Zinc and Autophagy in Age-Related Macular Degeneration

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
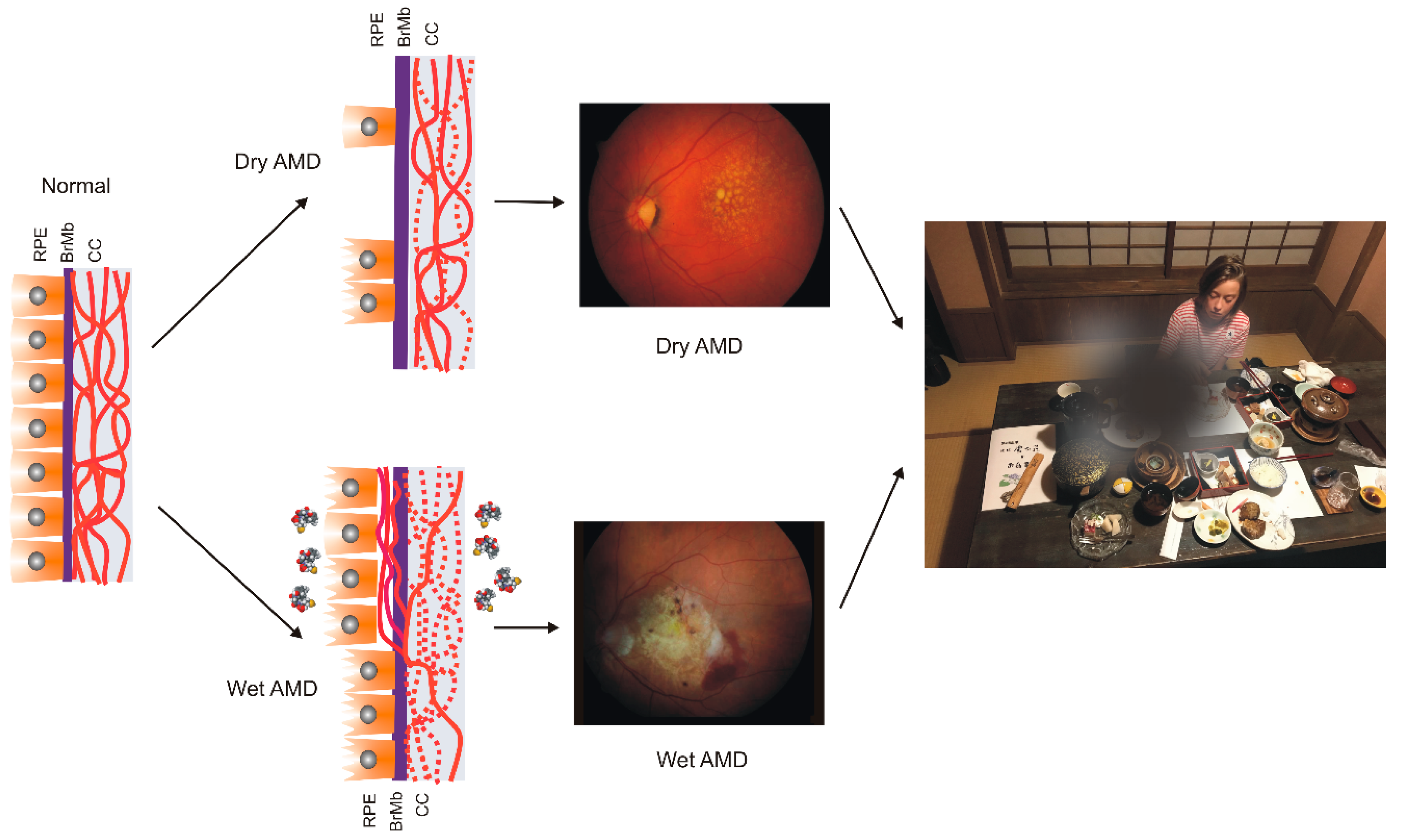
2. Age-Related Macular Degeneration
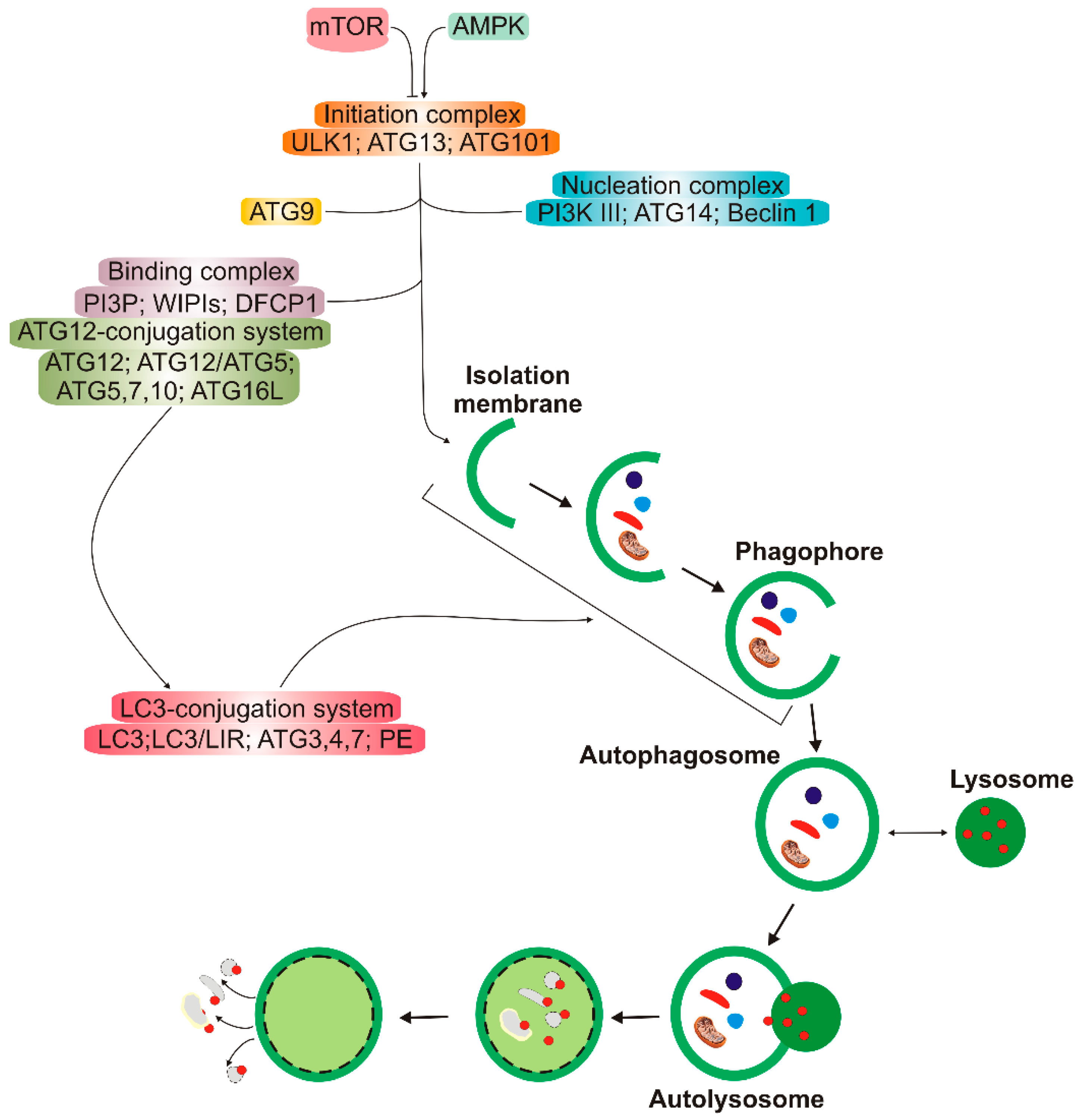
3. Autophagy in Age-Related Macular Degeneration
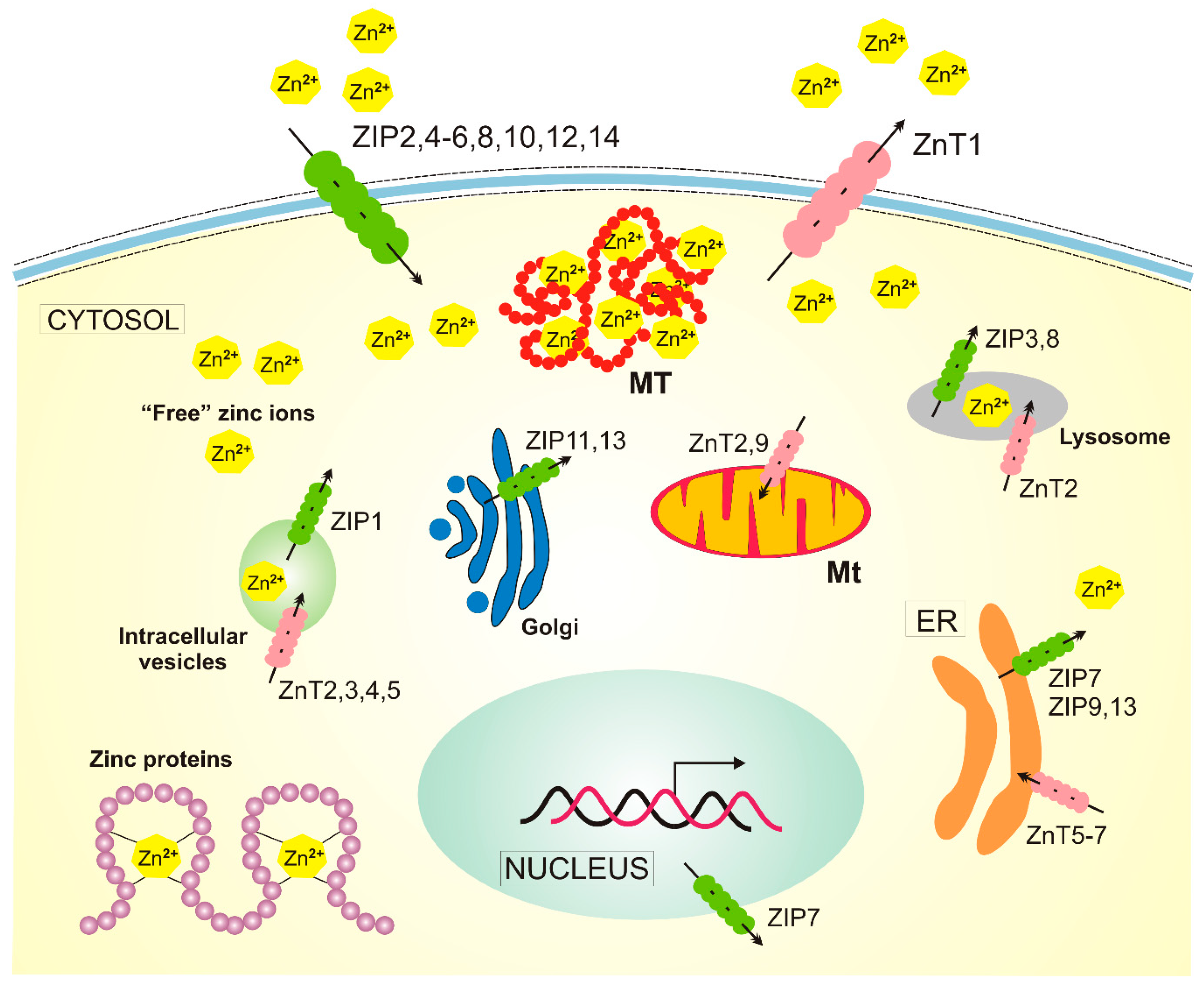
4. Zinc in Age-Related Macular Degeneration
5. Zinc and Autophagy
6. Zinc and Autophagy in AMD
7. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gilbert, R.; Peto, T.; Lengyel, I.; Emri, E. Zinc Nutrition and Inflammation in the Aging Retina. Mol. Nutr. Food Res. 2019, 63, e1801049. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.; Rubinsztein, D.C.; Walker, D.W. Autophagy as a promoter of longevity: Insights from model organisms. Nat. Rev. Mol. Cell Biol. 2018, 19, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Kaarniranta, K.; Sinha, D.; Blasiak, J.; Kauppinen, A.; Vereb, Z.; Salminen, A.; Boulton, M.E.; Petrovski, G. Autophagy and heterophagy dysregulation leads to retinal pigment epithelium dysfunction and development of age-related macular degeneration. Autophagy 2013, 9, 973–984. [Google Scholar] [CrossRef]
- Liuzzi, J.P.; Guo, L.; Yoo, C.; Stewart, T.S. Zinc and autophagy. Biomet. Int. J. Role Met. Ions Biol. Biochem. Med. 2014, 27, 1087–1096. [Google Scholar] [CrossRef]
- Colijn, J.M.; Buitendijk, G.H.S.; Prokofyeva, E.; Alves, D.; Cachulo, M.L.; Khawaja, A.P.; Cougnard-Gregoire, A.; Merle, B.M.J.; Korb, C.; Erke, M.G.; et al. Prevalence of Age-Related Macular Degeneration in Europe: The Past and the Future. Ophthalmology 2017, 124, 1753–1763. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116. [Google Scholar] [CrossRef]
- Ratnayaka, J.A.; Serpell, L.C.; Lotery, A.J. Dementia of the eye: The role of amyloid beta in retinal degeneration. Eye 2015, 29, 1013–1026. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Salminen, A.; Haapasalo, A.; Soininen, H.; Hiltunen, M. Age-related macular degeneration (AMD): Alzheimer’s disease in the eye? J. Alzheimers Dis. JAD 2011, 24, 615–631. [Google Scholar] [CrossRef]
- Yoneyama, S.; Sakurada, Y.; Kikushima, W.; Sugiyama, A.; Matsubara, M.; Fukuda, Y.; Tanabe, N.; Parikh, R.; Mabuchi, F.; Kashiwagi, K.; et al. Genetic factors associated with response to as-needed aflibercept therapy for typical neovascular age-related macular degeneration and polypoidal choroidal vasculopathy. Sci. Rep. 2020, 10, 7188. [Google Scholar] [CrossRef]
- Golestaneh, N.; Chu, Y.; Xiao, Y.Y.; Stoleru, G.L.; Theos, A.C. Dysfunctional autophagy in RPE, a contributing factor in age-related macular degeneration. Cell Death Dis. 2017, 8, e2537. [Google Scholar] [CrossRef]
- Nakatogawa, H. Mechanisms governing autophagosome biogenesis. Nat. Rev. Mol. Cell Biol. 2020. [Google Scholar] [CrossRef]
- Klionsky, D.J. Autophagy participates in, well, just about everything. Cell Death Differ. 2020, 27, 831–832. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Lipton, S.A. Molecular mechanisms of nitrosative stress-mediated protein misfolding in neurodegenerative diseases. Cell. Mol. Life Sci. CMLS 2007, 64, 1609–1620. [Google Scholar] [CrossRef] [PubMed]
- Zientara-Rytter, K.; Subramani, S. The Roles of Ubiquitin-Binding Protein Shuttles in the Degradative Fate of Ubiquitinated Proteins in the Ubiquitin-Proteasome System and Autophagy. Cells 2019, 8, 40. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, A.; Bertolotti, A. Regulation of proteasome assembly and activity in health and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 697–712. [Google Scholar] [CrossRef]
- Kauppinen, A.; Paterno, J.J.; Blasiak, J.; Salminen, A.; Kaarniranta, K. Inflammation and its role in age-related macular degeneration. Cell. Mol. Life Sci. CMLS 2016, 73, 1765–1786. [Google Scholar] [CrossRef] [PubMed]
