Strain-Level Metagenomic Data Analysis of Enriched In Vitro and In Silico Spiked Food Samples: Paving the Way towards a Culture-Free Foodborne Outbreak Investigation Using STEC as a Case Study

 , , , and
, , , and 
Abstract
1. Introduction
2. Results
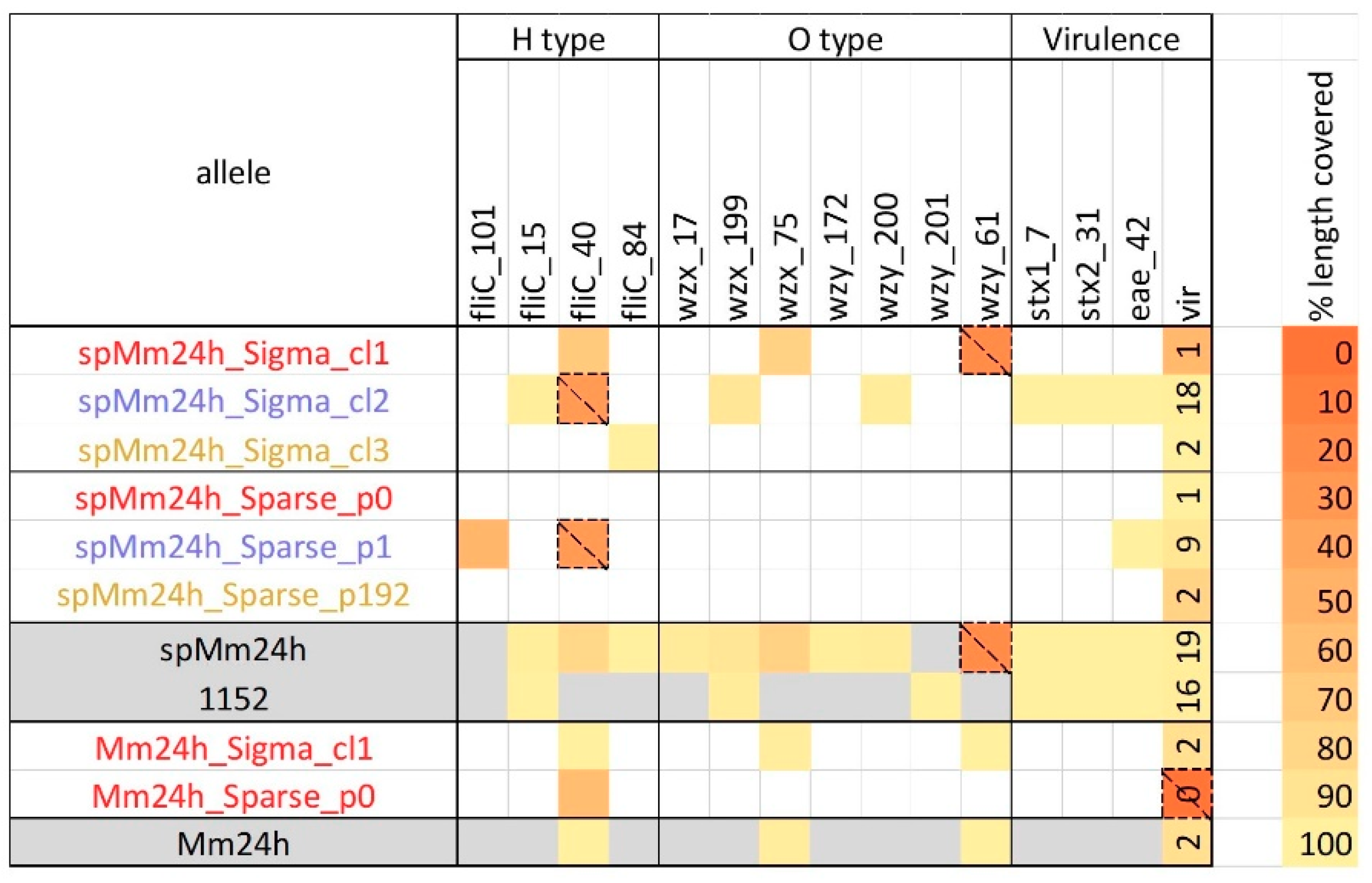
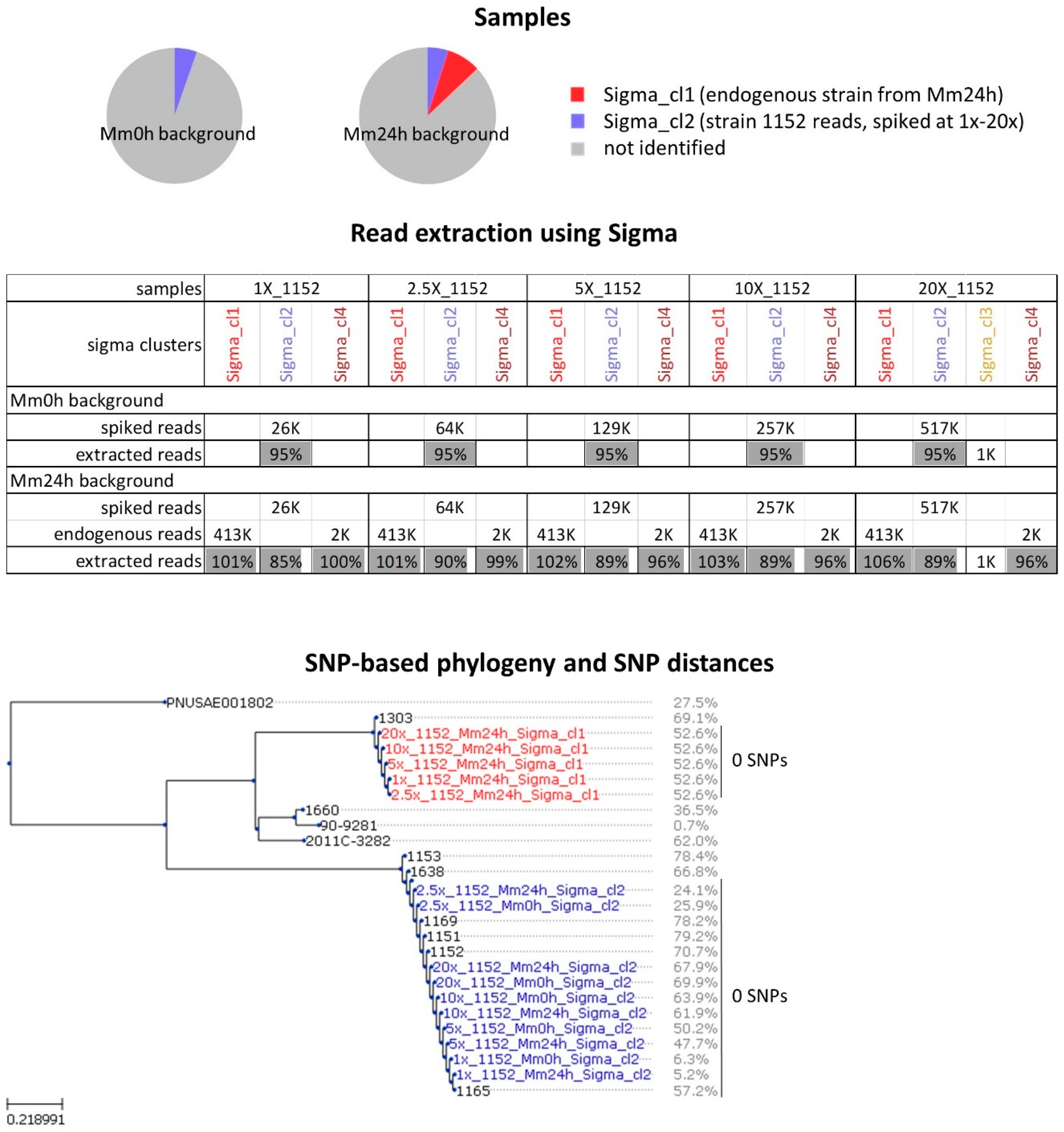
2.1. Analysis of In-Vitro Spiked Metagenomic Samples
2.2. In Silico Spiked Samples—Investigation of Different Coverage Ranges
2.3. In Silico Spiked Samples—Additional Test Cases
2.4. In Silico Spiked Samples—Mixed Samples
3. Discussion
4. Materials and Methods
4.1. Sequencing Data
4.2. In Silico Spiking
4.3. Metagenomic Analysis
4.4. Downstream Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ANI | Average Nucleotide Identity |
| CFU | Colony-Forming Unit |
| cgMLST | core genome Multilocus Sequence Typing |
| EAEC | Enteroaggregative E. coli |
| EHEC | Enterohaemorrhagic E. coli |
