Experimentally Determined Long Intrinsically Disordered Protein Regions Are Now Abundant in the Protein Data Bank

 , , ,
, , ,  , and
, and
Abstract
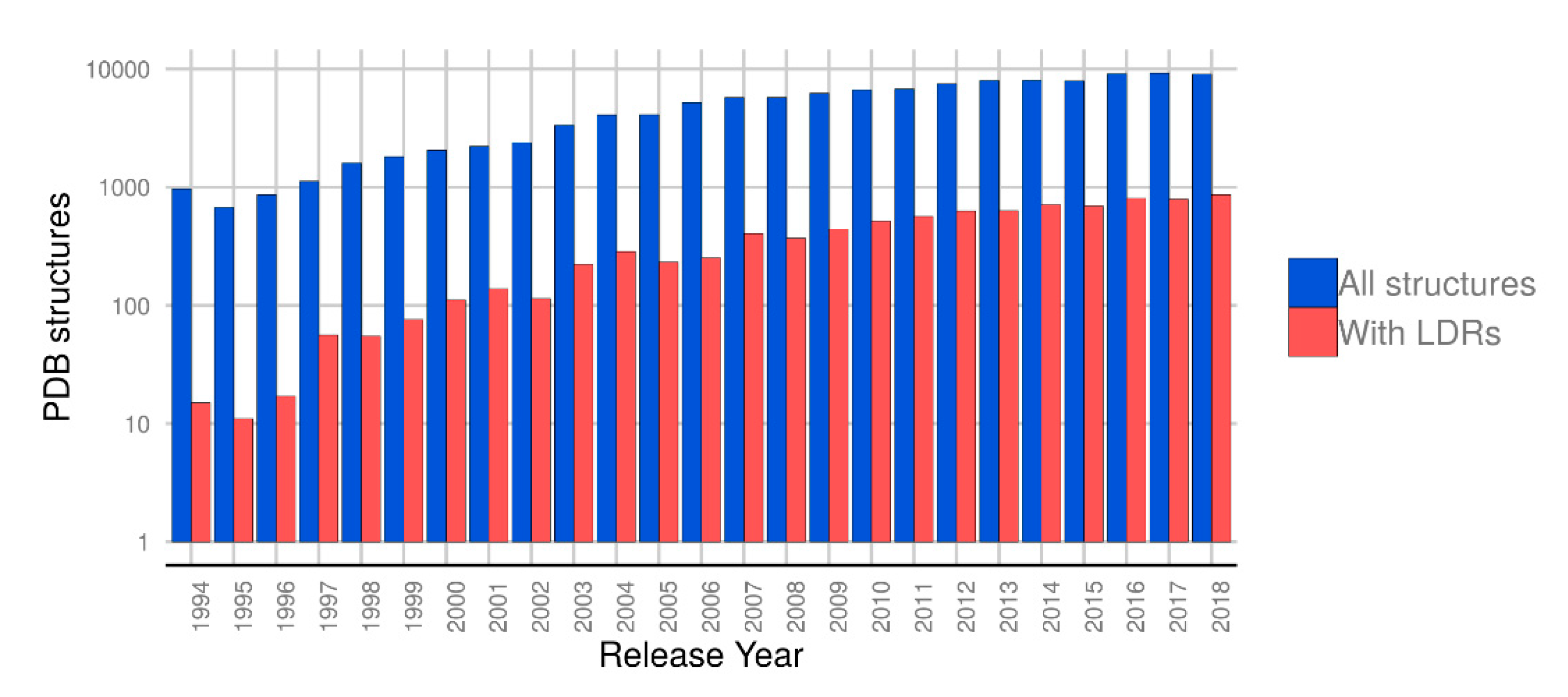
1. Introduction
2. Results
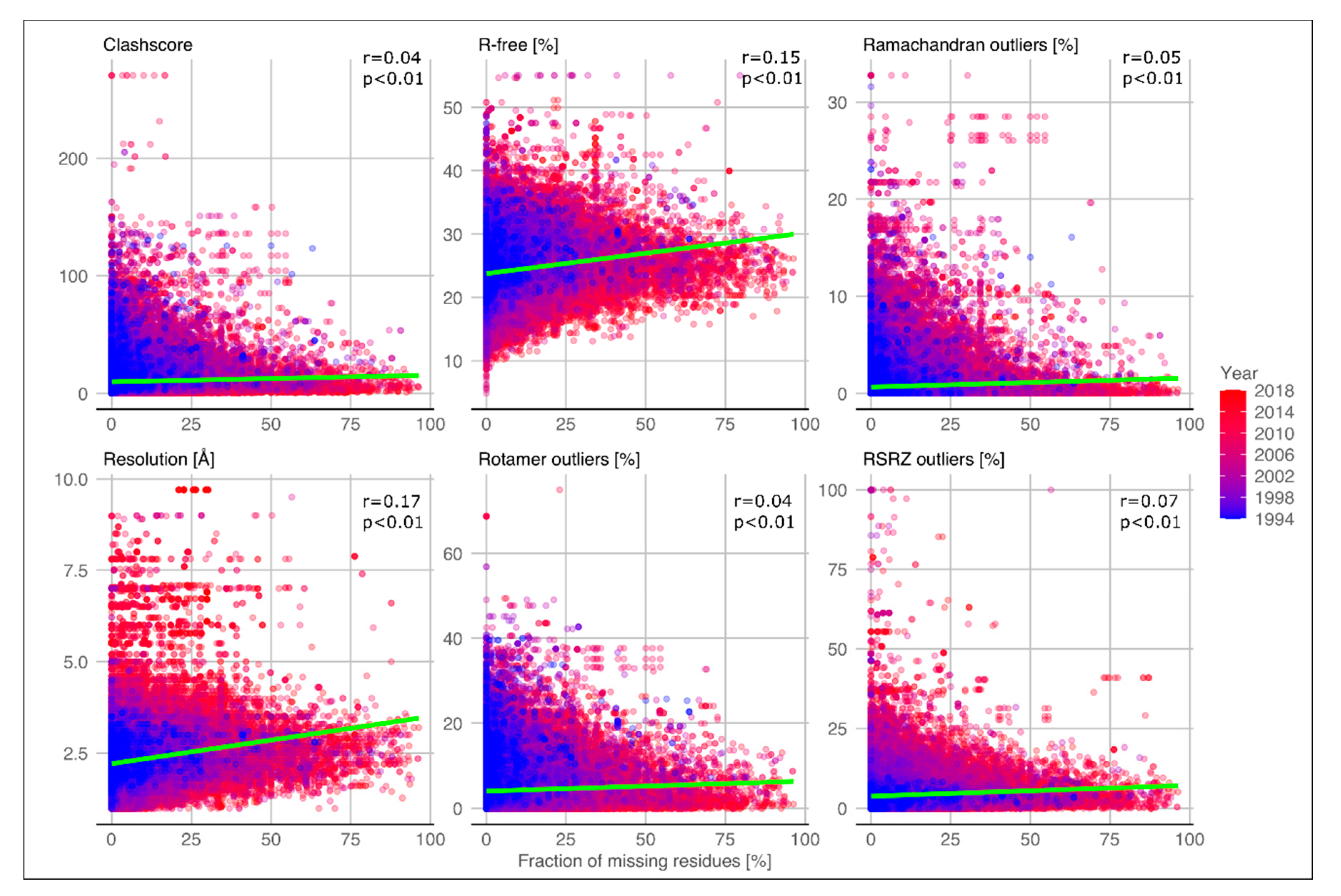
2.1. Quality of Structures with Disordered Regions
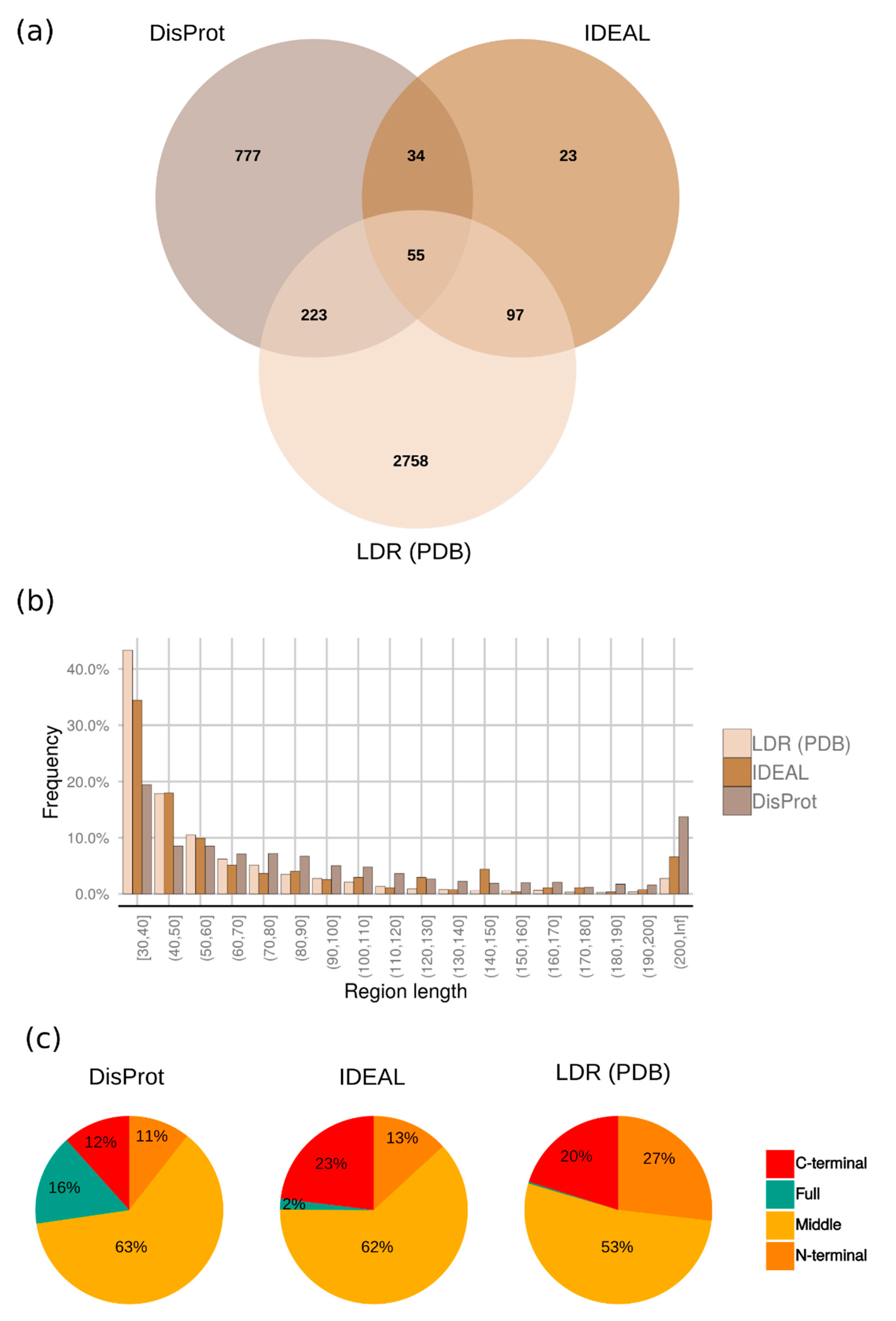
2.2. Distribution of Disordered Regions
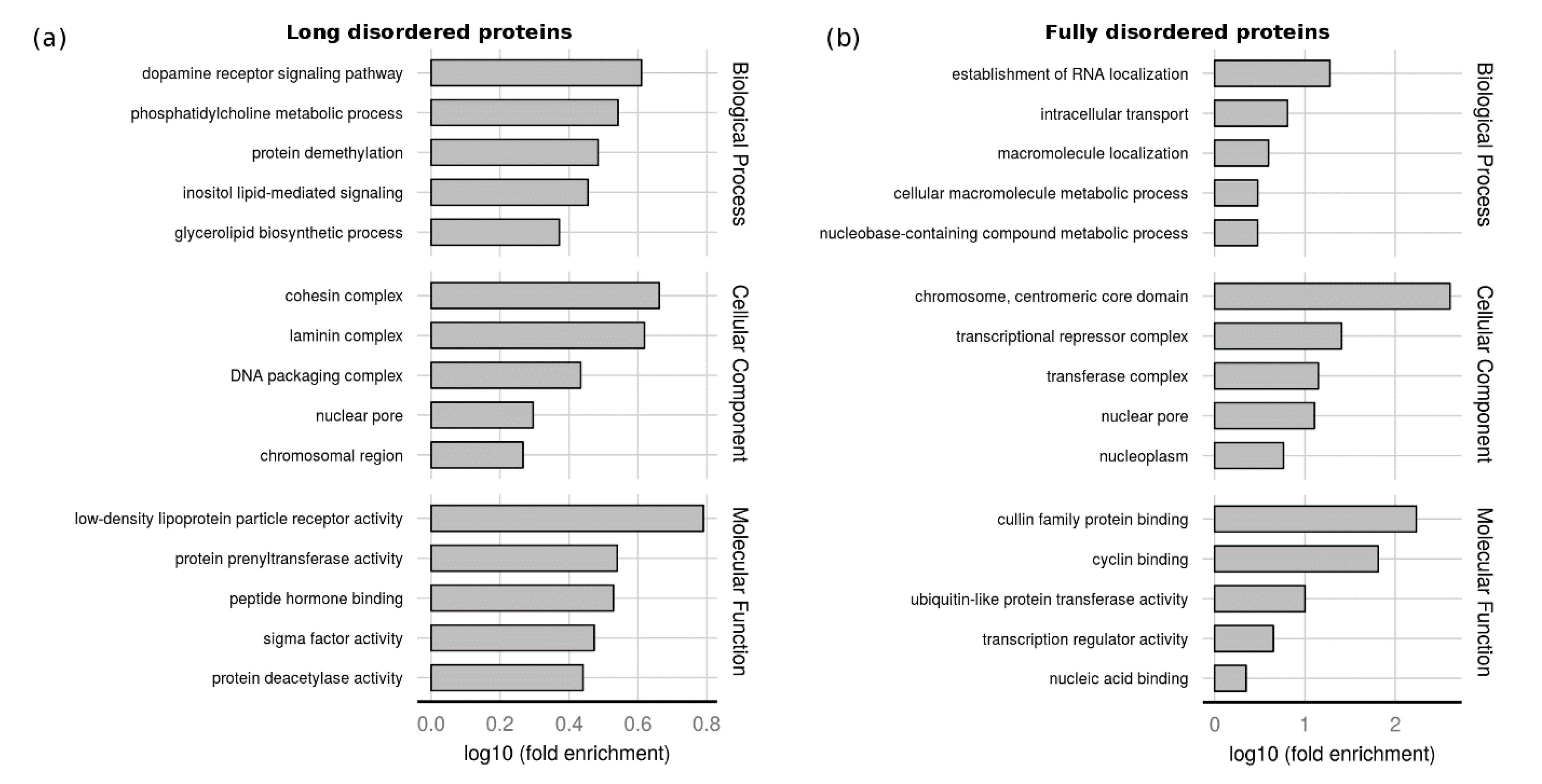
2.3. Function of Proteins with LDRs
2.4. Assessment of Disorder Predictors
3. Discussion
4. Materials and Methods
4.1. Long Disorder Data
4.2. GO-Terms Enrichment Analysis
4.3. Disorder Prediction and Evaluation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| IDR | Intrinsically disordered region |
| IDP | Intrinsically disordered protein |
| LDR | Long disordered region |
| SDR | Short disordered region |
References
- Habchi, J.; Tompa, P.; Longhi, S.; Uversky, V.N. Introducing protein intrinsic disorder. Chem. Rev. 2014, 114, 6561–6588. [Google Scholar] [CrossRef] [PubMed]
- Van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of Intrinsically Disordered Regions and Proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Vucetic, S.; Brown, C.J.; Dunker, A.K.; Obradovic, Z. Flavors of protein disorder. Proteins 2003, 52, 573–584. [Google Scholar] [CrossRef]