- Kosmidou, C.; Efstathiou, N.E.; Hoang, M.V.; Notomi, S.; Konstantinou, E.K.; Hirano, M.; Takahashi, K.; Maidana, D.E.; Tsoka, P.; Young, L.; et al. Issues with the Specificity of Immunological Reagents for NLRP3: Implications for Age-related Macular Degeneration. Sci. Rep. 2018, 8, 461. [Google Scholar] [CrossRef] [PubMed]
- Brunk, U.T.; Terman, A. Lipofuscin: Mechanisms of age-related accumulation and influence on cell function. Free Radic. Biol. Med. 2002, 33, 611–619. [Google Scholar] [CrossRef]
- Mitter, S.K.; Song, C.; Qi, X.; Mao, H.; Rao, H.; Akin, D.; Lewin, A.; Grant, M.; Dunn, W., Jr.; Ding, J.; et al. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy 2014, 10, 1989–2005. [Google Scholar] [CrossRef]
- Zhang, J.; Bai, Y.; Huang, L.; Qi, Y.; Zhang, Q.; Li, S.; Wu, Y.; Li, X. Protective effect of autophagy on human retinal pigment epithelial cells against lipofuscin fluorophore A2E: Implications for age-related macular degeneration. Cell Death Dis. 2015, 6, e1972. [Google Scholar] [CrossRef]
- Wang, L.; Ye, X.; Zhao, T. The physiological roles of autophagy in the mammalian life cycle. Biol. Rev. 2019, 94, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Denton, D.; Xu, T.; Kumar, S. Autophagy as a pro-death pathway. Immunol. Cell Biol. 2015, 93, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Bafaro, E.; Liu, Y.; Xu, Y.; Dempski, R.E. The emerging role of zinc transporters in cellular homeostasis and cancer. Signal Transduct. Target. Ther. 2017, 2, 17029. [Google Scholar] [CrossRef]
- King, J.C.; Shames, D.M.; Woodhouse, L.R. Zinc homeostasis in humans. J. Nutr. 2000, 130, 1360s–1366s. [Google Scholar] [CrossRef] [PubMed]
- Yuan, N.; Wang, Y.H.; Li, K.J.; Zhao, Y.; Hu, X.; Mao, L.; Zhao, W.J.; Lian, H.Z.; Zheng, W.J. Effects of exogenous zinc on the cellular zinc distribution and cell cycle of A549 cells. Biosci. Biotechnol. Biochem. 2012, 76, 2014–2020. [Google Scholar] [CrossRef][Green Version]
- Brown, K.H.; Rivera, J.A.; Bhutta, Z.; Gibson, R.S.; King, J.C.; Lönnerdal, B.; Ruel, M.T.; Sandtröm, B.; Wasantwisut, E.; Hotz, C. International Zinc Nutrition Consultative Group (IZiNCG) technical document #1. Assessment of the risk of zinc deficiency in populations and options for its control. Food Nutr. Bull. 2004, 25, S99–S203. [Google Scholar] [PubMed]
- Krężel, A.; Maret, W. The biological inorganic chemistry of zinc ions. Arch. Biochem. Biophys. 2016, 611, 3–19. [Google Scholar] [CrossRef]
- Maret, W. Zinc biochemistry: From a single zinc enzyme to a key element of life. Adv. Nutr. 2013, 4, 82–91. [Google Scholar] [CrossRef]
- Sekler, I.; Sensi, S.L.; Hershfinkel, M.; Silverman, W.F. Mechanism and regulation of cellular zinc transport. Mol. Med. 2007, 13, 337–343. [Google Scholar] [CrossRef]
- McCall, K.A.; Huang, C.; Fierke, C.A. Function and mechanism of zinc metalloenzymes. J. Nutr. 2000, 130, 1437s–1446s. [Google Scholar] [CrossRef]
- Andreini, C.; Banci, L.; Bertini, I.; Rosato, A. Counting the zinc-proteins encoded in the human genome. J. Proteom. Res. 2006, 5, 196–201. [Google Scholar] [CrossRef]
- Newsome, D.A.; Miceli, M.V.; Tate, D.J., Jr.; Alcock, N.W.; Oliver, P.D. Zinc content of human retinal pigment epithelium decreases with age and macular degeneration, but superoxide dismutase activity increases. J. Trace Elem. Exp. Med. 1996, 8, 193–199. [Google Scholar] [CrossRef]
- Ripps, H.; Chappell, R.L. Review: Zinc’s functional significance in the vertebrate retina. Mol. Vis. 2014, 20, 1067–1074. [Google Scholar]
- Organisciak, D.; Wong, P.; Rapp, C.; Darrow, R.; Ziesel, A.; Rangarajan, R.; Lang, J. Light-induced retinal degeneration is prevented by zinc, a component in the age-related eye disease study formulation. Photochem. Photobiol. 2012, 88, 1396–1407. [Google Scholar] [CrossRef]
