Integrative Bioinformatic Analyses of Global Transcriptome Data Decipher Novel Molecular Insights into Cardiac Anti-Fibrotic Therapies

 , , ,
, , , 
 and
and 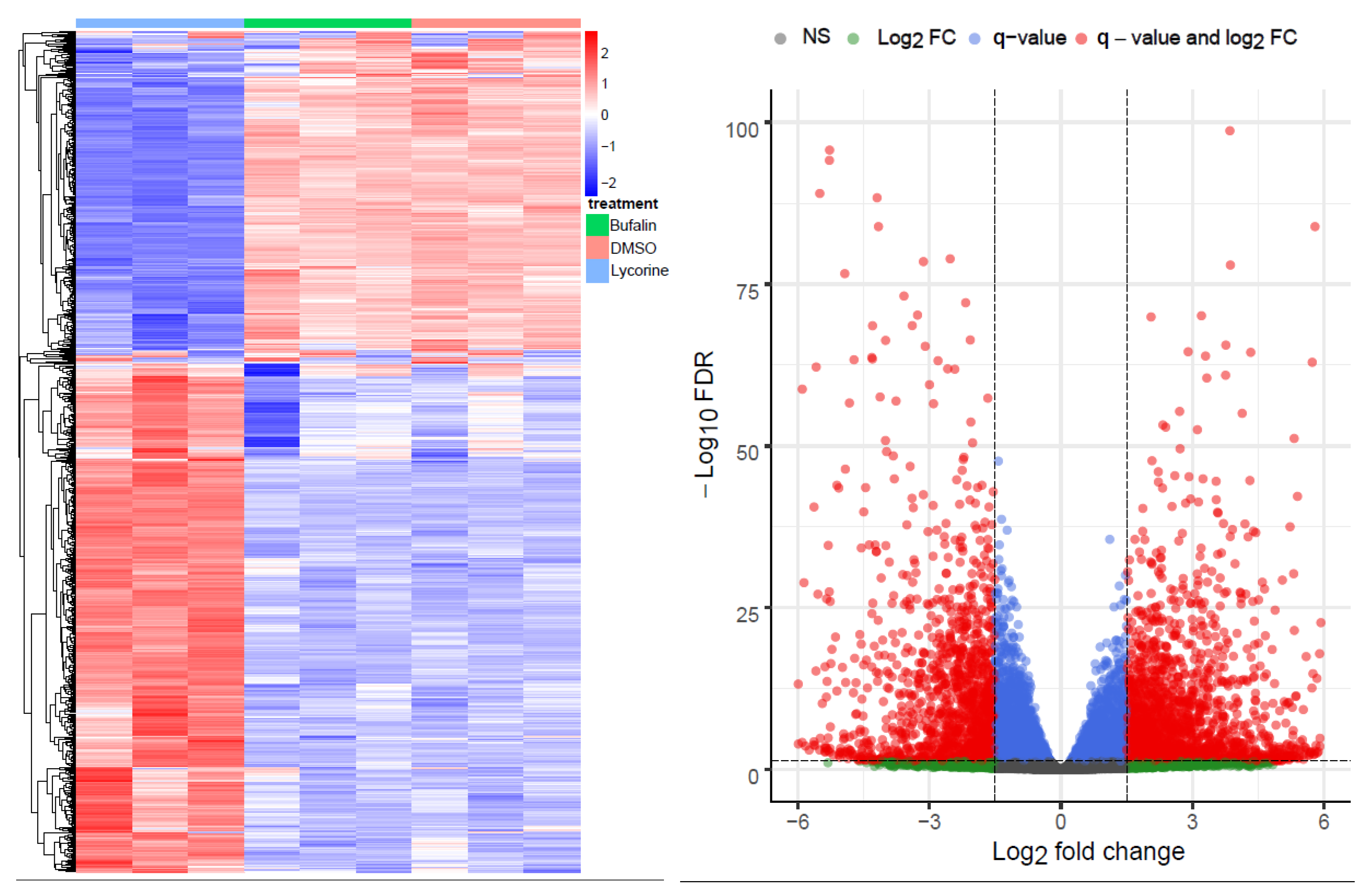{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Bioinformatics RNA-Seq Analysis Showed Deregulation of Lyc-s but Not of Buf-s Compared to DMSO
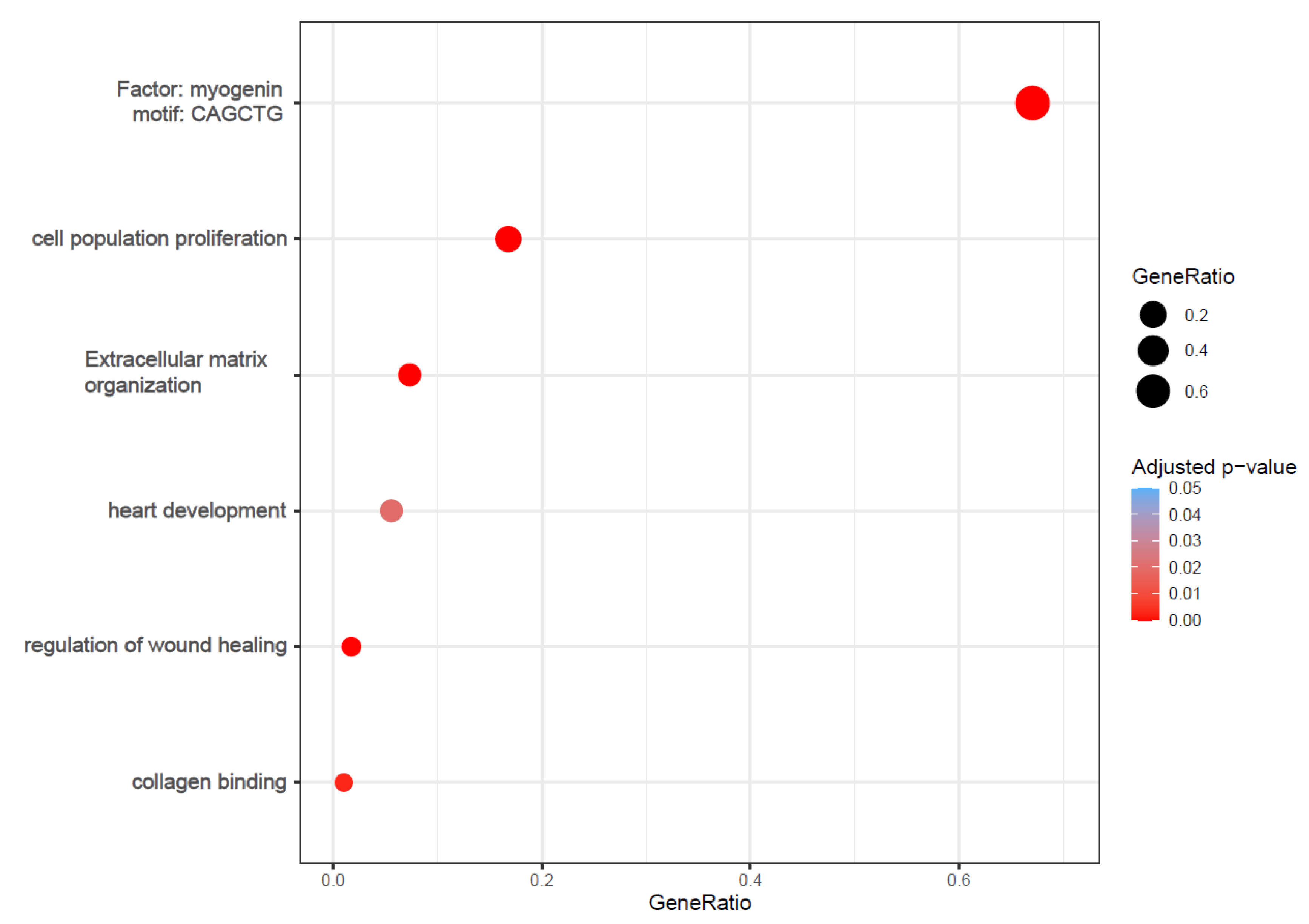
2.2. Functional Analysis Revealed Enrichment of Processes Involved in Cardiac Fibrosis
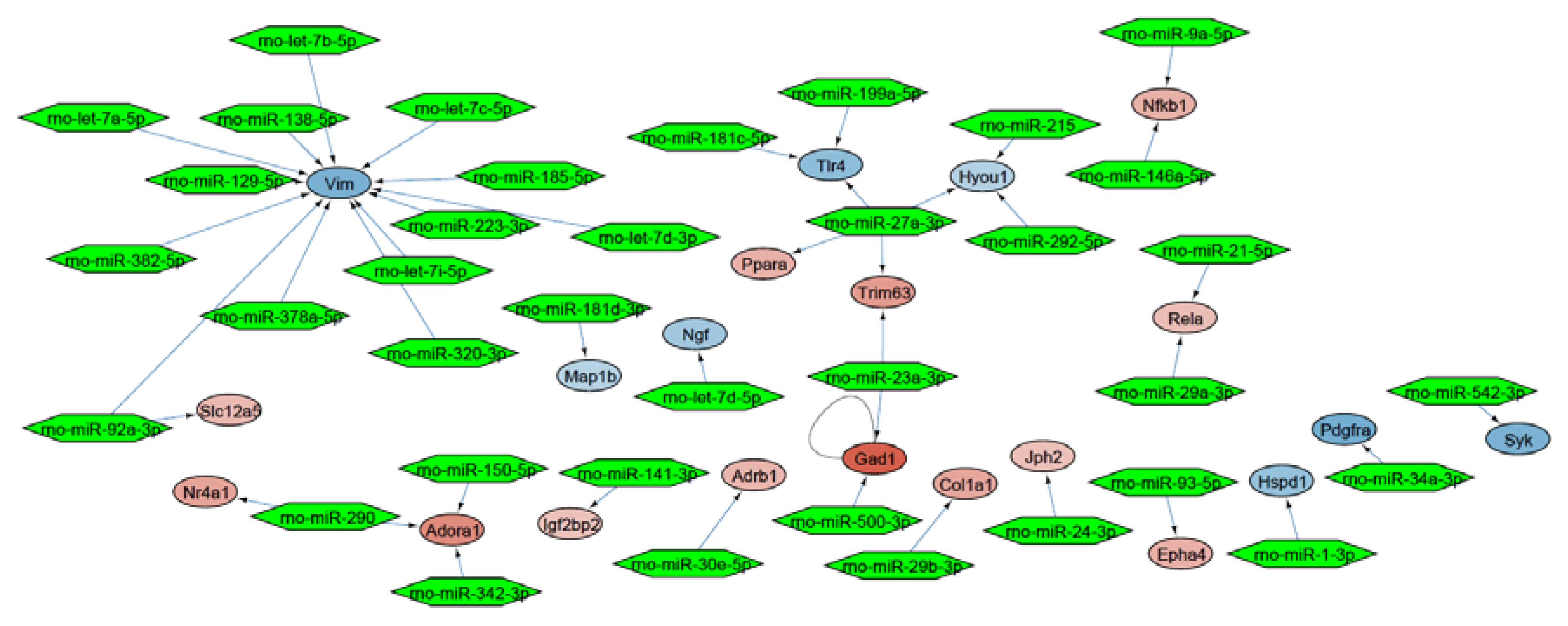
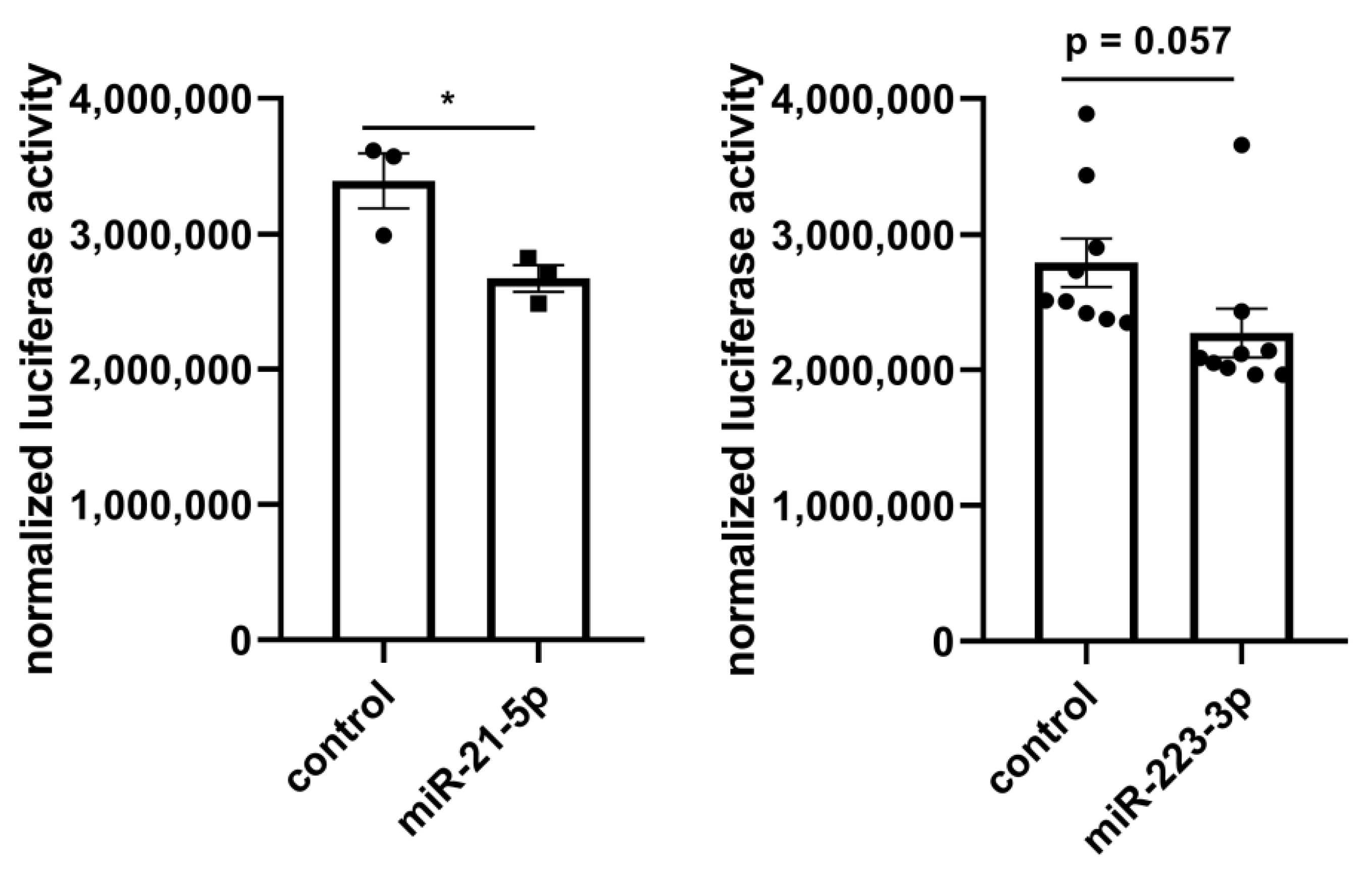
2.3. Molecular Network Analysis Identified miRNA-21-5p and miRNA-223-3p as Key Interaction Partners around the DEGs
3. Discussion
4. Materials and Methods
4.1. Animal Experiments
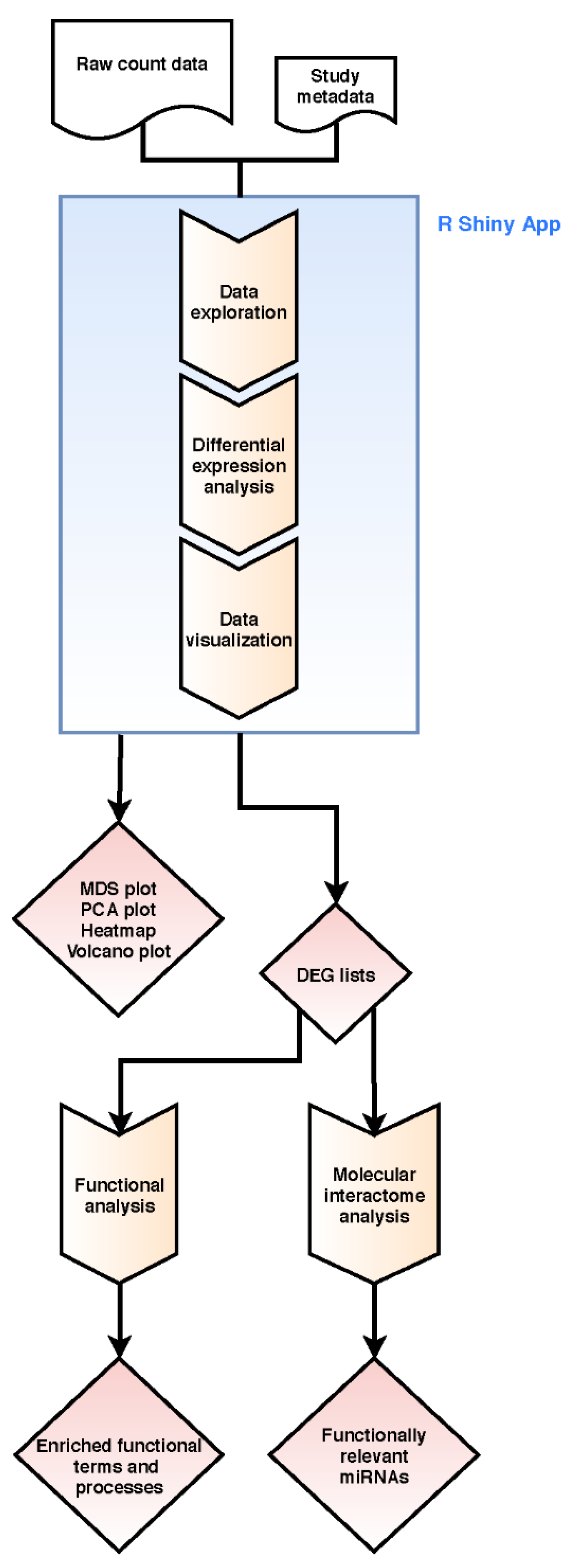
4.2. Bioinformatics Transcriptome Analysis Approach
4.3. Luciferase Reporter Assay
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. G:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using david bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Schurch, N.J.; Schofield, P.; Gierliński, M.; Cole, C.; Sherstnev, A.; Singh, V.; Wrobel, N.; Gharbi, K.; Simpson, G.G.; Owen-Hughes, T.; et al. How many biological replicates are needed in an rna-seq experiment and which differential expression tool should you use? RNA 2016, 22, 839–851. [Google Scholar] [CrossRef] [PubMed]
