Immunotherapies and Combination Strategies for Immuno-Oncology

 ,
,  ,
,
Abstract
1. Introduction
2. Types of Immunotherapy: Their Combinations and Limitations
2.1. Checkpoint Inhibitors
2.1.1. Programmed Cell Death-1 (PD-1) Blockade and Combinations
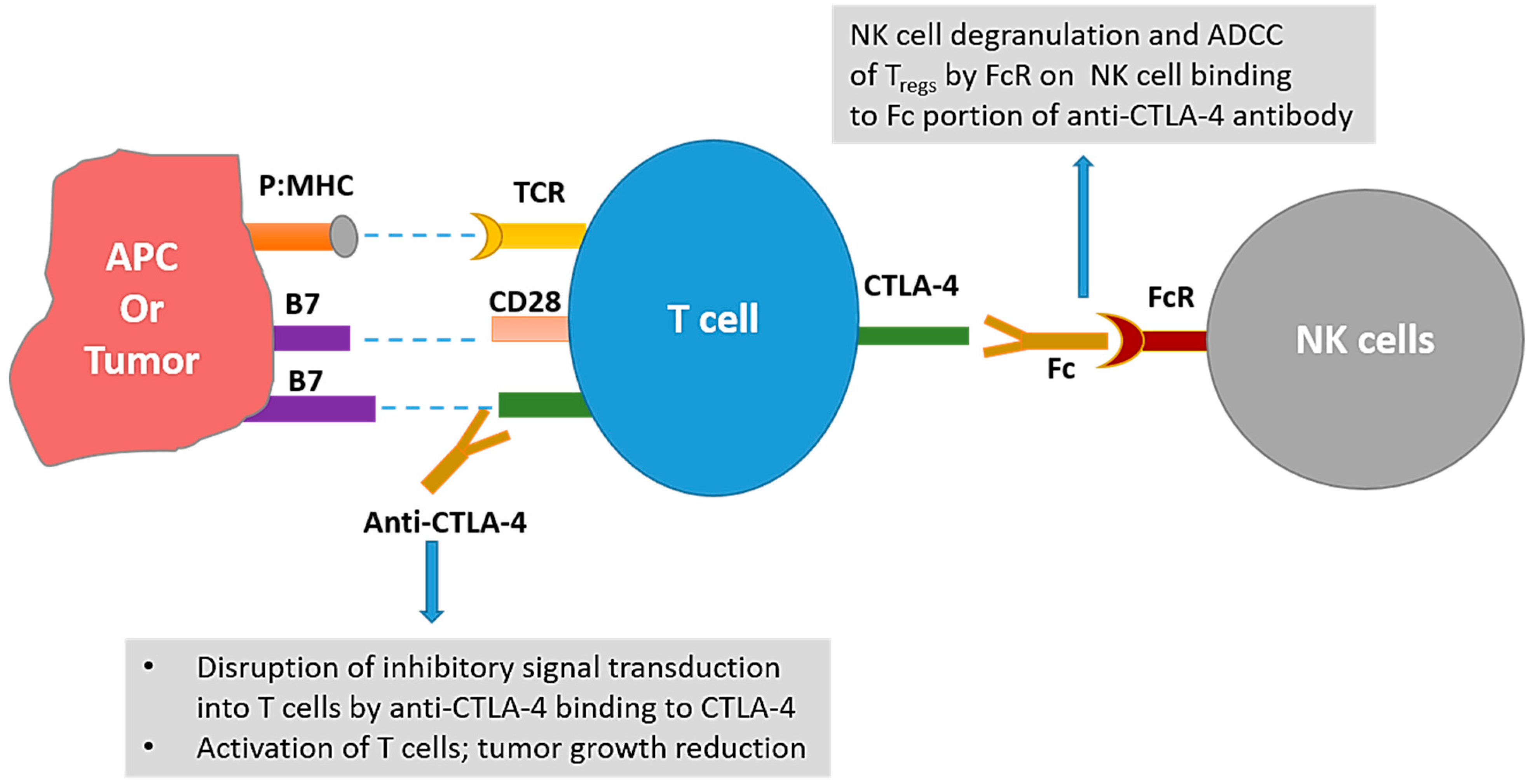
2.1.2. Cytotoxic T-Lymphocyte-Associated Protein-4 (CTLA-4) Blockade and Combinations
2.2. Combination of Anti-PD-1/PD-L1 and Anti-CTLA-4
2.3. Limitations of PD-1/PD-L1 and CTLA-4 Checkpoint Inhibitors and Their Combinations
2.4. Emerging Checkpoint Molecules as Targets for Immunotherapy
2.4.1. T Cell Immunoglobulin and Mucin Domain 3 (Tim-3)
2.4.2. V-Domain Ig Suppressor of T Cell Activation (VISTA)
2.5. Emerging Co-Stimulatory Molecules as Targets for Immunotherapy
2.5.1. 4-1BB
2.5.2. OX40
2.6. Cellular Immunotherapy
2.6.1. Tumor Infiltrating Lymphocyte (TIL) Therapy and Combinations
2.6.2. Chimeric Antigen Receptor (CAR) T Cell Therapy and Combinations
3. Combination of Immunotherapy and Radiation Therapy
3.1. Mechanistic Effects of Radiotherapy Alone and in Combination with Immunotherapy
3.2. Efficacy of Radiotherapy Concomitant with Immunotherapy
4. Combination of Cancer Vaccines with Chemotherapy
5. Biomarkers in Immunotherapy and Combinations
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACT | Adoptive cellular therapy |
| APC | Antigen presenting cell |
| CAR | Chimeric antigen receptor |
| CDK | Cyclin-dependent kinase |
| CI | Confidence interval |
| CML | Chronic myeloid leukemia |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| CTL | Cytotoxic T-lymphocyte |
| CTLA-4 | Cytotoxic T-lymphocyte-associated protein 4 |
| DC | Dendritic cell |
| DNA | Deoxyribonucleic acid |
| FasL | Fas ligand |
| HLA | Human leukocyte antigen |
| ICI | Immune checkpoint inhibitor |
| IFN | Interferon |
| Ig | Immunoglobulin |
| IL | Interleukin |
| ITIM | Immunoreceptor tyrosine-based inhibition motif |
| LAG-3 | Lymphocyte activation gene 3 protein |
| LFT | Liver function test |
| mAbs | Monoclonal antibodies |
| MHC | Major histocompatibility complex |
| NF-kB | Nuclear factor kB |
| NHL | Non-Hodgkin’s lymphoma |
| NK | Natural killer |
| NSCLC | Non-small cell lung cancer |
| OS | Overall survival |
| pCR | Pathologic complete response rates |
| PD-1 | Programmed cell death protein 1 |
| PD-L1 | Programmed death-ligand 1 |
| PFS | Progression free survival |
| RCC | Renal cell carcinoma |
| TCR | T cell receptor |
| TGF | Transforming growth factor |
| TIL | Tumor infiltrating lymphocyte |
| TIM-3 | T cell Ig mucin domain-containing 3 |
| TME | Tumor microenvironment |
| TNBC | Triple negative breast cancer |
| TNF | Tumor necrosis factor |
| TNFR | Tumor necrosis factor receptor |
| TNFRSF4 | Tumor necrosis factor receptor superfamily member 4 |
| TRAEs | Treatment-related adverse effects |
| Treg | Regulatory T cell |
| VEGF | Vascular endothelial growth factor |
| VEGFR | Vascular endothelial growth factor receptor |
| VISTA | V-domain Ig suppressor of t-cell activation |
References
- Cancer. Available online: https://www.cdc.gov/chronicdisease/resources/publications/factsheets/cancer.htm#:~:text=Cancer%20is%20the%20second%20leading,and%20drinking%20too%20much%20alcohol (accessed on 10 July 2020).