- Anastassov, I.; Ripps, H.; Chappell, R.L. Cytoprotection by endogenous zinc in the vertebrate retina. J. Neurochem. 2014, 129, 249–255. [Google Scholar] [CrossRef]
- Prasad, A.S. Impact of the discovery of human zinc deficiency on health. J. Trace Elem. Med. Biol. Organ Soc. Miner. Trace Elem. (GMS) 2014, 28, 357–363. [Google Scholar] [CrossRef]
- Carneiro, Â.; Andrade, J.P. Erratum to “Nutritional and Lifestyle Interventions for Age-Related Macular Degeneration: A Review”. Oxid. Med. Cell. Longev. 2017, 2017, 2435963. [Google Scholar] [CrossRef]
- Ugarte, M.; Osborne, N.N. Recent advances in the understanding of the role of zinc in ocular tissues. Metallomics Integr. Biomet. Sci. 2014, 6, 189–200. [Google Scholar] [CrossRef]
- Newsome, D.A.; Swartz, M.; Leone, N.C.; Elston, R.C.; Miller, E. Oral zinc in macular degeneration. Arch. Ophthal. 1988, 106, 192–198. [Google Scholar] [CrossRef]
- Chew, E.Y.; Clemons, T.E.; Agrón, E.; Sperduto, R.D.; Sangiovanni, J.P.; Kurinij, N.; Davis, M.D. Long-term effects of vitamins C and E, β-carotene, and zinc on age-related macular degeneration: AREDS report no. 35. Ophthalmology 2013, 120, 1604–1611.e1604. [Google Scholar] [CrossRef]
- Newsome, D.A. A randomized, prospective, placebo-controlled clinical trial of a novel zinc-monocysteine compound in age-related macular degeneration. Curr. Eye Res. 2008, 33, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.; Markey, M.; Rapp, C.M.; Darrow, R.M.; Ziesel, A.; Organisciak, D.T. Enhancing the efficacy of AREDS antioxidants in light-induced retinal degeneration. Mol. Vis. 2017, 23, 718–739. [Google Scholar] [PubMed]
- Gopinath, B.; Liew, G.; Russell, J.; Cosatto, V.; Burlutsky, G.; Mitchell, P. Intake of key micronutrients and food groups in patients with late-stage age-related macular degeneration compared with age-sex-matched controls. Br. J. Ophthalmol. 2017, 101, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.S.; Wang, J.J.; Flood, V.; Rochtchina, E.; Smith, W.; Mitchell, P. Dietary antioxidants and the long-term incidence of age-related macular degeneration: The Blue Mountains Eye Study. Ophthalmology 2008, 115, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Van Leeuwen, R.; Boekhoorn, S.; Vingerling, J.R.; Witteman, J.C.; Klaver, C.C.; Hofman, A.; de Jong, P.T. Dietary intake of antioxidants and risk of age-related macular degeneration. JAMA 2005, 294, 3101–3107. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Bedell, M.; Zhang, K. Age-related macular degeneration: Genetic and environmental factors of disease. Mol. Interv. 2010, 10, 271–281. [Google Scholar] [CrossRef]
- Ho, L.; van Leeuwen, R.; Witteman, J.C.; van Duijn, C.M.; Uitterlinden, A.G.; Hofman, A.; de Jong, P.T.; Vingerling, J.R.; Klaver, C.C. Reducing the genetic risk of age-related macular degeneration with dietary antioxidants, zinc, and ω-3 fatty acids: The Rotterdam study. Arch. Ophthalmol. 2011, 129, 758–766. [Google Scholar] [CrossRef] [PubMed]
- Awh, C.C.; Hawken, S.; Zanke, B.W. Treatment response to antioxidants and zinc based on CFH and ARMS2 genetic risk allele number in the Age-Related Eye Disease Study. Ophthalmology 2015, 122, 162–169. [Google Scholar] [CrossRef]
- Awh, C.C.; Lane, A.M.; Hawken, S.; Zanke, B.; Kim, I.K. CFH and ARMS2 genetic polymorphisms predict response to antioxidants and zinc in patients with age-related macular degeneration. Ophthalmology 2013, 120, 2317–2323. [Google Scholar] [CrossRef]
- Assel, M.J.; Li, F.; Wang, Y.; Allen, A.S.; Baggerly, K.A.; Vickers, A.J. Genetic Polymorphisms of CFH and ARMS2 Do Not Predict Response to Antioxidants and Zinc in Patients with Age-Related Macular Degeneration: Independent Statistical Evaluations of Data from the Age-Related Eye Disease Study. Ophthalmology 2018, 125, 391–397. [Google Scholar] [CrossRef]