| EPEC | Enteropathogenic E. coli |
| ETEC | Enterotoxigenic E. coli |
| MAEC | Mastitis-associated E. coli |
| Mm0h | Minced Meat sample enriched for 0 h |
| Mm24h | Minced Meat sample enriched for 24 h |
| SNP | Single Nucleotide Polymorphism |
| spMm24h | spiked Minced Meat sample enriched for 24 h |
| WGS | Whole Genome Sequencing |
References
- Van Goethem, N.; Descamps, T.; Devleesschauwer, B.; Roosens, N.H.C.; Boon, N.A.M.; Van Oyen, H.; Robert, A. Status and potential of bacterial genomics for public health practice: A scoping review. Implement. Sci. 2019, 14, 79. [Google Scholar] [CrossRef] [PubMed]
- Leopold, S.R.; Goering, R.V.; Witten, A.; Harmsen, D.; Mellmann, A. Bacterial whole-genome sequencing revisited: Portable, scalable, and standardized analysis for typing and detection of virulence and antibiotic resistance genes. J. Clin. Microbiol. 2014, 52, 2365–2370. [Google Scholar] [CrossRef] [PubMed]
- EFSA BIOHAZ Panel; Koutsoumanis, K.; Allende, A.; Alvarez-Ordóñez, A.; Bolton, D.; Bover-Cid, S.; Chemaly, M.; Davies, R.; De Cesare, A.; Hilbert, F. Whole genome sequencing and metagenomics for outbreak investigation, source attribution and risk assessment of food-borne microorganisms. EFSA. J. 2019, 17, e05898. [Google Scholar] [PubMed]
- Rantsiou, K.; Kathariou, S.; Winkler, A.; Skandamis, P.; Saint-Cyr, M.J.; Rouzeau-Szynalski, K.; Amézquita, A. Next generation microbiological risk assessment: Opportunities of whole genome sequencing (WGS) for foodborne pathogen surveillance, source tracking and risk assessment. Int. J. Food Microbiol. 2018, 287, 3–9. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Whole Genome Sequencing for Foodborne Disease Surveillance: Landscape Paper. Available online: http://origin.who.int/foodsafety/publications/foodborne_disease/wgs_landscape/en/ (accessed on 1 July 2020).