- Cancer Treatment. Available online: https://www.cancer.gov/about-cancer/treatment (accessed on 10 July 2020).
- How Immunotherapy Is Used to Treat Cancer. Available online: https://www.cancer.org/treatment/treatments-and-side-effects/treatment-types/immunotherapy/what-is-immunotherapy.html (accessed on 10 July 2020).
- Kaufman, H.L.; Atkins, M.B.; Subedi, P.; Wu, J.; Chambers, J.; Joseph Mattingly, T., II; Campbell, J.D.; Allen, J.; Ferris, A.E.; Schilsky, R.L.; et al. The promise of Immuno-oncology: Implications for defining the value of cancer treatment. J. Immunother. Cancer 2019, 7, 129. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol. Cancer 2019, 18, 10. [Google Scholar] [CrossRef]
- Xia, A.; Zhang, Y.; Xu, J.; Yin, T.; Lu, X.J. T Cell Dysfunction in Cancer Immunity and Immunotherapy. Front. Immunol. 2019, 10, 1719. [Google Scholar] [CrossRef]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharmacol. 2017, 8, 561. [Google Scholar] [CrossRef] [PubMed]
- Teets, A.; Pham, L.; Tran, E.L.; Hochmuth, L.; Deshmukh, R. Avelumab: A Novel Anti-PD-L1 Agent in the Treatment of Merkel Cell Carcinoma and Urothelial Cell Carcinoma. Crit. Rev. Immunol. 2018, 38, 159–206. [Google Scholar] [CrossRef]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef] [PubMed]
- Wills, S.; Hochmuth, L.K.; Bauer, K.S., Jr.; Deshmukh, R. Durvalumab: A newly approved checkpoint inhibitor for the treatment of urothelial carcinoma. Curr. Probl. Cancer 2019, 43, 181–194. [Google Scholar] [CrossRef]
- Barrueto, L.; Caminero, F.; Cash, L.; Makris, C.; Lamichhane, P.; Deshmukh, R.R. Resistance to Checkpoint Inhibition in Cancer Immunotherapy. Transl. Oncol. 2020, 13, 100738. [Google Scholar] [CrossRef] [PubMed]
- Toschi, L.; Rossi, S.; Finocchiaro, G.; Santoro, A. Non-small cell lung cancer treatment (r)evolution: Ten years of advances and more to come. Ecancermedicalscience 2017, 11, 787. [Google Scholar] [CrossRef] [PubMed]
- Chemotherapy for Non-Small Cell Lung Cancer. Available online: https://www.cancer.org/cancer/lung-cancer/treating-non-small-cell/chemotherapy.html (accessed on 10 July 2020).
- Lazzari, C.; Bulotta, A.; Ducceschi, M.; Vigano, M.G.; Brioschi, E.; Corti, F.; Gianni, L.; Gregorc, V. Historical Evolution of Second-Line Therapy in Non-Small Cell Lung Cancer. Front. Med. (Lausanne) 2017, 4, 4. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Luft, A.; Vicente, D.; Tafreshi, A.; Gumus, M.; Mazieres, J.; Hermes, B.; Cay Senler, F.; Csoszi, T.; Fulop, A.; et al. Pembrolizumab plus Chemotherapy for Squamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- Wirsdörfer, F.; de Leve, S.; Jendrossek, V. Combining Radiotherapy and Immunotherapy in Lung Cancer: Can We Expect Limitations Due to Altered Normal Tissue Toxicity? Int. J. Mol. Sci. 2018, 20, 24. [Google Scholar] [CrossRef]
- Marshall, H.T.; Djamgoz, M.B.A. Immuno-Oncology: Emerging Targets and Combination Therapies. Front. Oncol. 2018, 8, 315. [Google Scholar] [CrossRef] [PubMed]
- Immunotherapy. Timeline of Progress. Available online: https://www.cancerresearch.org/immunotherapy/timeline-of-progress (accessed on 10 July 2020).
- Mahoney, K.M.; Rennert, P.D.; Freeman, G.J. Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discov. 2015, 14, 561–584. [Google Scholar] [CrossRef] [PubMed]
- Casan, J.M.L.; Wong, J.; Northcott, M.J.; Opat, S. Anti-CD20 monoclonal antibodies: Reviewing a revolution. Hum. Vaccin. Immunother. 2018, 14, 2820–2841. [Google Scholar] [CrossRef]
- Jimenez Aguilar, E.; Ricciuti, B.; Gainor, J.F.; Nishino, M.; Adeni, A.E.; Subegdjo, S.; Khosrowjerdi, S.; Peterson, R.; Digumarthy, S.; Liu, C.; et al. Outcomes to first-line pembrolizumab in patients with non-small cell lung cancer and a PD-L1 tumor proportion score ≥90%. J. Clin. Oncol. 2019, 37, 9111. [Google Scholar] [CrossRef]
- Yi, M.; Jiao, D.; Xu, H.; Liu, Q.; Zhao, W.; Han, X.; Wu, K. Biomarkers for predicting efficacy of PD-1/PD-L1 inhibitors. Mol. Cancer 2018, 17, 129. [Google Scholar] [CrossRef]
- Marcus, L.; Lemery, S.J.; Keegan, P.; Pazdur, R. FDA Approval Summary: Pembrolizumab for the Treatment of Microsatellite Instability-High Solid Tumors. Clin. Cancer Res. 2019, 25, 3753. [Google Scholar] [CrossRef]
- Oiseth, S.; Aziz, M. Cancer immunotherapy: A brief review of the history, possibilities, and challenges ahead. J. Cancer Metastasis Treat. 2017, 3, 250–261. [Google Scholar] [CrossRef]
- Andrews, A. Treating with Checkpoint Inhibitors-Figure $1 Million per Patient. Am. Health Drug Benefits 2015, 8, 9. [Google Scholar]
- Verma, V.; Sprave, T.; Haque, W.; Simone, C.B., II; Chang, J.Y.; Welsh, J.W.; Thomas, C.R., Jr. A systematic review of the cost and cost-effectiveness studies of immune checkpoint inhibitors. J. Immunother. Cancer 2018, 6, 128. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Yusa, S.; Campbell, K.S. Src homology region 2-containing protein tyrosine phosphatase-2 (SHP-2) can play a direct role in the inhibitory function of killer cell Ig-like receptors in human NK cells. J. Immunol. 2003, 170, 4539–4547. [Google Scholar] [CrossRef] [PubMed]
- Fares, C.M.; Van Allen, E.M.; Drake, C.G.; Allison, J.P.; Hu-Lieskovan, S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Rossi, C.; Gilhodes, J.; Maerevoet, M.; Herbaux, C.; Morschhauser, F.; Brice, P.; Garciaz, S.; Borel, C.; Ysebaert, L.; Oberic, L.; et al. Efficacy of chemotherapy or chemo-anti-PD-1 combination after failed anti-PD-1 therapy for relapsed and refractory Hodgkin lymphoma: A series from Lysa centers. Am. J. Hematol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Sun, D.; Wang, J.; Han, C.; Qian, Y.; Chen, G.; Li, X.; Zhang, J.; Cui, P.; Du, W.; et al. Immune checkpoint inhibitors combined with chemotherapy for the treatment of advanced pancreatic cancer patients. Cancer Immunol. Immunother. 2020, 69, 365–372. [Google Scholar] [CrossRef]
- Sun, D.; Ma, J.; Wang, J.; Han, C.; Qian, Y.; Chen, G.; Li, X.; Zhang, J.; Cui, P.; Du, W.; et al. Anti-PD-1 therapy combined with chemotherapy in patients with advanced biliary tract cancer. Cancer Immunol. Immunother. 2019, 68, 1527–1535. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Horn, L.; Mansfield, A.S.; Szczesna, A.; Havel, L.; Krzakowski, M.; Hochmair, M.J.; Huemer, F.; Losonczy, G.; Johnson, M.L.; Nishio, M.; et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2220–2229. [Google Scholar] [CrossRef]