- Martin, A.J.M.; Walsh, I.; Tosatto, S.C.E. MOBI: A web server to define and visualize structural mobility in NMR protein ensembles. Bioinformatics 2010, 26, 2916–2917. [Google Scholar] [CrossRef]
- Piovesan, D.; Tosatto, S.C.E. Mobi 2.0: An improved method to define intrinsic disorder, mobility and linear binding regions in protein structures. Bioinforma. Oxf. Engl. 2018, 34, 122–123. [Google Scholar] [CrossRef]
- Bellay, J.; Han, S.; Michaut, M.; Kim, T.; Costanzo, M.; Andrews, B.J.; Boone, C.; Bader, G.D.; Myers, C.L.; Kim, P.M. Bringing order to protein disorder through comparative genomics and genetic interactions. Genome Biol. 2011, 12, R14. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Coupling of folding and binding for unstructured proteins. Curr. Opin. Struct. Biol. 2002, 12, 54–60. [Google Scholar] [CrossRef]
- Tompa, P.; Fuxreiter, M. Fuzzy complexes: Polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci. 2008, 33, 2–8. [Google Scholar] [CrossRef]
- Necci, M.; Piovesan, D.; Dosztanyi, Z.; Tompa, P.; Tosatto, S.C.E. A comprehensive assessment of long intrinsic protein disorder from the DisProt database. Bioinformatics 2017. [Google Scholar] [CrossRef]