- Gill, A. The importance of bacterial culture to food microbiology in the age of genomics. Front. Microbiol. 2017, 8, 777. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.D.; Knox, N.C.; Ronholm, J.; Pagotto, F.; Reimer, A. Metagenomics: The Next Culture-Independent Game Changer. Front. Microbiol. 2017, 8, 01069. [Google Scholar] [CrossRef] [PubMed]
- Schneeberger, P.H.H.; Becker, S.L.; Pothier, J.F.; Duffy, B.; N’Goran, E.K.; Beuret, C.; Frey, J.E.; Utzinger, J. Metagenomic diagnostics for the simultaneous detection of multiple pathogens in human stool specimens from Côte d’Ivoire: A proof-of-concept study. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2016, 40, 389–397. [Google Scholar] [CrossRef]
- Grützke, J.; Malorny, B.; Hammerl, J.A.; Busch, A.; Tausch, S.H.; Tomaso, H.; Deneke, C. Fishing in the soup–Pathogen detection in food safety using Metabarcoding and Metagenomic sequencing. Front. Microbiol. 2019, 10, 1805. [Google Scholar] [CrossRef]
- Sekse, C.; Holst-Jensen, A.; Dobrindt, U.; Johannessen, G.S.; Li, W.; Spilsberg, B.; Shi, J. High Throughput Sequencing for Detection of Foodborne Pathogens. Front. Microbiol. 2017, 8, 2029. [Google Scholar] [CrossRef]
- Ottesen, A.R.; Gonzalez, A.; Bell, R.; Arce, C.; Rideout, S.; Allard, M.; Evans, P.; Strain, E.; Musser, S.; Knight, R. Co-enriching microflora associated with culture based methods to detect Salmonella from tomato phyllosphere. PLoS ONE 2013, 8, e73079. [Google Scholar] [CrossRef]
- Ottesen, A.; Ramachandran, P.; Reed, E.; White, J.R.; Hasan, N.; Subramanian, P.; Ryan, G.; Jarvis, K.; Grim, C.; Daquiqan, N.; et al. Enrichment dynamics of Listeria monocytogenes and the associated microbiome from naturally contaminated ice cream linked to a listeriosis outbreak. BMC Microbiol. 2016, 16, 275. [Google Scholar] [CrossRef] [PubMed]
- Leonard, S.R.; Mammel, M.K.; Lacher, D.W.; Elkins, C.A. Application of Metagenomic Sequencing to Food Safety: Detection of Shiga Toxin-Producing Escherichia coli on Fresh Bagged Spinach. Drake HL, editor. Appl. Environ. Microbiol. 2015, 81, 8183–8191. [Google Scholar] [CrossRef] [PubMed]
- Leonard, S.R.; Mammel, M.K.; Lacher, D.W.; Elkins, C.A. Strain-level discrimination of Shiga toxin-producing Escherichia coli in spinach using metagenomic sequencing. PLoS ONE 2016, 11, e0167870. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, K.G.; White, J.R.; Grim, C.J.; Ewing, L.; Ottesen, A.R.; Beaubrun, J.J.-G.; Pettengill, J.B.; Brown, E.; Hanes, D.E. Cilantro microbiome before and after nonselective pre-enrichment for Salmonella using 16S rRNA and metagenomic sequencing. BMC Microbiol. 2015, 15, 160. [Google Scholar] [CrossRef]
- Walsh, A.M.; Crispie, F.; Daari, K.; O’Sullivan, O.; Martin, J.C.; Arthur, C.T.; Claesson, M.J.; Scott, K.P.; Cotter, P.D. Strain-level metagenomic analysis of the fermented dairy beverage nunu highlights potential food safety risks. Appl. Environ. Microbiol. 2017, 83, e01144-17. [Google Scholar] [CrossRef]
- Walsh, A.M.; Crispie, F.; O’Sullivan, O.; Finnegan, L.; Claesson, M.J.; Cotter, P.D. Species classifier choice is a key consideration when analysing low-complexity food microbiome data. Microbiome 2018, 6, 50. [Google Scholar] [CrossRef]
- Croxen, M.A.; Law, R.J.; Scholz, R.; Keeney, K.M.; Wlodarska, M.; Finlay, B.B. Recent advances in understanding enteric pathogenic Escherichia coli. Clin. Microbiol. Rev. 2013, 26, 822–880. [Google Scholar] [CrossRef]