- Drew, Y.; Kaufman, B.; Banerjee, S.; Lortholary, A.; Hong, S.H.; Park, Y.H.; Zimmermann, S.; Roxburgh, P.; Ferguson, M.; Alvarez, R.H.; et al. Phase II study of olaparib + durvalumab (MEDIOLA): Updated results in germline BRCA-mutated platinumsensitive relapsed (PSR) ovarian cancer (OC) [ESMO abstract 1190PD]. Ann. Oncol. 2019, 30. [Google Scholar] [CrossRef]
- Bang, Y.-J.; Kaufman, B.; Geva, R.; Stemmer, S.M.; Hong, S.-H.; Lee, J.-S.; Domchek, S.M.; Lanasa, M.C.; Tang, M.; Gresty, C.; et al. An open-label, phase II basket study of olaparib and durvalumab (MEDIOLA): Results in patients with relapsed gastric cancer. J. Clin. Oncol. 2019. [Google Scholar] [CrossRef]
- Peng, J.; Hamanishi, J.; Matsumura, N.; Abiko, K.; Murat, K.; Baba, T.; Yamaguchi, K.; Horikawa, N.; Hosoe, Y.; Murphy, S.K.; et al. Chemotherapy Induces Programmed Cell Death-Ligand 1 Overexpression via the Nuclear Factor-kappaB to Foster an Immunosuppressive Tumor Microenvironment in Ovarian Cancer. Cancer Res. 2015, 75, 5034–5045. [Google Scholar] [CrossRef]
- Yu, Y.; Ma, X.; Zhang, Y.; Zhang, Y.; Ying, J.; Zhang, W.; Zhong, Q.; Zhou, A.; Zeng, Y. Changes in Expression of Multiple Checkpoint Molecules and Infiltration of Tumor Immune Cells after Neoadjuvant Chemotherapy in Gastric Cancer. J. Cancer 2019, 10, 2754–2763. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.Y.; Li, J.; Tao, L.; Lam, A.K.; Chan, K.W.; Ko, J.M.Y.; Yu, V.Z.; Wong, M.; Li, B.; Lung, M.L. Chemotherapeutic Treatments Increase PD-L1 Expression in Esophageal Squamous Cell Carcinoma through EGFR/ERK Activation. Transl. Oncol. 2018, 11, 1323–1333. [Google Scholar] [CrossRef] [PubMed]
- Clinical Trial Evaluating FOLFIRI + Durvalumab vs FOLFIRI + Durvalumab and Tremelimumab in Second-line Treatment of Patients With Advanced Gastric or Gastro-oesophageal Junction Adenocarcinoma. Available online: https://ClinicalTrials.gov/show/NCT03959293 (accessed on 10 July 2020).
- Study For Patients With NSCLC EGFR Mutations (Del 19 or L858R +/- T790M). Available online: https://ClinicalTrials.gov/show/NCT02349633 (accessed on 10 July 2020).
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Nirschl, C.J.; Drake, C.G. Molecular pathways: Coexpression of immune checkpoint molecules: Signaling pathways and implications for cancer immunotherapy. Clin. Cancer Res. 2013, 19, 4917–4924. [Google Scholar] [CrossRef]
- Rotte, A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J. Exp. Clin. Cancer Res. 2019, 38, 255. [Google Scholar] [CrossRef] [PubMed]
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med. 2013, 210, 1695–1710. [Google Scholar] [CrossRef]
- Pol, J.; Kroemer, G. Anti-CTLA-4 immunotherapy: Uncoupling toxicity and efficacy. Cell Res. 2018, 28, 501–502. [Google Scholar] [CrossRef]
- Sharma, A.; Subudhi, S.K.; Blando, J.; Scutti, J.; Vence, L.; Wargo, J.; Allison, J.P.; Ribas, A.; Sharma, P. Anti-CTLA-4 Immunotherapy Does Not Deplete FOXP3(+) Regulatory T Cells (Tregs) in Human Cancers. Clin. Cancer Res. 2019, 25, 1233–1238. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. Immune checkpoint targeting in cancer therapy: Toward combination strategies with curative potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Maio, M.; Grob, J.J.; Aamdal, S.; Bondarenko, I.; Robert, C.; Thomas, L.; Garbe, C.; Chiarion-Sileni, V.; Testori, A.; Chen, T.T.; et al. Five-year survival rates for treatment-naive patients with advanced melanoma who received ipilimumab plus dacarbazine in a phase III trial. J. Clin. Oncol. 2015, 33, 1191–1196. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; Lee, S.; McDermott, D.F.; Rao, U.N.; Butterfield, L.H.; Tarhini, A.A.; Leming, P.; Puzanov, I.; Shin, D.; Kirkwood, J.M. Ipilimumab plus sargramostim vs ipilimumab alone for treatment of metastatic melanoma: A randomized clinical trial. JAMA 2014, 312, 1744–1753. [Google Scholar] [CrossRef]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef]
- Hid Cadena, R.; Abdulahad, W.H.; Hospers, G.A.P.; Wind, T.T.; Boots, A.M.H.; Heeringa, P.; Brouwer, E. Checks and Balances in Autoimmune Vasculitis. Front. Immunol. 2018, 9, 315. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Ciuleanu, T.E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef]
- Chen, E.X.; Jonker, D.J.; Loree, J.M.; Kennecke, H.F.; Berry, S.R.; Couture, F.; Ahmad, C.E.; Goffin, J.R.; Kavan, P.; Harb, M.; et al. Effect of Combined Immune Checkpoint Inhibition vs Best Supportive Care Alone in Patients With Advanced Colorectal Cancer: The Canadian Cancer Trials Group CO.26 Study. JAMA Oncol. 2020. [Google Scholar] [CrossRef]
- Amaria, R.N.; Reddy, S.M.; Tawbi, H.A.; Davies, M.A.; Ross, M.I.; Glitza, I.C.; Cormier, J.N.; Lewis, C.; Hwu, W.J.; Hanna, E.; et al. Neoadjuvant immune checkpoint blockade in high-risk resectable melanoma. Nat. Med. 2018, 24, 1649–1654. [Google Scholar] [CrossRef]
- Blank, C.U.; Rozeman, E.A.; Fanchi, L.F.; Sikorska, K.; van de Wiel, B.; Kvistborg, P.; Krijgsman, O.; van den Braber, M.; Philips, D.; Broeks, A.; et al. Neoadjuvant versus adjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma. Nat. Med. 2018, 24, 1655–1661. [Google Scholar] [CrossRef]
- Sharma, P.; Siefker-Radtke, A.; de Braud, F.; Basso, U.; Calvo, E.; Bono, P.; Morse, M.A.; Ascierto, P.A.; Lopez-Martin, J.; Brossart, P.; et al. Nivolumab Alone and With Ipilimumab in Previously Treated Metastatic Urothelial Carcinoma: CheckMate 032 Nivolumab 1 mg/kg Plus Ipilimumab 3 mg/kg Expansion Cohort Results. J. Clin. Oncol. 2019, 37, 1608–1616. [Google Scholar] [CrossRef] [PubMed]
- Lebbe, C.; Meyer, N.; Mortier, L.; Marquez-Rodas, I.; Robert, C.; Rutkowski, P.; Menzies, A.M.; Eigentler, T.; Ascierto, P.A.; Smylie, M.; et al. Evaluation of Two Dosing Regimens for Nivolumab in Combination With Ipilimumab in Patients With Advanced Melanoma: Results From the Phase IIIb/IV CheckMate 511 Trial. J. Clin. Oncol. 2019, 37, 867–875. [Google Scholar] [CrossRef] [PubMed]
- FDA Grants Accelerated Approval to Nivolumab and Ipilimumab Combination for Hepatocellular Carcinoma. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-nivolumab-and-ipilimumab-combination-hepatocellular-carcinoma (accessed on 19 June 2020).