- Papatheodorou, I.; Fonseca, N.A.; Keays, M.; Tang, Y.A.; Barrera, E.; Bazant, W.; Burke, M.; Füllgrabe, A.; Fuentes, A.M.-P.; George, N.; et al. Expression atlas: Gene and protein expression across multiple studies and organisms. Nucleic Acids Res. 2018, 46, D246–D251. [Google Scholar] [CrossRef] [PubMed]
- Petryszak, R.; Fonseca, N.A.; Füllgrabe, A.; Huerta, L.; Keays, M.; Tang, Y.A.; Brazma, A. The rnaseq-er api—A gateway to systematically updated analysis of public rna-seq data. Bioinformatics 2017, 33, 2218–2220. [Google Scholar] [CrossRef] [PubMed]
- Roth, G.A.; Johnson, C.; Abajobir, A.; Abd-Allah, F.; Abera, S.F.; Abyu, G.; Ahmed, M.; Aksut, B.; Alam, T.; Alam, K.; et al. Global, regional, and national burden of cardiovascular diseases for 10 causes, 1990 to 2015. J. Am. Coll. Cardiol. 2017, 70, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; Gonzalez-Juanatey, J.R.; Harjola, V.P.; Jankowska, E.A.; et al. 2016 esc guidelines for the diagnosis and treatment of acute and chronic heart failure: The task force for