- Ward, J.J.; Sodhi, J.S.; McGuffin, L.J.; Buxton, B.F.; Jones, D.T. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 2004, 337, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Vucetic, S.; Iakoucheva, L.M.; Oldfield, C.J.; Dunker, A.K.; Uversky, V.N.; Obradovic, Z. Functional Anthology of Intrinsic Disorder. I. Biological Processes and Functions of Proteins with Long Disordered Regions. J. Proteome Res. 2007, 6, 1882–1898. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.J.; Takayama, S.; Campen, A.M.; Vise, P.; Marshall, T.W.; Oldfield, C.J.; Williams, C.J.; Dunker, A.K. Evolutionary rate heterogeneity in proteins with long disordered regions. J. Mol. Evol. 2002, 55, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P.; Fuxreiter, M.; Oldfield, C.J.; Simon, I.; Dunker, A.K.; Uversky, V.N. Close encounters of the third kind: Disordered domains and the interactions of proteins. BioEssays News Rev. Mol. Cell. Dev. Biol. 2009, 31, 328–335. [Google Scholar] [CrossRef]
- Le Gall, T.; Romero, P.R.; Cortese, M.S.; Uversky, V.N.; Dunker, A.K. Intrinsic disorder in the Protein Data Bank. J. Biomol. Struct. Dyn. 2007, 24, 325–342. [Google Scholar] [CrossRef]
- Oldfield, C.J.; Xue, B.; Van, Y.-Y.; Ulrich, E.L.; Markley, J.L.; Dunker, A.K.; Uversky, V.N. Utilization of protein intrinsic disorder knowledge in structural proteomics. Biochim. Biophys. Acta 2013, 1834, 487–498. [Google Scholar] [CrossRef][Green Version]