- FDA Approves Nivolumab Plus Ipilimumab Combination for Intermediate or Poor-Risk Advanced Renal Cell Carcinoma. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-nivolumab-plus-ipilimumab-combination-intermediate-or-poor-risk-advanced-renal-cell (accessed on 19 June 2020).
- FDA Approves Nivolumab Plus Ipilimumab for First-Line mNSCLC (PD-L1 Tumor Expression ≥1%). Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-nivolumab-plus-ipilimumab-first-line-mnsclc-pd-l1-tumor-expression-1 (accessed on 19 June 2020).
- FDA Grants Accelerated Approval to Ipilimumab for MSI-H or dMMR Metastatic Colorectal Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-ipilimumab-msi-h-or-dmmr-metastatic-colorectal-cancer (accessed on 19 June 2020).
- FDA Approves Nivolumab Plus Ipilimumab and Chemotherapy for First-Line Treatment of Metastatic NSCLC. Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-nivolumab-plus-ipilimumab-and-chemotherapy-first-line-treatment-metastatic-nsclc (accessed on 19 June 2020).
- Lamichhane, P.; Deshmukh, R.; Brown, J.A.; Jakubski, S.; Parajuli, P.; Nolan, T.; Raja, D.; Badawy, M.; Yoon, T.; Zmiyiwsky, M.; et al. Novel Delivery Systems for Checkpoint Inhibitors. Medicines 2019, 6, 74. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Y.; Salem, J.E.; Cohen, J.V.; Chandra, S.; Menzer, C.; Ye, F.; Zhao, S.; Das, S.; Beckermann, K.E.; Ha, L.; et al. Fatal Toxic Effects Associated With Immune Checkpoint Inhibitors: A Systematic Review and Meta-analysis. JAMA Oncol. 2018, 4, 1721–1728. [Google Scholar] [CrossRef] [PubMed]
- Denis, M.; Duruisseaux, M.; Brevet, M.; Dumontet, C. How Can Immune Checkpoint Inhibitors Cause Hyperprogression in Solid Tumors? Front. Immunol. 2020, 11, 492. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Thummalapalli, R.; Carter, J.; Canadas, I.; Barbie, D.A. Molecular and Genomic Determinants of Response to Immune Checkpoint Inhibition in Cancer. Annu. Rev. Med. 2018, 69, 333–347. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef]
- Li, X.; Shao, C.; Shi, Y.; Han, W. Lessons learned from the blockade of immune checkpoints in cancer immunotherapy. J. Hematol. Oncol. 2018, 11, 31. [Google Scholar] [CrossRef]
- Wang, Y.; Deng, W.; Li, N.; Neri, S.; Sharma, A.; Jiang, W.; Lin, S.H. Combining Immunotherapy and Radiotherapy for Cancer Treatment: Current Challenges and Future Directions. Front. Pharmacol. 2018, 9, 185. [Google Scholar] [CrossRef]
- Bang, A.; Schoenfeld, J.D. Immunotherapy and radiotherapy for metastatic cancers. Ann. Palliat. Med. 2019, 8, 312–325. [Google Scholar] [CrossRef]
- Ngwa, W.; Irabor, O.C.; Schoenfeld, J.D.; Hesser, J.; Demaria, S.; Formenti, S.C. Using immunotherapy to boost the abscopal effect. Nat. Rev. Cancer 2018, 18, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Grass, G.D.; Krishna, N.; Kim, S. The immune mechanisms of abscopal effect in radiation therapy. Curr. Probl. Cancer 2016, 40, 10–24. [Google Scholar] [CrossRef]
- Lamichhane, P.; Amin, N.P.; Agarwal, M.; Lamichhane, M. Checkpoint Inhibition: Will Combination with Radiotherapy and Nanoparticle-Mediated Delivery Improve Efficacy? Medicines 2018, 5, 114. [Google Scholar] [CrossRef] [PubMed]
- Bhalla, N.; Brooker, R.; Brada, M. Combining immunotherapy and radiotherapy in lung cancer. J. Thorac. Dis. 2018, 10, S1447–S1460. [Google Scholar] [CrossRef]
- Tang, C.; Wang, X.; Soh, H.; Seyedin, S.; Cortez, M.A.; Krishnan, S.; Massarelli, E.; Hong, D.; Naing, A.; Diab, A.; et al. Combining radiation and immunotherapy: A new systemic therapy for solid tumors? Cancer Immunol. Res. 2014, 2, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Demaria, S.; Formenti, S. Current clinical trials testing the combination of immunotherapy with radiotherapy. J. Immunother. Cancer 2016, 4, 51. [Google Scholar] [CrossRef] [PubMed]
- Bang, A.; Wilhite, T.J.; Pike, L.R.G.; Cagney, D.N.; Aizer, A.A.; Taylor, A.; Spektor, A.; Krishnan, M.; Ott, P.A.; Balboni, T.A.; et al. Multicenter Evaluation of the Tolerability of Combined Treatment With PD-1 and CTLA-4 Immune Checkpoint Inhibitors and Palliative Radiation Therapy. Int. J. Radiat. Oncol. Biol. Phys. 2017, 98, 344–351. [Google Scholar] [CrossRef]
- Antonia, S.J.; Mirza, N.; Fricke, I.; Chiappori, A.; Thompson, P.; Williams, N.; Bepler, G.; Simon, G.; Janssen, W.; Lee, J.H.; et al. Combination of p53 cancer vaccine with chemotherapy in patients with extensive stage small cell lung cancer. Clin. Cancer Res. 2006, 12, 878–887. [Google Scholar] [CrossRef]
- Walter, S.; Weinschenk, T.; Stenzl, A.; Zdrojowy, R.; Pluzanska, A.; Szczylik, C.; Staehler, M.; Brugger, W.; Dietrich, P.Y.; Mendrzyk, R.; et al. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat. Med. 2012, 18, 1254–1261. [Google Scholar] [CrossRef]
- Gatti-Mays, M.E.; Redman, J.M.; Collins, J.M.; Bilusic, M. Cancer vaccines: Enhanced immunogenic modulation through therapeutic combinations. Hum. Vaccin. Immunother. 2017, 13, 2561–2574. [Google Scholar] [CrossRef]
- Rodriguez, P.C.; Sanchez, B. Challenges and opportunities for cancer vaccines in the current NSCLC clinical scenario. Curr. Top. Med. Chem. 2013, 13, 2551–2561. [Google Scholar] [CrossRef] [PubMed]