- Cho, E.; Stampfer, M.J.; Seddon, J.M.; Hung, S.; Spiegelman, D.; Rimm, E.B.; Willett, W.C.; Hankinson, S.E. Prospective study of zinc intake and the risk of age-related macular degeneration. Ann. Epidemiol. 2001, 11, 328–336. [Google Scholar] [CrossRef]
- Chong, E.W.; Wong, T.Y.; Kreis, A.J.; Simpson, J.A.; Guymer, R.H. Dietary antioxidants and primary prevention of age related macular degeneration: Systematic review and meta-analysis. BMJ (Clin. Res. Ed.) 2007, 335, 755. [Google Scholar] [CrossRef]
- Evans, J.R. Antioxidant vitamin and mineral supplements for slowing the progression of age-related macular degeneration. Cochrane Database Systematic Rev. 2006. [Google Scholar] [CrossRef]
- Evans, J.R.; Henshaw, K. Antioxidant vitamin and mineral supplements for preventing age-related macular degeneration. Cochrane Database Systematic Rev. 2008. [Google Scholar] [CrossRef]
- Blom, A.M.; Kask, L.; Ramesh, B.; Hillarp, A. Effects of zinc on factor I cofactor activity of C4b-binding protein and factor H. Arch. Biochem. Biophys. 2003, 418, 108–118. [Google Scholar] [CrossRef]
- Evans, B.D.; Amiraian, K. Effect of zinc on lysis of sheep erythrocytes by guinea pig C4-deficient serum. Mol. Immunol. 1987, 24, 1345–1350. [Google Scholar] [CrossRef]
- Jarosz, M.; Olbert, M.; Wyszogrodzka, G.; Młyniec, K.; Librowski, T. Antioxidant and anti-inflammatory effects of zinc. Zinc-dependent NF-κB signaling. Inflammopharmacology 2017, 25, 11–24. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Flinn, J.M.; Kakalec, P.; Tappero, R.; Jones, B.; Lengyel, I. Correlations in distribution and concentration of calcium, copper and iron with zinc in isolated extracellular deposits associated with age-related macular degeneration. Metallomics Integr. Biomet. Sci. 2014, 6, 1223–1228. [Google Scholar] [CrossRef]
- Van Kuijk, F.; McPherson, S.W.; Roehrich, H. Enhanced Detection of Sub-Retinal Pigment Epithelial Cell Layer Deposits in Human and Murine Tissue: Imaging Zinc as a Biomarker for Age-Related Macular Degeneration (An American Ophthalmological Society Thesis). Trans. Am. Ophthalmol. Soc. 2017, 115, T3. [Google Scholar] [PubMed]
- Vishwanathan, R.; Chung, M.; Johnson, E.J. A Systematic Review on Zinc for the Prevention and Treatment of Age-Related Macular Degeneration. Investig. Ophthal. Vis. Sci. 2013, 54, 3985–3998. [Google Scholar] [CrossRef] [PubMed]
- Wills, N.K.; Ramanujam, V.M.; Kalariya, N.; Lewis, J.R.; van Kuijk, F.J. Copper and zinc distribution in the human retina: Relationship to cadmium accumulation, age, and gender. Exp. Eye Res. 2008, 87, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.S. Zinc: An antioxidant and anti-inflammatory agent: Role of zinc in degenerative disorders of aging. J. Trace Elem. Med. Biol. 2014, 28, 364–371. [Google Scholar] [CrossRef]
- Domenech, E.B.; Marfany, G. The Relevance of Oxidative Stress in the Pathogenesis and Therapy of Retinal Dystrophies. Antioxidants 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Zhang, Q.; Xu, L.; Lin, X.; Fu, J.; Wang, X.; Liu, Y.; Lin, Y.; Li, B.; Wang, R.; et al. Zinc is essential for the transcription function of the PGC-1α/Nrf2 signaling pathway in human primary endometrial stromal cells. Am. J. Physiol. Cell Physiol. 2020, 318, C640–C648. [Google Scholar] [CrossRef]
- Felszeghy, S.; Viiri, J.; Paterno, J.J.; Hyttinen, J.M.T.; Koskela, A.; Chen, M.; Leinonen, H.; Tanila, H.; Kivinen, N.; Koistinen, A.; et al. Loss of NRF-2 and PGC-1alpha genes leads to retinal pigment epithelium damage resembling dry age-related macular degeneration. Redox Biol. 2019, 20, 1–12. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Kajdanek, J.; Morawiec, J.; Pawlowska, E.; Blasiak, J. PGC-1α Protects RPE Cells of the Aging Retina against Oxidative Stress-Induced Degeneration through the Regulation of Senescence and Mitochondrial Quality Control. The Significance for AMD Pathogenesis. Int. J. Mol. Sci. 2018, 19, 2317. [Google Scholar] [CrossRef]