- Zhang, Y.; Stec, B.; Godzik, A. Between order and disorder in protein structures–analysis of “dual personality” fragments in proteins. Structure 2007, 15, 1141–1147. [Google Scholar] [CrossRef]
- Burley, S.K.; Berman, H.M.; Bhikadiya, C.; Bi, C.; Chen, L.; Di Costanzo, L.; Christie, C.; Dalenberg, K.; Duarte, J.M.; Dutta, S.; et al. RCSB Protein Data Bank: Biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2019, 47, D464–D474. [Google Scholar] [CrossRef]
- Sormanni, P.; Piovesan, D.; Heller, G.T.; Bonomi, M.; Kukic, P.; Camilloni, C.; Fuxreiter, M.; Dosztanyi, Z.; Pappu, R.V.; Babu, M.M.; et al. Simultaneous quantification of protein order and disorder. Nat. Chem. Biol. 2017, 13, 339–342. [Google Scholar] [CrossRef]
- Nogales, E. The development of cryo-EM into a mainstream structural biology technique. Nat. Methods 2016, 13, 24–27. [Google Scholar] [CrossRef]
- Nwanochie, E.; Uversky, V.N. Structure Determination by Single-Particle Cryo-Electron Microscopy: Only the Sky (and Intrinsic Disorder) is the Limit. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; Radivojac, P.; Vucetic, S.; Dunker, A.K.; Obradovic, Z. Length-dependent prediction of protein intrinsic disorder. BMC Bioinform. 2006, 7, 208. [Google Scholar] [CrossRef]