- Vinay, D.S.; Kwon, B.S. 4-1BB (CD137), an inducible costimulatory receptor, as a specific target for cancer therapy. BMB Rep. 2014, 47, 122–129. [Google Scholar] [CrossRef]
- Chu, D.T.; Bac, N.D.; Nguyen, K.H.; Tien, N.L.B.; Thanh, V.V.; Nga, V.T.; Ngoc, V.T.N.; Anh Dao, D.T.; Hoan, L.N.; Hung, N.P.; et al. An Update on Anti-CD137 Antibodies in Immunotherapies for Cancer. Int. J. Mol. Sci. 2019, 20, 1822. [Google Scholar] [CrossRef] [PubMed]
- Claus, C.; Ferrara, C.; Xu, W.; Sam, J.; Lang, S.; Uhlenbrock, F.; Albrecht, R.; Herter, S.; Schlenker, R.; Husser, T.; et al. Tumor-targeted 4-1BB agonists for combination with T cell bispecific antibodies as off-the-shelf therapy. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Qi, X.; Li, F.; Wu, Y.; Cheng, C.; Han, P.; Wang, J.; Yang, X. Optimization of 4-1BB antibody for cancer immunotherapy by balancing agonistic strength with FcgammaR affinity. Nat. Commun. 2019, 10, 2141. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Lin, Q.; Zhang, Z.; Zhang, L. Therapeutic strategies for the costimulatory molecule OX40 in T-cell-mediated immunity. Acta Pharm. Sin. B 2020, 10, 414–433. [Google Scholar] [CrossRef] [PubMed]
- Glisson, B.; Leidner, R.; Ferris, R.L.; Powderly, J.; Rizvi, N.A.; Keam, B.; Schneider, R.; Goel, S.; Ohr, J.P.; Zheng, Y.; et al. Safety and clinical activity of MEDI0562, a humanized OX40 agonist monoclonal antibody, in adult patients with advanced solid tumors. Ann. Oncol. 2018, 29 (Suppl. 8), VIII410. [Google Scholar] [CrossRef]
- Curti, B.D.; Kovacsovics-Bankowski, M.; Morris, N.; Walker, E.; Chisholm, L.; Floyd, K.; Walker, J.; Gonzalez, I.; Meeuwsen, T.; Fox, B.A.; et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res. 2013, 73, 7189–7198. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yannelli, J.R.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Weber, J.S.; Parkinson, D.R.; Seipp, C.A.; Einhorn, J.H.; White, D.E. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. J. Natl. Cancer Inst. 1994, 86, 1159–1166. [Google Scholar] [CrossRef]
- Wrzesinski, C.; Paulos, C.M.; Kaiser, A.; Muranski, P.; Palmer, D.C.; Gattinoni, L.; Yu, Z.; Rosenberg, S.A.; Restifo, N.P. Increased intensity lymphodepletion enhances tumor treatment efficacy of adoptively transferred tumor-specific T cells. J. Immunother. 2010, 33, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Dudley, M.E.; Wunderlich, J.R.; Yang, J.C.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Sherry, R.M.; Marincola, F.M.; Leitman, S.F.; Seipp, C.A.; et al. A phase I study of nonmyeloablative chemotherapy and adoptive transfer of autologous tumor antigen-specific T lymphocytes in patients with metastatic melanoma. J. Immunother. 2002, 25, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Forget, M.A.; Haymaker, C.; Hess, K.R.; Meng, Y.J.; Creasy, C.; Karpinets, T.; Fulbright, O.J.; Roszik, J.; Woodman, S.E.; Kim, Y.U.; et al. Prospective Analysis of Adoptive TIL Therapy in Patients with Metastatic Melanoma: Response, Impact of Anti-CTLA4, and Biomarkers to Predict Clinical Outcome. Clin. Cancer Res. 2018, 24, 4416–4428. [Google Scholar] [CrossRef] [PubMed]
- Khammari, A.; Knol, A.C.; Nguyen, J.M.; Bossard, C.; Denis, M.G.; Pandolfino, M.C.; Quereux, G.; Bercegeay, S.; Dreno, B. Adoptive TIL transfer in the adjuvant setting for melanoma: Long-term patient survival. J. Immunol. Res. 2014, 2014, 186212. [Google Scholar] [CrossRef] [PubMed]
- Deniger, D.C.; Kwong, M.L.; Pasetto, A.; Dudley, M.E.; Wunderlich, J.R.; Langhan, M.M.; Lee, C.R.; Rosenberg, S.A. A Pilot Trial of the Combination of Vemurafenib with Adoptive Cell Therapy in Patients with Metastatic Melanoma. Clin. Cancer Res. 2017, 23, 351–362. [Google Scholar] [CrossRef]
- Mullinax, J.E.; Hall, M.; Prabhakaran, S.; Weber, J.; Khushalani, N.; Eroglu, Z.; Brohl, A.S.; Markowitz, J.; Royster, E.; Richards, A.; et al. Combination of Ipilimumab and Adoptive Cell Therapy with Tumor-Infiltrating Lymphocytes for Patients with Metastatic Melanoma. Front. Oncol. 2018, 8, 44. [Google Scholar] [CrossRef]
- Shank, B.R.; Do, B.; Sevin, A.; Chen, S.E.; Neelapu, S.S.; Horowitz, S.B. Chimeric Antigen Receptor T Cells in Hematologic Malignancies. Pharmacotherapy 2017, 37, 334–345. [Google Scholar] [CrossRef]
- Levine, B.L.; Miskin, J.; Wonnacott, K.; Keir, C. Global Manufacturing of CAR T Cell Therapy. Mol. Ther. Methods Clin. Dev. 2017, 4, 92–101. [Google Scholar] [CrossRef]
- Holzinger, A.; Barden, M.; Abken, H. The growing world of CAR T cell trials: A systematic review. Cancer Immunol. Immunother. 2016, 65, 1433–1450. [Google Scholar] [CrossRef]
- Maude, S.L.; Rheingold, D.T.; Rheingold, S.R.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Barker, C.S.; Callahan, C.; Frey, N.V.; Nazimuddin, F.; et al. Sustained remissions with CD19-specific chimeric antigen receptor (CAR)-modified T cells in children with relapsed/refractory ALL. J. Clin. Oncol. 2016, 34, 3011. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 2014, 6, 224ra25. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, P. How Cellular Immunotherapies Are Changing the Outlook for Cancer Patients. Available online: https://www.cancerresearch.org/immunotherapy/treatment-types/adoptive-cell-therapy (accessed on 19 June 2020).