- Ding, B.; Zhong, Q. Zinc deficiency: An unexpected trigger for autophagy. J. Biol. Chem. 2017, 292, 8531–8532. [Google Scholar] [CrossRef]
- Kawamata, T.; Horie, T.; Matsunami, M.; Sasaki, M.; Ohsumi, Y. Zinc starvation induces autophagy in yeast. J. Biol. Chem. 2017, 292, 8520–8530. [Google Scholar] [CrossRef]
- Shinozaki, D.; Merkulova, E.A.; Naya, L.; Horie, T.; Kanno, Y.; Seo, M.; Ohsumi, Y.; Masclaux-Daubresse, C.; Yoshimoto, K. Autophagy Increases Zinc Bioavailability to Avoid Light-Mediated Reactive Oxygen Species Production under Zinc Deficiency. Plant Physiol. 2020, 182, 1284–1296. [Google Scholar] [CrossRef]
- Hwang, J.J.; Kim, H.N.; Kim, J.; Cho, D.H.; Kim, M.J.; Kim, Y.S.; Kim, Y.; Park, S.J.; Koh, J.Y. Zinc(II) ion mediates tamoxifen-induced autophagy and cell death in MCF-7 breast cancer cell line. Biomet. Int. J. Role Met. Ions Biol. Biochem. Med. 2010, 23, 997–1013. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Koh, J.Y. Roles of zinc and metallothionein-3 in oxidative stress-induced lysosomal dysfunction, cell death, and autophagy in neurons and astrocytes. Mol. Brain 2010, 3, 30. [Google Scholar] [CrossRef] [PubMed]
- Hung, H.H.; Huang, W.P.; Pan, C.Y. Dopamine- and zinc-induced autophagosome formation facilitates PC12 cell survival. Cell Biol. Toxicol. 2013, 29, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Liuzzi, J.P.; Yoo, C. Role of zinc in the regulation of autophagy during ethanol exposure in human hepatoma cells. Biol. Trace Elem. Res. 2013, 156, 350–356. [Google Scholar] [CrossRef]
- Botti, J.; Djavaheri-Mergny, M.; Pilatte, Y.; Codogno, P. Autophagy signaling and the cogwheels of cancer. Autophagy 2006, 2, 67–73. [Google Scholar] [CrossRef]
- Wang, J.; Whiteman, M.W.; Lian, H.; Wang, G.; Singh, A.; Huang, D.; Denmark, T. A non-canonical MEK/ERK signaling pathway regulates autophagy via regulating Beclin 1. J. Biol. Chem. 2009, 284, 21412–21424. [Google Scholar] [CrossRef]
- Grzywacz, A.; Gdula-Argasińska, J.; Muszyńska, B.; Tyszka-Czochara, M.; Librowski, T.; Opoka, W. Metal responsive transcription factor 1 (MTF-1) regulates zinc dependent cellular processes at the molecular level. Acta Biochim. Polonica 2015, 62, 491–498. [Google Scholar] [CrossRef]
- Petr, M.A.; Tulika, T.; Carmona-Marin, L.M.; Scheibye-Knudsen, M. Protecting the Aging Genome. Trends Cell Biol. 2020, 30, 117–132. [Google Scholar] [CrossRef]
- Hewitt, G.; Korolchuk, V.I. Repair, Reuse, Recycle: The Expanding Role of Autophagy in Genome Maintenance. Trends Cell Biol. 2017, 27, 340–351. [Google Scholar] [CrossRef]
- Yildiz, A.; Kaya, Y.; Tanriverdi, O. Effect of the Interaction Between Selenium and Zinc on DNA Repair in Association With Cancer Prevention. J. Cancer Prev. 2019, 24, 146–154. [Google Scholar] [CrossRef]
- Sharif, R.; Thomas, P.; Zalewski, P.; Fenech, M. Zinc supplementation influences genomic stability biomarkers, antioxidant activity, and zinc transporter genes in an elderly Australian population with low zinc status. Mol. Nutr. Food Res. 2015, 59, 1200–1212. [Google Scholar] [CrossRef]
- Joerger, A.C.; Fersht, A.R. The p53 Pathway: Origins, Inactivation in Cancer, and Emerging Therapeutic Approaches. Annu. Rev. Biochem. 2016, 85, 375–404. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Persp. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [PubMed]