- Jagadeesan, B.; Gerner-Smidt, P.; Allard, M.W.; Leuillet, S.; Winkler, A.; Xiao, Y.; Chaffron, S.; Van Der Vossen, J.; Tang, S.; Katase, M.; et al. The use of next generation sequencing for improving food safety: Translation into practice. Food Microbiol. 2019, 79, 96–115. [Google Scholar] [CrossRef]
- Zolfo, M.; Tett, A.; Jousson, O.; Donati, C.; Segata, N. MetaMLST: Multi-locus strain-level bacterial typing from metagenomic samples. Nucleic. Acids Res. 2017, 45, e7. [Google Scholar] [CrossRef]
- Truong, D.T.; Tett, A.; Pasolli, E.; Huttenhower, C.; Segata, N. Microbial strain-level population structure and genetic diversity from metagenomes. Genome Res. 2017, 27, 626–638. [Google Scholar] [CrossRef]
- Scholz, M.; Ward, D.V.; Pasolli, E.; Tolio, T.; Zolfo, M.; Asnicar, F.; Truong, D.T.; Tett, A.; Morrow, A.L.; Segata, N. Strain-level microbial epidemiology and population genomics from shotgun metagenomics. Nat. Methods. 2016, 13, 435–438. [Google Scholar] [CrossRef] [PubMed]
- Albanese, D.; Donati, C. Strain profiling and epidemiology of bacterial species from metagenomic sequencing. Nat. Commun. 2017, 8, 2260. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef] [PubMed]
- Smillie, C.S.; Sauk, J.; Gevers, D.; Friedman, J.; Sung, J.; Youngster, I.; Hohmann, E.L.; Staley, C.; Khoruts, A.; Sadowsky, M.J. Strain tracking reveals the determinants of bacterial engraftment in the human gut following fecal microbiota transplantation. Cell Host Microbe. 2018, 23, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Knight, R.; Siljander, H.; Knip, M.; Xavier, R.J.; Gevers, D. ConStrains identifies microbial strains in metagenomic datasets. Nat. Biotechnol. 2015, 33, 1045. [Google Scholar] [CrossRef]
- Quince, C.; Delmont, T.O.; Raguideau, S.; Alneberg, J.; Darling, A.E.; Collins, G.; Eren, A.M. DESMAN: A new tool for de novo extraction of strains from metagenomes. Genome Biol. 2017, 18, 181. [Google Scholar] [CrossRef]
- O’Brien, J.D.; Didelot, X.; Iqbal, Z.; Amenga-Etego, L.; Ahiska, B.; Falush, D. A Bayesian approach to inferring the phylogenetic structure of communities from metagenomic data. Genetics 2014, 197, 925–937. [Google Scholar] [CrossRef]
- Pulido-Tamayo, S.; Sánchez-Rodríguez, A.; Swings, T.; Van den Bergh, B.; Dubey, A.; Steenackers, H.; Michiels, J.; Fostier, J.; Marchal, K. Frequency-based haplotype reconstruction from deep sequencing data of bacterial populations. Nucleic Acids Res. 2015, 43, e105. [Google Scholar] [CrossRef]
- Fischer, M.; Strauch, B.; Renard, B.Y. Abundance estimation and differential testing on strain level in metagenomics data. Bioinformatics 2017, 33, i124–i132. [Google Scholar] [CrossRef]
- Nicholls, S.M.; Aubrey, W.; Edwards, A.; De Grave, K.; Huws, S.; Schietgat, L.; Soares, A.; Creevey, C.J.; Clare, A. Recovery of gene haplotypes from a metagenome. BioRxiv 2019, 223404. [Google Scholar] [CrossRef]
- Tyakht, A.V.; Manolov, A.I.; Kanygina, A.V.; Ischenko, D.S.; Kovarsky, B.A.; Popenko, A.S.; Pavlenko, A.V.; Elizarova, A.V.; Rakitina, D.V.; Baikova, J.P.; et al. Genetic diversity of Escherichia coli in gut microbiota of patients with Crohn’s disease discovered using metagenomic and genomic analyses. BMC Genomics 2018, 19, 968. [Google Scholar] [CrossRef] [PubMed]