- D’Aloia, M.M.; Zizzari, I.G.; Sacchetti, B.; Pierelli, L.; Alimandi, M. CAR-T cells: The long and winding road to solid tumors. Cell Death Dis. 2018, 9, 282. [Google Scholar] [CrossRef]
- Xu, X.; Sun, Q.; Liang, X.; Chen, Z.; Zhang, X.; Zhou, X.; Li, M.; Tu, H.; Liu, Y.; Tu, S.; et al. Mechanisms of Relapse After CD19 CAR T-Cell Therapy for Acute Lymphoblastic Leukemia and Its Prevention and Treatment Strategies. Front. Immunol. 2019, 10, 2664. [Google Scholar] [CrossRef] [PubMed]
- Bonifant, C.L.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncol. 2016, 3, 16011. [Google Scholar] [CrossRef]
- Lamers, C.H.; Sleijfer, S.; Vulto, A.G.; Kruit, W.H.; Kliffen, M.; Debets, R.; Gratama, J.W.; Stoter, G.; Oosterwijk, E. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: First clinical experience. J. Clin. Oncol. 2006, 24, e20–e22. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Hucks, G.E.; Seif, A.E.; Talekar, M.K.; Teachey, D.T.; Baniewicz, D.; Callahan, C.; Gonzalez, V.; Nazimuddin, F.; Gupta, M.; et al. The effect of pembrolizumab in combination with CD19-targeted chimeric antigen receptor (CAR) T cells in relapsed acute lymphoblastic leukemia (ALL). J. Clin. Oncol. 2017, 35 (Suppl. 15), 103. [Google Scholar] [CrossRef]
- Li, A.M.; Hucks, G.E.D.; Dinofia, A.M.; Seif, A.E.; Teachey, D.T.; Baniewicz, D.; Callahan, C.; Fasano, C.; McBride, B.; Gonzalez, V.; et al. Checkpoint Inhibitors Augment CD19-Directed Chimeric Antigen Receptor (CAR) T Cell Therapy in Relapsed B-Cell Acute Lymphoblastic Leukemia. Blood 2018, 132 (Suppl. 1), 556. [Google Scholar] [CrossRef]
- Yoon, D.H.; Osborn, M.J.; Tolar, J.; Kim, C.J. Incorporation of Immune Checkpoint Blockade into Chimeric Antigen Receptor T Cells (CAR-Ts): Combination or Built-In CAR-T. Int. J. Mol. Sci. 2018, 19, 340. [Google Scholar] [CrossRef]
- Chong, E.A.; Melenhorst, J.J.; Lacey, S.F.; Ambrose, D.E.; Gonzalez, V.; Levine, B.L.; June, C.H.; Schuster, S.J. PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: Refueling the CAR. Blood 2017, 129, 1039–1041. [Google Scholar] [CrossRef] [PubMed]
- El Chaer, F.; Holtzman, N.G.; Sausville, E.A.; Law, J.Y.; Lee, S.T.; Duong, V.H.; Baer, M.R.; Koka, R.; Singh, Z.N.; Hardy, N.M.; et al. Relapsed Philadelphia Chromosome-Positive Pre-B-ALL after CD19-Directed CAR-T Cell Therapy Successfully Treated with Combination of Blinatumomab and Ponatinib. Acta Haematol. 2019, 141, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, J.; Hirayama, A.V.; Purushe, J.; Hay, K.A.; Lymp, J.; Li, D.H.; Yeung, C.C.S.; Sheih, A.; Pender, B.S.; Hawkins, R.M.; et al. Feasibility and efficacy of CD19-targeted CAR T cells with concurrent ibrutinib for CLL after ibrutinib failure. Blood 2020, 135, 1650–1660. [Google Scholar] [CrossRef] [PubMed]
- Geukes Foppen, M.H.; Donia, M.; Svane, I.M.; Haanen, J.B. Tumor-infiltrating lymphocytes for the treatment of metastatic cancer. Mol. Oncol. 2015, 9, 1918–1935. [Google Scholar] [CrossRef]
- Banerjee, H.; Kane, L.P. Immune regulation by Tim-3. F1000 Research 2018, 7, 316. [Google Scholar] [CrossRef]
- Du, W.; Yang, M.; Turner, A.; Xu, C.; Ferris, R.L.; Huang, J.; Kane, L.P.; Lu, B. TIM-3 as a Target for Cancer Immunotherapy and Mechanisms of Action. Int. J. Mol. Sci. 2017, 18, 645. [Google Scholar] [CrossRef]
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 2010, 207, 2175–2186. [Google Scholar] [CrossRef]
- Sakuishi, K.; Apetoh, L.; Sullivan, J.M.; Blazar, B.R.; Kuchroo, V.K.; Anderson, A.C. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 2010, 207, 2187–2194. [Google Scholar] [CrossRef]
- Shayan, G.; Srivastava, R.; Li, J.; Schmitt, N.; Kane, L.P.; Ferris, R.L. Adaptive resistance to anti-PD1 therapy by Tim-3 upregulation is mediated by the PI3K-Akt pathway in head and neck cancer. Oncoimmunology 2017, 6, e1261779. [Google Scholar] [CrossRef]
- Gao, X.; Zhu, Y.; Li, G.; Huang, H.; Zhang, G.; Wang, F.; Sun, J.; Yang, Q.; Zhang, X.; Lu, B. TIM-3 expression characterizes regulatory T cells in tumor tissues and is associated with lung cancer progression. PLoS ONE 2012, 7, e30676. [Google Scholar] [CrossRef]
- Ngiow, S.F.; von Scheidt, B.; Akiba, H.; Yagita, H.; Teng, M.W.; Smyth, M.J. Anti-TIM3 antibody promotes T cell IFN-gamma-mediated antitumor immunity and suppresses established tumors. Cancer Res. 2011, 71, 3540–3551. [Google Scholar] [CrossRef]
- Li, J.; Shayan, G.; Avery, L.; Jie, H.B.; Gildener-Leapman, N.; Schmitt, N.; Lu, B.F.; Kane, L.P.; Ferris, R.L. Tumor-infiltrating Tim-3(+) T cells proliferate avidly except when PD-1 is co-expressed: Evidence for intracellular cross talk. Oncoimmunology 2016, 5, e1200778. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Cao, J.; Zhao, C.; Li, X.; Zhou, C.; Hirsch, F.R. TIM-3, a promising target for cancer immunotherapy. Oncol. Targets Ther. 2018, 11, 7005–7009. [Google Scholar] [CrossRef] [PubMed]
- Harding, J.J.; Patnaik, A.; Moreno, V.; Stein, M.; Jankowska, A.M.; Velez de Mendizabal, N.; Tina Liu, Z.; Koneru, M.; Calvo, E. A phase Ia/Ib study of an anti-TIM-3 antibody (LY3321367) monotherapy or in combination with an anti-PD-L1 antibody (LY3300054): Interim safety, efficacy, and pharmacokinetic findings in advanced cancers. JCO 2019, 37, 12. [Google Scholar] [CrossRef]
- Curigliano, G.G.H.; Mach, N.; Doi, T.; Tai, W.M.D.; Forde, P.; Sarantopoulos, J.; Bedard, P.L.; Lin, C.-C.; Hodi, S.; Wilgenhof, S.; et al. Abstract CT183: Phase (Ph) I/II study of MBG453± spartalizumab (PDR001) in patients (pts) with advanced malignancies. Cancer Res. 2019, 79 (Suppl. 13), CT183. [Google Scholar]