- Garufi, A.; Trisciuoglio, D.; Porru, M.; Leonetti, C.; Stoppacciaro, A.; D’Orazi, V.; Avantaggiati, M.; Crispini, A.; Pucci, D.; D’Orazi, G. A fluorescent curcumin-based Zn(II)-complex reactivates mutant (R175H and R273H) p53 in cancer cells. J. Exp. Clin. Cancer Res. 2013, 32, 72. [Google Scholar] [CrossRef] [PubMed]
- Garufi, A.; Pucci, D.; D’Orazi, V.; Cirone, M.; Bossi, G.; Avantaggiati, M.L.; D’Orazi, G. Degradation of mutant p53H175 protein by Zn(II) through autophagy. Cell Death Dis. 2014, 5, e1271. [Google Scholar] [CrossRef]
- Bosomworth, H.J.; Thornton, J.K.; Coneyworth, L.J.; Ford, D.; Valentine, R.A. Efflux function, tissue-specific expression and intracellular trafficking of the Zn transporter ZnT10 indicate roles in adult Zn homeostasis. Metallomics Integr. Biomet. Sci. 2012, 4, 771–779. [Google Scholar] [CrossRef]
- Djajadikerta, A.; Keshri, S.; Pavel, M.; Prestil, R.; Ryan, L.; Rubinsztein, D.C. Autophagy Induction as a Therapeutic Strategy for Neurodegenerative Diseases. J. Mol. Biol. 2020, 432, 2799–2821. [Google Scholar] [CrossRef]
- Koh, J.-Y.; Kim, H.N.; Hwang, J.J.; Kim, Y.-H.; Park, S.E. Lysosomal dysfunction in proteinopathic neurodegenerative disorders: Possible therapeutic roles of cAMP and zinc. Mol. Brain 2019, 12, 18. [Google Scholar] [CrossRef]
- Lee, M.-G.; Choi, M.-A.; Chae, S.; Kang, M.-A.; Jo, H.; Baek, J.-M.; In, K.-R.; Park, H.; Heo, H.; Jang, D.; et al. Loss of the dermis zinc transporter ZIP13 promotes the mildness of fibrosarcoma by inhibiting autophagy. Sci. Rep. 2019, 9, 15042. [Google Scholar] [CrossRef]
- Ni, H.; Feng, X.; Xiao, Z.J.; Tao, L.Y.; Jin, M.F. Dynamic pattern of gene expression of ZnT-4, caspase-3, LC3, and PRG-3 in rat cerebral cortex following flurothyl-induced recurrent neonatal seizures. Biol. Trace Elem. Res. 2011, 143, 1607–1615. [Google Scholar] [CrossRef]
- Kusanaga, M.; Oe, S.; Ogino, N.; Minami, S.; Miyagawa, K.; Honma, Y.; Harada, M. Zinc Attenuates the Cytotoxicity of Some Stimuli by Reducing Endoplasmic Reticulum Stress in Hepatocytes. Int. J. Mol. Sci. 2019, 20, 2192. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Yu, Z.; Chen, Z.; Yang, L.; Ma, M.; Lu, S.; Wang, C.; Teng, C.; Nie, Y. Zinc chelator TPEN induces pancreatic cancer cell death through causing oxidative stress and inhibiting cell autophagy. J. Cell. Physiol. 2019, 234, 20648–20661. [Google Scholar] [CrossRef] [PubMed]
- Hadj Abdallah, N.; Baulies, A.; Bouhlel, A.; Bejaoui, M.; Zaouali, M.A.; Ben Mimouna, S.; Messaoudi, I.; Fernandez-Checa, J.C.; García Ruiz, C.; Ben Abdennebi, H. Zinc mitigates renal ischemia-reperfusion injury in rats by modulating oxidative stress, endoplasmic reticulum stress, and autophagy. J. Cell. Physiol. 2018, 233, 8677–8690. [Google Scholar] [CrossRef]
- Bian, X.; Teng, T.; Zhao, H.; Qin, J.; Qiao, Z.; Sun, Y.; Liun, Z.; Xu, Z. Zinc prevents mitochondrial superoxide generation by inducing mitophagy in the setting of hypoxia/reoxygenation in cardiac cells. Free Radic. Res. 2018, 52, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Li, Y.; Li, C. Autophagy plays a positive role in zinc-induced apoptosis in intestinal porcine epithelial cells. Toxicol. In Vitro Int. J. Publ. Assoc. BIBRA 2017, 44, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Chang, Y.; Feng, Y.; Jian, H.; Wu, X.; Zheng, R.; Xu, K.; Zhang, H. Bismuth Sulfide Nanorods with Retractable Zinc Protoporphyrin Molecules for Suppressing Innate Antioxidant Defense System and Strengthening Phototherapeutic Effects. Adv. Mater. 2019, 31, e1806808. [Google Scholar] [CrossRef] [PubMed]
- Blasiak, J.; Szaflik, J.; Szaflik, J.P. Implications of altered iron homeostasis for age-related macular degeneration. Front. Biosci. (Landmark Ed.) 2011, 16, 1551–1559. [Google Scholar] [CrossRef]
- Shu, W.; Dunaief, J.L. Potential Treatment of Retinal Diseases with Iron Chelators. Pharmaceuticals 2018, 11, 112. [Google Scholar] [CrossRef] [PubMed]
