R.; Huang, H.; Yu, Y.; Mei, L.; Bai, L.; Ding, Y.; Zheng, P. Effect of Lactobacillus paracasei N1115 and Fructooligosaccharides in Nonalcoholic Fatty Liver Disease. Arch. Med. Sci. 2019, 15, 1336–1344. [Google Scholar] [CrossRef] [PubMed]
- Alves, C.C.; Waitzberg, D.L.; de Andrade, L.S.; Dos Santos Aguiar, L.; Reis, M.B.; Guanabara, C.C.; Júnior, O.A.; Ribeiro, D.A.; Sala, P. Prebiotic and Synbiotic Modifications of Beta Oxidation and Lipogenic Gene Expression After Experimental Hypercholesterolemia in Rat Liver. Front. Microbiol. 2017, 8, 2010. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, M.; Vacante, M.; Antic, T.; Giordano, M.; Chisari, G.; Acquaviva, R.; Mastrojeni, S.; Malaguarnera, G.; Mistretta, A.; Volti, G.L.; et al. Bifidobacterium Longum With Fructo-Oligosaccharides in Patients With Non Alcoholic Steatohepatitis. Dig. Dis. Sci. 2012, 57, 545–553. [Google Scholar] [CrossRef]
- Scorletti, E.; Afolabi, P.R.; Miles, E.A.; Smith, D.E.; Almehmadi, A.; Alshathry, A.; Childs, C.E.; Del Fabbro, S.; Bilson, J.; Moyses, H.E.; et al. Synbiotics Alter Fecal Microbiomes, But Not Liver Fat or Fibrosis, in a Randomized Trial of Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2020, 158, 1597–1610. [Google Scholar] [CrossRef]
- Eiseman, B.; Silen, W.; Bascom, G.S.; KAUVAR, A.J. Fecal Enema as an Adjunct in the Treatment of Pseudomembranous Enterocolitis. Surgery 1958, 44, 854–859. [Google Scholar]
- Zhou, D.; Pan, Q.; Shen, F.; Cao, H.X.; Ding, W.J.; Chen, Y.W.; Fan, J.G. Total Fecal Microbiota Transplantation Alleviates High-Fat Diet-Induced Steatohepatitis in Mice via Beneficial Regulation of Gut Microbiota. Sci. Rep. 2017, 7, 1529. [Google Scholar] [CrossRef]
- Vrieze, A.; Nood, E.V.; Holleman, F.; Salojärvi, J.; Kootte, R.S.; Bartelsman, J.F.; Dallinga-Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of Intestinal Microbiota From Lean Donors Increases Insulin Sensitivity in Individuals With Metabolic Syndrome. Gastroenterology 2012, 143, 913–916. [Google Scholar] [CrossRef]
- Wei, M.; Brandhorst, S.; Shelehchi, M.; Mirzaei, H.; Cheng, C.W.; Budniak, J.; Groshen, S.; Mack, W.J.; Guen, E.; Di Biase, S.; et al. Fasting-Mimicking Diet and Markers/Risk Factors for Aging, Diabetes, Cancer, and Cardiovascular Disease. Sci Transl. Med. 2017, 9, eaai8700. [Google Scholar] [CrossRef]
- Louala, S.; Lamri-Senhadji, M. Beneficial Effects of Low-Calorie-Carbohydrate/High-Agar Diet on Cardiometabolic Disorders Associated With Non-Alcoholic Fatty Liver Disease in Obese Rats. Prev. Nutr. Food Sci. 2019, 24, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.W.; Villani, V.; Buono, R.; Wei, M.; Kumar, S.; Yilmaz, O.H.; Cohen, P.; Sneddon, J.B.; Perin, L.; Longo, V.D. Fasting-Mimicking Diet Promotes Ngn3-Driven β-Cell Regeneration to Reverse Diabetes. Cell 2017, 168, 775–788. [Google Scholar] [CrossRef]
- Wei, S.; Han, R.; Zhao, J.; Wang, S.; Huang, M.; Wang, Y.; Chen, Y. Intermittent Administration of a Fasting-Mimicking Diet Intervenes in Diabetes Progression, Restores β Cells and Reconstructs Gut Microbiota in Mice. Nutr. Metab. Lond. 2018, 15, 80. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.L.; Jia, X.B.; Sun, M.F.; Zhu, Y.L.; Qiao, C.M.; Zhang, B.P.; Zhao, L.P.; Yang, Q.; Cui, C.; Chen, X.; et al. Neuroprotection of Fasting Mimicking Diet on MPTP-Induced Parkinson’s Disease Mice via Gut Microbiota and Metabolites. Neurotherapeutics 2019, 16, 741–760. [Google Scholar] [CrossRef] [PubMed]
- Asghari, S.; Asghari-Jafarabadi, M.; Somi, M.H.; Ghavami, S.M.; Rafraf, M. Comparison of Calorie-Restricted Diet and Resveratrol Supplementation on Anthropometric Indices, Metabolic Parameters, and Serum Sirtuin-1 Levels in Patients With Nonalcoholic Fatty Liver Disease: A Randomized Controlled Clinical Trial. J. Am. Coll. Nutr. 2018, 37, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Sofi, F. Fasting-Mimicking Diet a Clarion Call for Human Nutrition Research or an Additional Swan Song for a Commercial Diet? Int. J. Food Sci. Nutr. 2020, 1–8. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Vitetta, L. Gut Microbiota Metabolites in NAFLD Pathogenesis and Therapeutic Implications. Int. J. Mol. Sci. 2020, 21, 5214. https://doi.org/10.3390/ijms21155214
Chen J, Vitetta L. Gut Microbiota Metabolites in NAFLD Pathogenesis and Therapeutic Implications. International Journal of Molecular Sciences. 2020; 21(15):5214. https://doi.org/10.3390/ijms21155214
Chicago/Turabian StyleChen, Jiezhong, and Luis Vitetta. 2020. "Gut Microbiota Metabolites in NAFLD Pathogenesis and Therapeutic Implications" International Journal of Molecular Sciences 21, no. 15: 5214. https://doi.org/10.3390/ijms21155214
APA StyleChen, J., & Vitetta, L. (2020). Gut Microbiota Metabolites in NAFLD Pathogenesis and Therapeutic Implications. International Journal of Molecular Sciences, 21(15), 5214. https://doi.org/10.3390/ijms21155214