- Lines, J.L.; Sempere, L.F.; Broughton, T.; Wang, L.; Noelle, R. VISTA is a novel broad-spectrum negative checkpoint regulator for cancer immunotherapy. Cancer Immunol. Res. 2014, 2, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Lines, J.L.; Pantazi, E.; Mak, J.; Sempere, L.F.; Wang, L.; O’Connell, S.; Ceeraz, S.; Suriawinata, A.A.; Yan, S.; Ernstoff, M.S.; et al. VISTA is an immune checkpoint molecule for human T cells. Cancer Res. 2014, 74, 1924–1932. [Google Scholar] [CrossRef] [PubMed]
- Mulati, K.; Hamanishi, J.; Matsumura, N.; Chamoto, K.; Mise, N.; Abiko, K.; Baba, T.; Yamaguchi, K.; Horikawa, N.; Murakami, R.; et al. VISTA expressed in tumour cells regulates T cell function. Br. J. Cancer 2019, 120, 115–127. [Google Scholar] [CrossRef]
- Wang, L.; Jia, B.; Claxton, D.F.; Ehmann, W.C.; Rybka, W.B.; Mineishi, S.; Naik, S.; Khawaja, M.R.; Sivik, J.; Han, J.; et al. VISTA is highly expressed on MDSCs and mediates an inhibition of T cell response in patients with AML. Oncoimmunology 2018, 7, e1469594. [Google Scholar] [CrossRef]
- Nowak, E.C.; Lines, J.L.; Varn, F.S.; Deng, J.; Sarde, A.; Mabaera, R.; Kuta, A.; Le Mercier, I.; Cheng, C.; Noelle, R.J. Immunoregulatory functions of VISTA. Immunol. Rev. 2017, 276, 66–79. [Google Scholar] [CrossRef]
- Le Mercier, I.; Chen, W.; Lines, J.L.; Day, M.; Li, J.; Sergent, P.; Noelle, R.J.; Wang, L. VISTA Regulates the Development of Protective Antitumor Immunity. Cancer Res 2014, 74, 1933–1944. [Google Scholar] [CrossRef] [PubMed]
- Curis Announces FDA Clearance of IND Application for CI-8993, the First-In-Class Monoclonal Anti-VISTA Antibody. Available online: https://www.prnewswire.com/news-releases/curis-announces-fda-clearance-of-ind-application-for-ci-8993-the-first-in-class-monoclonal-anti-vista-antibody-301073426.html (accessed on 19 June 2020).
- Bukczynski, J.; Wen, T.; Ellefsen, K.; Gauldie, J.; Watts, T.H. Costimulatory ligand 4-1BBL (CD137L) as an efficient adjuvant for human antiviral cytotoxic T cell responses. Proc. Natl. Acad. Sci. USA 2004, 101, 1291–1296. [Google Scholar] [CrossRef] [PubMed]
- Menk, A.V.; Scharping, N.E.; Rivadeneira, D.B.; Calderon, M.J.; Watson, M.J.; Dunstane, D.; Watkins, S.C.; Delgoffe, G.M. 4-1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. J. Exp. Med. 2018, 215, 1091–1100. [Google Scholar] [CrossRef] [PubMed]
- Maus, M.V.; Thomas, A.K.; Leonard, D.G.; Allman, D.; Addya, K.; Schlienger, K.; Riley, J.L.; June, C.H. Ex vivo expansion of polyclonal and antigen-specific cytotoxic T lymphocytes by artificial APCs expressing ligands for the T-cell receptor, CD28 and 4-1BB. Nat. Biotechnol. 2002, 20, 143–148. [Google Scholar] [CrossRef]
- Ying, Z.; He, T.; Wang, X.; Zheng, W.; Lin, N.; Tu, M.; Xie, Y.; Ping, L.; Zhang, C.; Liu, W.; et al. Parallel Comparison of 4-1BB or CD28 Co-stimulated CD19-Targeted CAR-T Cells for B Cell Non-Hodgkin’s Lymphoma. Mol. Ther. Oncol. 2019, 15, 60–68. [Google Scholar] [CrossRef]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, Ö.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef]
- Schuster, S.J.; Svoboda, J.; Nasta, S.D.; Chong, E.A.; Winchell, N.; Landsburg, D.J.; Porter, D.; Mato, A.R.; Strauser, H.T.; Schrank-Hacker, A.M.; et al. Treatment with chimeric antigen receptor modified T cells directed against CD19 (CTL019) results in durable remissions in patients with relapsed or refractory diffuse large B cell lymphomas of germinal center and non-germinal center origin, “double hit” diffuse large B cell lymphomas, and transformed follicular to diffuse large B cell lymphomas [Abstract 3026]. Blood 2016, 128, 3026. [Google Scholar] [CrossRef]
- Guedan, S.; Posey, A.D., Jr.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 2018, 3, e96976. [Google Scholar] [CrossRef]
- Polesso, F.; Sarker, M.; Weinberg, A.D.; Murray, S.E.; Moran, A.E. OX40 Agonist Tumor Immunotherapy Does Not Impact Regulatory T Cell Suppressive Function. J. Immunol. 2019, 203, 2011–2019. [Google Scholar] [CrossRef]
- Taraban, V.Y.; Rowley, T.F.; O’Brien, L.; Chan, H.T.; Haswell, L.E.; Green, M.H.; Tutt, A.L.; Glennie, M.J.; Al-Shamkhani, A. Expression and costimulatory effects of the TNF receptor superfamily members CD134 (OX40) and CD137 (4-1BB), and their role in the generation of anti-tumor immune responses. Eur. J. Immunol. 2002, 32, 3617–3627. [Google Scholar] [CrossRef]
- Deng, J.; Zhao, S.; Zhang, X.; Jia, K.; Wang, H.; Zhou, C.; He, Y. OX40 (CD134) and OX40 ligand, important immune checkpoints in cancer. Oncol. Targets Ther. 2019, 12, 7347–7353. [Google Scholar] [CrossRef] [PubMed]
- Vu, M.D.; Xiao, X.; Gao, W.; Degauque, N.; Chen, M.; Kroemer, A.; Killeen, N.; Ishii, N.; Li, X.C. OX40 costimulation turns off Foxp3+ Tregs. Blood 2007, 110, 2501–2510. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, J.A.; Reidy, A.; Mirandola, L.; Trotter, K.; Suvorava, N.; Figueroa, A.; Konala, V.; Aulakh, A.; Littlefield, L.; Grizzi, F.; et al. Chimeric antigen receptor engineering: A right step in the evolution of adoptive cellular immunotherapy. Int. Rev. Immunol. 2015, 34, 154–187. [Google Scholar] [CrossRef] [PubMed]
- Knochelmann, H.M.; Smith, A.S.; Dwyer, C.J.; Wyatt, M.M.; Mehrotra, S.; Paulos, C.M. CAR T Cells in Solid Tumors: Blueprints for Building Effective Therapies. Front. Immunol. 2018, 9, 1740. [Google Scholar] [CrossRef]
- Barrett, D.M.; Grupp, S.A.; June, C.H. Chimeric Antigen Receptor- and TCR-Modified T Cells Enter Main Street and Wall Street. J. Immunol. 2015, 195, 755–761. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A.; et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef]
- Clinical Trials Using Tumor Infiltrating Lymphocyte Therapy. Available online: https://www.cancer.gov/about-cancer/treatment/clinical-trials/intervention/tumor-infiltrating-lymphocyte-therapy (accessed on 19 June 2020).