the diagnosis and treatment of acute and chronic heart failure of the european society of cardiology (esc)developed with the special contribution of the heart failure association (hfa) of the esc. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar]
- Hara, H.; Takeda, N.; Komuro, I. Pathophysiology and therapeutic potential of cardiac fibrosis. Inflamm. Regen. 2017, 37, 13. [Google Scholar] [CrossRef]
- Thum, T.; Gross, C.; Fiedler, J.; Fischer, T.; Kissler, S.; Bussen, M.; Galuppo, P.; Just, S.; Rottbauer, W.; Frantz, S.; et al. Microrna-21 contributes to myocardial disease by stimulating map kinase signalling in fibroblasts. Nature 2008, 456, 980–984. [Google Scholar] [CrossRef]
- Lorenzen, J.M.; Schauerte, C.; Hubner, A.; Kolling, M.; Martino, F.; Scherf, K.; Batkai, S.; Zimmer, K.; Foinquinos, A.; Kaucsar, T.; et al. Osteopontin is indispensible for ap1-mediated angiotensin ii-related mir-21 transcription during cardiac fibrosis. Eur. Heart J. 2015, 36, 2184–2196. [Google Scholar] [CrossRef] [PubMed]
- Kolling, M.; Kaucsar, T.; Schauerte, C.; Hubner, A.; Dettling, A.; Park, J.K.; Busch, M.; Wulff, X.; Meier, M.; Scherf, K.; et al. Therapeutic mir-21 silencing ameliorates diabetic kidney disease in mice. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 165–180. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, M.T.; Gupta, S.K.; Viereck, J.; Foinquinos, A.; Samolovac, S.; Kramer, F.L.; Garg, A.; Remke, J.; Zimmer, K.; Batkai, S.; et al. Inhibition of the cardiac fibroblast-enriched lncrna meg3 prevents cardiac fibrosis and diastolic dysfunction. Circ. Res. 2017, 121, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Schimmel, K.; Jung, M.; Foinquinos, A.; Jose, G.S.; Beaumont, J.; Bock, K.; Grote-Levi, L.; Xiao, K.; Bar, C.; Pfanne, A.; et al. Natural compound library screening identifies new molecules for the treatment of cardiac fibrosis and diastolic dysfunction. Circulation 2020, 141, 751–767. [Google Scholar] [CrossRef]
- Watson, S.A.; Scigliano, M.; Bardi, I.; Ascione, R.; Terracciano, C.M.; Perbellini, F. Preparation of viable adult ventricular myocardial slices from large and small mammals. Nat. Protoc. 2017, 12, 2623–2639. [Google Scholar] [CrossRef]
- Watson, S.A.; Duff, J.; Bardi, I.; Zabielska, M.; Atanur, S.S.; Jabbour, R.J.; Simon, A.; Tomas, A.; Smolenski, R.T.; Harding, S.E.; et al. Biomimetic electromechanical stimulation to maintain adult myocardial slices in vitro. Nat. Commun. 2019, 10, 2168. [Google Scholar] [CrossRef]