- Hao, S.; Yang, P.; Han, M.; Xu, J.; Yu, S.; Chen, C.; Chen, W.-H.; Zhang, H.; Ning, K. Data-mining of antibiotic resistance genes provides insight into the community structure of ocean microbiome. BioRxiv 2018, 246033. [Google Scholar] [CrossRef]
- Zagordi, O.; Bhattacharya, A.; Eriksson, N.; Beerenwinkel, N. ShoRAH: Estimating the genetic diversity of a mixed sample from next-generation sequencing data. BMC Bioinformatics 2011, 12, 119. [Google Scholar] [CrossRef]
- Hong, C.; Manimaran, S.; Shen, Y.; Perez-Rogers, J.F.; Byrd, A.L.; Castro-Nallar, E.; Crandall, K.A.; Johnson, W.E. PathoScope 2.0: A complete computational framework for strain identification in environmental or clinical sequencing samples. Microbiome 2014, 2, 33. [Google Scholar] [CrossRef]
- Zhou, Z.; Luhmann, N.; Alikhan, N.F.; Quince, C.; Achtman, M. Accurate reconstruction of microbial strains from metagenomic sequencing using representative reference genomes. International Conference on Research in Computational. Mol. Biol. 2018, 10812, 225–240. [Google Scholar]
- Ahn, T.H.; Chai, J.; Pan, C. Sigma: Strain-level inference of genomes from metagenomic analysis for biosurveillance. Bioinformatics 2015, 31, 170–177. [Google Scholar] [CrossRef]
- Braeye, T.; Denayer, S.; De Rauw, K.; Forier, A.; Verluyten, J.; Fourie, L.; Dierick, K.; Botteldoorn, N.; Quoilin, S.; Cosse, P. Lessons learned from a textbook outbreak: EHEC-O157: H7 infections associated with the consumption of raw meat products. Arch. public Heal. 2014, 72, 44. [Google Scholar] [CrossRef]
- Nouws, S.; Bogaerts, B.; Verhaegen, B.; Denayer, S.; Piérard, D.; Marchal, K.; Roosens, N.H.C.; Vanneste, K.; De Keersmaecker, S.C.J. Impact of DNA extraction on Whole Genome Sequencing analysis for characterization and relatedness of Shiga toxin-producing Escherichia coli isolates. Sci. Rep. 2020. [Google Scholar]
- Scheutz, F.; Nielsen, E.M.; Frimodt-Møller, J.; Boisen, N.; Morabito, S.; Tozzoli, R.; Nataro, J.P.; Caprioli, A. Characteristics of the Enteroaggregative Shiga Toxin/Verotoxin-Producing Escherichia coli O104: H4 Strain Causing the Outbreak of Haemolytic Uraemic Syndrome; European Centre for Disease Prevention and Control: Solna, Sweden, 2011; Volume 16, p. 19889. [Google Scholar]
- Rasko, D.A.; Webster, D.R.; Sahl, J.W.; Bashir, A.; Boisen, N.; Scheutz, F.; Paxinos, E.E.; Sebra, R.; Chin, C.-S.; Iliopoulos, D.; et al. Origins of the E. coli strain causing an outbreak of hemolytic-uremic syndrome in Germany. NEJM 2011, 365, 709–717. [Google Scholar] [CrossRef]
- Karnisova, L.; Marejkova, M.; Hrbackova, H.; Mellmann, A.; Karch, H.; Fruth, A.; Drevinek, P.; Blahova, K.; Bielaszewska, M.; Nunvar, J. Attack of the clones: Whole genome-based characterization of two closely related enterohemorrhagic Escherichia coli O26 epidemic lineages. BMC Genomics 2018, 19, 647. [Google Scholar] [CrossRef] [PubMed]
- Dallman, T.J.; Chattaway, M.A.; Cowley, L.A.; Doumith, M.; Tewolde, R.; Wooldridge, D.J.; Underwood, A.; Ready, D.; Wain, J.; Foster, K. An investigation of the diversity of strains of enteroaggregative Escherichia coli isolated from cases associated with a large multi-pathogen foodborne outbreak in the UK. PLoS ONE 2014, 9, e98103. [Google Scholar] [CrossRef] [PubMed]