- Njaria, P.M.; Okombo, J.; Njuguna, N.M.; Chibale, K. Chloroquine-containing compounds: A patent review (2010–2014). Expert Opin. Ther. Pat. 2015, 25, 1003–1024. [Google Scholar] [CrossRef] [PubMed]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Saito, Y.; Kuse, Y.; Inoue, Y.; Nakamura, S.; Hara, H.; Shimazawa, M. Transient acceleration of autophagic degradation by pharmacological Nrf2 activation is important for retinal pigment epithelium cell survival. Redox Biol. 2018, 19, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhao, X.; Cai, Y.; Li, Y.; Yu, X.; Lu, L. Protection of retina by mini-αA in NaIO3-induced retinal pigment epithelium degeneration mice. Int. J. Mol. Sci. 2015, 16, 1644–1656. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Ng, T.K.; Brelén, M.E.; Wu, D.; Wang, J.X.; Chan, K.P.; Yung, J.S.Y.; Cao, D.; Wang, Y.; Zhang, S.; et al. Continuous exposure to non-lethal doses of sodium iodate induces retinal pigment epithelial cell dysfunction. Sci. Rep. 2016, 6, 37279. [Google Scholar] [CrossRef]
- Michaelides, M.; Stover, N.; Francis, P.; Weleber, R. Retinal Toxicity Associated With Hydroxychloroquine and Chloroquine Risk Factors, Screening, and Progression Despite Cessation of Therapy. Arch. Ophthalmol. 2011, 129, 30–39. [Google Scholar] [CrossRef]
- Peters, S.; Reinthal, E.; Blitgen-Heinecke, P.; Bartz-Schmidt, K.U.; Schraermeyer, U. Inhibition of Lysosomal Degradation in Retinal Pigment Epithelium Cells Induces Exocytosis of Phagocytic Residual Material at the Basolateral Plasma Membrane. Ophthalmic Res. 2006, 38, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Mecklenburg, L.; Schraermeyer, U. An Overview on the Toxic Morphological Changes in the Retinal Pigment Epithelium after Systemic Compound Administration. Toxicol. Pathol. 2007, 35, 252–267. [Google Scholar] [CrossRef] [PubMed]
- Toler, S.M. Oxidative stress plays an important role in the pathogenesis of drug-induced retinopathy. Exp. Biol. Med. (Maywood) 2004, 229, 607–615. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Uusitalo, H.; Blasiak, J.; Felszeghy, S.; Kannan, R.; Kauppinen, A.; Salminen, A.; Sinha, D.; Ferrington, D. Mechanisms of mitochondrial dysfunction and their impact on age-related macular degeneration. Prog. Retin. Eye Res 2020. [Google Scholar] [CrossRef]
- Da Costa, B.; Pippi, B.; Andrzejewski Kaminski, T.F.; Andrade, S.F.; Fuentefria, A.M. In vitro antidermatophytic synergism of double and triple combination of clioquinol with ciclopirox and terbinafine. Mycoses 2020. [Google Scholar] [CrossRef]
- Park, M.H.; Lee, S.J.; Byun, H.R.; Kim, Y.; Oh, Y.J.; Koh, J.Y.; Hwang, J.J. Clioquinol induces autophagy in cultured astrocytes and neurons by acting as a zinc ionophore. Neurobiol. Dis. 2011, 42, 242–251. [Google Scholar] [CrossRef]
- Seo, B.R.; Lee, S.J.; Cho, K.S.; Yoon, Y.H.; Koh, J.Y. The zinc ionophore clioquinol reverses autophagy arrest in chloroquine-treated ARPE-19 cells and in APP/mutant presenilin-1-transfected Chinese hamster ovary cells. Neurobiol. Aging 2015, 36, 3228–3238. [Google Scholar] [CrossRef]
- Friedman, D.S.; Katz, J.; Bressler, N.M.; Rahmani, B.; Tielsch, J.M. Racial differences in the prevalence of age-related macular degeneration: The Baltimore Eye Survey. Ophthalmology 1999, 106, 1049–1055. [Google Scholar] [CrossRef]
- Weiter, J.J.; Delori, F.C.; Wing, G.L.; Fitch, K.A. Relationship of senile macular degeneration to ocular pigmentation. Am. J. Ophthalmol. 1985, 99, 185–187. [Google Scholar] [CrossRef]
- Weiter, J.J.; Delori, F.C.; Wing, G.L.; Fitch, K.A. Retinal pigment epithelial lipofuscin and melanin and choroidal melanin in human eyes. Investig. Ophthal. Vis. Sci. 1986, 27, 145–152. [Google Scholar]