- Shinde, A.V.; Humeres, C.; Frangogiannis, N.G. The role of alpha-smooth muscle actin in fibroblast-mediated matrix contraction and remodeling. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 298–309. [Google Scholar] [CrossRef]
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. Tgf-beta signaling in fibrosis. Growth Factors 2011, 29, 196–202. [Google Scholar] [CrossRef]
- Gyongyosi, M.; Winkler, J.; Ramos, I.; Do, Q.T.; Firat, H.; McDonald, K.; Gonzalez, A.; Thum, T.; Diez, J.; Jaisser, F.; et al. Myocardial fibrosis: Biomedical research from bench to bedside. Eur. J. Heart Fail. 2017, 19, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Khalil, H.; Kanisicak, O.; Boyer, J.G.; Vagnozzi, R.J.; Maliken, B.D.; Sargent, M.A.; Prasad, V.; Valiente-Alandi, I.; Blaxall, B.C.; et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J. Clin. Investig. 2018, 128, 2127–2143. [Google Scholar] [CrossRef]
- Jang, Y.-N.; Baik, E.J. Jak-stat pathway and myogenic differentiation. JAK-STAT 2013, 2, e23282. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Friggeri, A.; Yang, Y.; Milosevic, J.; Ding, Q.; Thannickal, V.J.; Kaminski, N.; Abraham, E. Mir-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J. Exp. Med. 2010, 207, 1589–1597. [Google Scholar] [CrossRef] [PubMed]
- Chau, B.N.; Xin, C.; Hartner, J.; Ren, S.; Castano, A.P.; Linn, G.; Li, J.; Tran, P.T.; Kaimal, V.; Huang, X.; et al. Microrna-21 promotes fibrosis of the kidney by silencing metabolic pathways. Sci. Transl. Med. 2012, 4, ra118–ra121. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Patrikeev, I.; Ochoa, L.; Vargas, G.; Belanger, K.K.; Litvinov, J.; Boldogh, I.; Ameredes, B.T.; Motamedi, M.; Brasier, A.R. Nf-kappab mediates mesenchymal transition, remodeling, and pulmonary fibrosis in response to chronic inflammation by viral rna patterns. Am. J. Respir. Cell Mol. Biol. 2017, 56, 506–520. [Google Scholar] [CrossRef] [PubMed]
- Jimenez Calvente, C.; Del Pilar, H.; Tameda, M.; Johnson, C.D.; Feldstein, A.E. Microrna 223 3p negatively regulates the nlrp3 inflammasome in acute and chronic liver injury. Mol. Ther. 2020, 28, 653–663. [Google Scholar] [CrossRef]