- Leimbach, A.; Poehlein, A.; Witten, A.; Scheutz, F.; Schukken, Y.; Daniel, R.; Dobrindt, U. Complete Genome Sequences of Escherichia coli Strains 1303 and ECC-1470 Isolated from Bovine Mastitis. Genome Announc. 2015, 3, e00182-15. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.; Lindsey, R.L.; Rowe, L.A.; Batra, D.; Stripling, D.; Garcia-Toledo, L.; Drapeau, D.; Knipe, K.; Strockbine, N. High-Quality Whole-Genome Sequences for 21 Enterotoxigenic Escherichia coli Strains Generated with PacBio Sequencing. Genome Announc. 2018, 6, e01311-17. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Garcia, F. Escherichia coli O104: H4 pathogenesis: An enteroaggregative E. coli/Shiga toxin-producing E. coli explosive cocktail of high virulence. Enterohemorrhagic Escherichia coli and Other Shiga Toxin-Producing E coli. Am. Soc. Microbiol. 2015, 533–539. [Google Scholar]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. PNAS 2009, 106, 19126–19131. [Google Scholar] [CrossRef]
- Baylis, C.L. Growth of pure cultures of Verocytotoxin-producing Escherichia coli in a range of enrichment media. J. Appl. Microbiol. 2008, 105, 1259–1265. [Google Scholar] [CrossRef]
- Leimbach, A.; Poehlein, A.; Vollmers, J.; Görlich, D.; Daniel, R.; Dobrindt, U. No evidence for a bovine mastitis Escherichia coli pathotype. BMC Genomics 2017, 18, 359. [Google Scholar] [CrossRef]
- Barbau-Piednoir, E.; Denayer, S.; Botteldoorn, N.; Dierick, K.; De Keersmaecker, S.C.J.; Roosens, N.H. Detection and discrimination of five E. coli pathotypes using a combinatory SYBR® Green qPCR screening system. Appl. Microbiol. Biotechnol. 2018, 102, 3267–3285. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Ponsting, H.; Ning, Z.; SMALT, A. New Mapper for DNA Sequencing Reads. F1000Posters 2010. Available online: https://f1000research.com/posters/327 (accessed on 1 July 2020).
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef] [PubMed]
- Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner; Lawrence Berkeley National Lab.(LBNL): Berkeley, CA, USA, 2014. [Google Scholar]
- Talevich, E.; Invergo, B.M.; Cock, P.J.A.; Chapman, B.A. Bio.Phylo: A unified toolkit for processing, analyzing and visualizing phylogenetic trees in Biopython. BMC Bioinformatics 2012, 13, 209. [Google Scholar] [CrossRef] [PubMed]
- Inouye, M.; Dashnow, H.; Raven, L.A.; Schultz, M.B.; Pope, B.J.; Tomita, T.; Zobel, J.; Holt, K.E. SRST2: Rapid genomic surveillance for public health and hospital microbiology labs. Genome Med. 2014, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Joensen, K.G.; Scheutz, F.; Lund, O.; Hasman, H.; Kaas, R.S.; Nielsen, E.M.; Aarestrup, F.M. Real-Time Whole-Genome Sequencing for Routine Typing, Surveillance, and Outbreak Detection of Verotoxigenic Escherichia coli. Carroll KC, editor. J. Clin. Microbiol. 2014, 52, 1501–1510. [Google Scholar] [CrossRef]
- Joensen, K.G.; Tetzschner, A.M.M.; Iguchi, A.; Aarestrup, F.M.; Scheutz, F. Rapid and Easy In Silico Serotyping of Escherichia coli Isolates by Use of Whole-Genome Sequencing Data. Carroll KC. J. Clin. Microbiol. 2015, 53, 2410–2426. [Google Scholar] [CrossRef]
- ISO. ISO/TS 13136:2012. Microbiology of food and animal feed-Real-time polymerase chain reaction (PCR)-based method for the detection of food-borne pathogens-Horizontal method for the detection of Shiga toxin-producing Escherichia coli (STEC). Available online: https://www.iso.org/standard/53328.html (accessed on 1 July 2020).