- Hellinen, L.; Hagström, M.; Knuutila, H.; Ruponen, M.; Urtti, A.; Reinisalo, M. Characterization of artificially re-pigmented ARPE-19 retinal pigment epithelial cell model. Sci. Rep. 2019, 9, 13761. [Google Scholar] [CrossRef] [PubMed]
- Herrling, T.; Jung, K.; Fuchs, J. The role of melanin as protector against free radicals in skin and its role as free radical indicator in hair. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2008, 69, 1429–1435. [Google Scholar] [CrossRef] [PubMed]
- Sulzer, D.; Cassidy, C.; Horga, G.; Kang, U.J.; Fahn, S.; Casella, L.; Pezzoli, G.; Langley, J.; Hu, X.P.; Zucca, F.A.; et al. Neuromelanin detection by magnetic resonance imaging (MRI) and its promise as a biomarker for Parkinson’s disease. NPJ Parkinsons Dis. 2018, 4, 11. [Google Scholar] [CrossRef] [PubMed]
- Hyodo, F.; Naganuma, T.; Eto, H.; Murata, M.; Utsumi, H.; Matsuo, M. In vivo melanoma imaging based on dynamic nuclear polarization enhancement in melanin pigment of living mice using in vivo dynamic nuclear polarization magnetic resonance imaging. Free Radic. Biol. Med. 2019, 134, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Rózanowska, M.; Bober, A.; Burke, J.M.; Sarna, T. The role of retinal pigment epithelium melanin in photoinduced oxidation of ascorbate. Photochem. Photobiol. 1997, 65, 472–479. [Google Scholar] [CrossRef]
- Chatterjee, S.; Prados-Rosales, R.; Tan, S.; Phan, V.C.; Chrissian, C.; Itin, B.; Wang, H.; Khajo, A.; Magliozzo, R.S.; Casadevall, A.; et al. The melanization road more traveled by: Precursor substrate effects on melanin synthesis in cell-free and fungal cell systems. J. Biol. Chem. 2018, 293, 20157–20168. [Google Scholar] [CrossRef]
- Horcicko, J.; Borovanský, J.; Duchon, J.; Procházková, B. Distribution of zinc and copper in pigmented tissues. Hoppe Seylers Z. Physiol. Chem. 1973, 354, 203–204. [Google Scholar] [PubMed]
- Julien, S.; Biesemeier, A.; Kokkinou, D.; Eibl, O.; Schraermeyer, U. Zinc deficiency leads to lipofuscin accumulation in the retinal pigment epithelium of pigmented rats. PLoS ONE 2011, 6, e29245. [Google Scholar] [CrossRef] [PubMed]
- Ikram, M.A.; Brusselle, G.G.O.; Murad, S.D.; van Duijn, C.M.; Franco, O.H.; Goedegebure, A.; Klaver, C.C.W.; Nijsten, T.E.C.; Peeters, R.P.; Stricker, B.H.; et al. The Rotterdam Study: 2018 update on objectives, design and main results. Eur. J. Epidemiol. 2017, 32, 807–850. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Zhang, Q.; Shen, Y.; Bai, Y.; Ma, X.; Zhang, B.; Qi, Y.; Zhang, J.; Hu, Q.; Du, W.; et al. ube3d, a New Gene Associated with Age-Related Macular Degeneration, Induces Functional Changes in Both In Vivo and In Vitro Studies. Mol. Ther. Nucleic Acids 2020, 20, 217–230. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blasiak, J.; Pawlowska, E.; Chojnacki, J.; Szczepanska, J.; Chojnacki, C.; Kaarniranta, K. Zinc and Autophagy in Age-Related Macular Degeneration. Int. J. Mol. Sci. 2020, 21, 4994. https://doi.org/10.3390/ijms21144994
Blasiak J, Pawlowska E, Chojnacki J, Szczepanska J, Chojnacki C, Kaarniranta K. Zinc and Autophagy in Age-Related Macular Degeneration. International Journal of Molecular Sciences. 2020; 21(14):4994. https://doi.org/10.3390/ijms21144994
Chicago/Turabian StyleBlasiak, Janusz, Elzbieta Pawlowska, Jan Chojnacki, Joanna Szczepanska, Cezary Chojnacki, and Kai Kaarniranta. 2020. "Zinc and Autophagy in Age-Related Macular Degeneration" International Journal of Molecular Sciences 21, no. 14: 4994. https://doi.org/10.3390/ijms21144994
APA StyleBlasiak, J., Pawlowska, E., Chojnacki, J., Szczepanska, J., Chojnacki, C., & Kaarniranta, K. (2020). Zinc and Autophagy in Age-Related Macular Degeneration. International Journal of Molecular Sciences, 21(14), 4994. https://doi.org/10.3390/ijms21144994