- Schaum, N.; Karkanias, J.; Neff, N.F.; May, A.P.; Quake, S.R.; Wyss-Coray, T.; Darmanis, S.; Batson, J.; Botvinnik, O.; Chen, M.B.; et al. Single-cell transcriptomics of 20 mouse organs creates a tabula muris. Nature 2018, 562, 367–372. [Google Scholar]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. Star: Ultrafast universal rna-seq aligner. Bioinformatics 2012, 29, 15–21. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Saini, H.K.; van Dongen, S.; Enright, A.J. Mirbase: Tools for microrna genomics. Nucleic Acids Res. 2007, 36, D154–D158. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. Featurecounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for rna-seq data with deseq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2 Elegant Graphics for Data Analysis. In Use R! 2nd ed.; Springer: Berlin, Germany, 2016; Volume XVI, p. 260. [Google Scholar]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Pagès, H.; Carlson, M.; Falcon, S.; Li, N. Annotationdbi: Manipulation of Sqlite-Based Annotations in Bioconductor. R package version 1.48.0. Available online: http://bioconductor.org/packages/release/bioc/html/AnnotationDbi.html (accessed on 7 May 2020).
- Kolde, R. Pheatmap: Pretty Heatmaps. R package version 1.0.12. 2019. Available online: https://rdrr.io/cran/pheatmap/#vignettes (accessed on 7 May 2020).