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N. The sequence alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef]
- Page, A.J.; Taylor, B.; Delaney, A.J.; Soares, J.; Seemann, T.; Keane, J.A.; Harris, S.R. SNP-sites: Rapid efficient extraction of SNPs from multi-FASTA alignments. Microb. Genom. 2016, 2, e000056. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolate | Serotype | Pathotype | Additional Information | Reference | Accession Number |
|---|---|---|---|---|---|
| TIAC1151 | O157:H7 | STEC | Beef meat, outbreak | [39,40] | SRR10201483 |
| TIAC1152 | O157:H7 | STEC | Beef meat, outbreak | [39,40] | SRR10201465 |
| TIAC1153 | O157:H7 | STEC | Bovine Carcass swab, sporadic | [39,40] | SRR10201452 |
| TIAC1165 | O157:H7 | STEC | Human faeces, outbreak | [39,40] | SRR10201427 |
| TIAC1169 | O157:H7 | STEC | Human faeces, outbreak | [39,40] | SRR10201416 |
| TIAC1638 | O157:H7 | STEC | Human faeces, sporadic | [39,40] | SRR10201408 |
| TIAC1660 | O113:H21 | STEC | Human faeces, sporadic | [39,40] | SRR10201398 |
| C227-11 | O104:H4 | EAEC | stx-negative O104:H4 strain | [42] | ERR883742 |
| PNUSAE001801 | O167 | EPEC | PulseNet STEC genome reference library | PRJNA218110 | SRR2982117 |
| PNUSAE001802 | O167 | EPEC | PulseNet STEC genome reference library | PRJNA218110 | SRR2982118 |
| 2011C-3282 | O26:H11 | STEC | PulseNet STEC genome reference library | [43] | SRR3360214 |
| 2011C-3274 | O26:H11 | STEC | PulseNet STEC genome reference library | [43] | SRR6373714 |
| 0216-13 | O104:H4 | EAEC | stx-negative O104:H4 strain | [44] | SRX522695 |
| 1303 | O70:H32 | MAEC | E. coli strain isolated from bovine mastitis | [45] | SRR3492218 |
| 90-9281 | O128:H27 | ETEC | Enterotoxigenic E. coli strain collected in 1988 in Bangladesh | [46] | NZ_CP024243.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saltykova, A.; Buytaers, F.E.; Denayer, S.; Verhaegen, B.; Piérard, D.; Roosens, N.H.C.; Marchal, K.; De Keersmaecker, S.C.J. Strain-Level Metagenomic Data Analysis of Enriched In Vitro and In Silico Spiked Food Samples: Paving the Way towards a Culture-Free Foodborne Outbreak Investigation Using STEC as a Case Study. Int. J. Mol. Sci. 2020, 21, 5688. https://doi.org/10.3390/ijms21165688
Saltykova A, Buytaers FE, Denayer S, Verhaegen B, Piérard D, Roosens NHC, Marchal K, De Keersmaecker SCJ. Strain-Level Metagenomic Data Analysis of Enriched In Vitro and In Silico Spiked Food Samples: Paving the Way towards a Culture-Free Foodborne Outbreak Investigation Using STEC as a Case Study. International Journal of Molecular Sciences. 2020; 21(16):5688. https://doi.org/10.3390/ijms21165688
Chicago/Turabian StyleSaltykova, Assia, Florence E. Buytaers, Sarah Denayer, Bavo Verhaegen, Denis Piérard, Nancy H. C. Roosens, Kathleen Marchal, and Sigrid C. J. De Keersmaecker. 2020. "Strain-Level Metagenomic Data Analysis of Enriched In Vitro and In Silico Spiked Food Samples: Paving the Way towards a Culture-Free Foodborne Outbreak Investigation Using STEC as a Case Study" International Journal of Molecular Sciences 21, no. 16: 5688. https://doi.org/10.3390/ijms21165688
APA StyleSaltykova, A., Buytaers, F. E., Denayer, S., Verhaegen, B., Piérard, D., Roosens, N. H. C., Marchal, K., & De Keersmaecker, S. C. J. (2020). Strain-Level Metagenomic Data Analysis of Enriched In Vitro and In Silico Spiked Food Samples: Paving the Way towards a Culture-Free Foodborne Outbreak Investigation Using STEC as a Case Study. International Journal of Molecular Sciences, 21(16), 5688. https://doi.org/10.3390/ijms21165688