- Rohaan, M.W.; van den Berg, J.H.; Kvistborg, P.; Haanen, J. Adoptive transfer of tumor-infiltrating lymphocytes in melanoma: A viable treatment option. J. Immunother. Cancer 2018, 6, 102. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ (accessed on 19 June 2020).
- Zhao, Z.; Chen, Y.; Francisco, N.M.; Zhang, Y.; Wu, M. The application of CAR-T cell therapy in hematological malignancies: Advantages and challenges. Acta Pharm. Sin. B 2018, 8, 539–551. [Google Scholar] [CrossRef]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef]
- Styczyński, J. A brief history of CAR-T cells: From laboratory to the bedside. Acta Haematol. Pol. 2020, 51. [Google Scholar] [CrossRef]
- Levine, B.L. Performance-enhancing drugs: Design and production of redirected chimeric antigen receptor (CAR) T cells. Cancer Gene Ther. 2015, 22, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Elahi, R.; Khosh, E.; Tahmasebi, S.; Esmaeilzadeh, A. Immune Cell Hacking: Challenges and Clinical Approaches to Create Smarter Generations of Chimeric Antigen Receptor T Cells. Front. Immunol. 2018, 9, 1717. [Google Scholar] [CrossRef] [PubMed]
- Maus, M.V.; Levine, B.L. Chimeric Antigen Receptor T-Cell Therapy for the Community Oncologist. Oncologist 2016, 21, 608–617. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Lin, Q.; Song, Y.; Liu, D. Universal CARs, universal T cells, and universal CAR T cells. J. Hematol. Oncol. 2018, 11, 132. [Google Scholar] [CrossRef]
- Filley, A.C.; Henriquez, M.; Dey, M. CART Immunotherapy: Development, Success, and Translation to Malignant Gliomas and Other Solid Tumors. Front. Oncol. 2018, 8, 453. [Google Scholar] [CrossRef]
- Kalbasi, A.; June, C.H.; Haas, N.; Vapiwala, N. Radiation and immunotherapy: A synergistic combination. J. Clin. Investig. 2013, 123, 2756–2763. [Google Scholar] [CrossRef]
- Klopp, A.H.; Spaeth, E.L.; Dembinski, J.L.; Woodward, W.A.; Munshi, A.; Meyn, R.E.; Cox, J.D.; Andreeff, M.; Marini, F.C. Tumor irradiation increases the recruitment of circulating mesenchymal stem cells into the tumor microenvironment. Cancer Res. 2007, 67, 11687–11695. [Google Scholar] [CrossRef]
- Morgan, G.W.; Breit, S.N. Radiation and the lung: A reevaluation of the mechanisms mediating pulmonary injury. Int. J. Radiat. Oncol. Biol. Phys. 1995, 31, 361–369. [Google Scholar] [CrossRef]
- Cox, J.D. Are the results of RTOG 0617 mysterious? Int. J. Radiat. Oncol. Biol. Phys. 2012, 82, 1042–1044. [Google Scholar] [CrossRef]
- Davis, A.A.; Patel, V.G. The role of PD-L1 expression as a predictive biomarker: An analysis of all US Food and Drug Administration (FDA) approvals of immune checkpoint inhibitors. J. Immunother. Cancer 2019, 7, 278. [Google Scholar] [CrossRef]
- Pu, X.; Wu, L.; Su, D.; Mao, W.; Fang, B. Immunotherapy for non-small cell lung cancers: Biomarkers for predicting responses and strategies to overcome resistance. BMC Cancer 2018, 18, 1082. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [PubMed]
- George, A.P.; Kuzel, T.M.; Zhang, Y.; Zhang, B. The Discovery of Biomarkers in Cancer Immunotherapy. Comput. Struct. Biotechnol. J. 2019, 17, 484–497. [Google Scholar] [CrossRef] [PubMed]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Immunotherapy | Combination | Successes/Advantages | Limitations | References |
|---|---|---|---|---|
| Checkpoint Inhibitors | Checkpoint Inhibitors |
|
| [11,29,52,53,54,55,56,57,58,59,65,66,67,68,69,70] |
| Chemotherapy |
|
| [30,31,32,33,35,36,50,64] | |
| Radiotherapy |
|
| [71,72,73,74,75,76,77,78,79] | |
| Cancer Vaccines | Checkpoint Inhibitors |
|
| [48,80,81,82,83] |
| Co-stimulatory Molecule Agonists | Checkpoint Inhibitors |
|
| [84,85,86,87,88,89,90] |
| Tumor Infiltrating Lymphocyte (TIL) and Chimeric Antigen Receptor (CAR) T Cell Therapy | Checkpoint Inhibitors, Chemotherapy |
|
| [91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbari, C.; Fontaine, T.; Parajuli, P.; Lamichhane, N.; Jakubski, S.; Lamichhane, P.; Deshmukh, R.R. Immunotherapies and Combination Strategies for Immuno-Oncology. Int. J. Mol. Sci. 2020, 21, 5009. https://doi.org/10.3390/ijms21145009
Barbari C, Fontaine T, Parajuli P, Lamichhane N, Jakubski S, Lamichhane P, Deshmukh RR. Immunotherapies and Combination Strategies for Immuno-Oncology. International Journal of Molecular Sciences. 2020; 21(14):5009. https://doi.org/10.3390/ijms21145009
Chicago/Turabian StyleBarbari, Cody, Tyler Fontaine, Priyanka Parajuli, Narottam Lamichhane, Silvia Jakubski, Purushottam Lamichhane, and Rahul R. Deshmukh. 2020. "Immunotherapies and Combination Strategies for Immuno-Oncology" International Journal of Molecular Sciences 21, no. 14: 5009. https://doi.org/10.3390/ijms21145009
APA StyleBarbari, C., Fontaine, T., Parajuli, P., Lamichhane, N., Jakubski, S., Lamichhane, P., & Deshmukh, R. R. (2020). Immunotherapies and Combination Strategies for Immuno-Oncology. International Journal of Molecular Sciences, 21(14), 5009. https://doi.org/10.3390/ijms21145009