- Blighe, K.; Rana, S.; Lewis, M. Enhancedvolcano: Publication-Ready Volcano Plots with Enhanced Colouring and Labeling. R package version 1.4.0. 2019. Available online: https://bioconductor.org/packages/release/bioc/html/EnhancedVolcano.html (accessed on 7 May 2020).
- Chang, W.; Cheng, J.; Allaire, J.; Xie, Y.; McPherson, J. Shiny: Web Application Framework for R. R Package Version 1.4.0.2. 2020. Available online: https://cran.R-project.Org/package=shiny (accessed on 7 May 2020).
- Chang, W.; Borges Ribeiro, B. Shinydashboard: Create Dashboards with ‘Shiny’. R Package Version 0.7.1. 2018. Available online: https://cran.R-project.Org/package=shinydashboard (accessed on 7 May 2020).
- Orchard, S.; Kerrien, S.; Abbani, S.; Aranda, B.; Bhate, J.; Bidwell, S.; Bridge, A.; Briganti, L.; Brinkman, F.S.L.; Cesareni, G.; et al. Protein interaction data curation: The international molecular exchange (imex) consortium. Nat. Methods 2012, 9, 345–350. [Google Scholar] [CrossRef]
- Chou, C.H.; Shrestha, S.; Yang, C.D.; Chang, N.W.; Lin, Y.L.; Liao, K.W.; Huang, W.C.; Sun, T.H.; Tu, S.J.; Lee, W.H.; et al. Mirtarbase update 2018: A resource for experimentally validated microrna-target interactions. Nucleic Acids Res. 2018, 46, D296–D302. [Google Scholar] [CrossRef]
- Kutmon, M.; Ehrhart, F.; Willighagen, E.; Evelo, C.; Coort, S. Cytargetlinker app update: A flexible solution for network extension in cytoscape [version 2; peer review: 2 approved]. F1000Research 2019, 7, ELIXIR-743. [Google Scholar] [CrossRef]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microrna targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fuchs, M.; Kreutzer, F.P.; Kapsner, L.A.; Mitzka, S.; Just, A.; Perbellini, F.; Terracciano, C.M.; Xiao, K.; Geffers, R.; Bogdan, C.; et al. Integrative Bioinformatic Analyses of Global Transcriptome Data Decipher Novel Molecular Insights into Cardiac Anti-Fibrotic Therapies. Int. J. Mol. Sci. 2020, 21, 4727. https://doi.org/10.3390/ijms21134727
Fuchs M, Kreutzer FP, Kapsner LA, Mitzka S, Just A, Perbellini F, Terracciano CM, Xiao K, Geffers R, Bogdan C, et al. Integrative Bioinformatic Analyses of Global Transcriptome Data Decipher Novel Molecular Insights into Cardiac Anti-Fibrotic Therapies. International Journal of Molecular Sciences. 2020; 21(13):4727. https://doi.org/10.3390/ijms21134727
Chicago/Turabian StyleFuchs, Maximilian, Fabian Philipp Kreutzer, Lorenz A. Kapsner, Saskia Mitzka, Annette Just, Filippo Perbellini, Cesare M. Terracciano, Ke Xiao, Robert Geffers, Christian Bogdan, and et al. 2020. "Integrative Bioinformatic Analyses of Global Transcriptome Data Decipher Novel Molecular Insights into Cardiac Anti-Fibrotic Therapies" International Journal of Molecular Sciences 21, no. 13: 4727. https://doi.org/10.3390/ijms21134727
APA StyleFuchs, M., Kreutzer, F. P., Kapsner, L. A., Mitzka, S., Just, A., Perbellini, F., Terracciano, C. M., Xiao, K., Geffers, R., Bogdan, C., Prokosch, H.-U., Fiedler, J., Thum, T., & Kunz, M. (2020). Integrative Bioinformatic Analyses of Global Transcriptome Data Decipher Novel Molecular Insights into Cardiac Anti-Fibrotic Therapies. International Journal of Molecular Sciences, 21(13), 4727. https://doi.org/10.3390/ijms21134727