- Shoemaker, S.C.; Ando, N. X-rays in the Cryo-EM Era: Structural Biology’s Dynamic Future. Biochemistry 2018, 57, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Wlodawer, A.; Li, M.; Dauter, Z. High-Resolution Cryo-EM Maps and Models: A Crystallographer’s Perspective. Structure 2017, 25, 1589–1597.e1. [Google Scholar] [CrossRef]
- Brzezinski, D.; Dauter, Z.; Minor, W.; Jaskolski, M. On the evolution of the quality of macromolecular models in the PDB. FEBS J. 2020. [Google Scholar] [CrossRef]
- Shao, C.; Yang, H.; Westbrook, J.D.; Young, J.Y.; Zardecki, C.; Burley, S.K. Multivariate Analyses of Quality Metrics for Crystal Structures in the PDB Archive. Structure 2017, 25, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Gore, S.; Sanz García, E.; Hendrickx, P.M.S.; Gutmanas, A.; Westbrook, J.D.; Yang, H.; Feng, Z.; Baskaran, K.; Berrisford, J.M.; Hudson, B.P.; et al. Validation of Structures in the Protein Data Bank. Structure 2017, 25, 1916–1927. [Google Scholar] [CrossRef]
- Hatos, A.; Hajdu-Soltész, B.; Monzon, A.M.; Palopoli, N.; Álvarez, L.; Aykac-Fas, B.; Bassot, C.; Benítez, G.I.; Bevilacqua, M.; Chasapi, A.; et al. DisProt: Intrinsic protein disorder annotation in 2020. Nucleic Acids Res. 2020, 48, D269–D276,. [Google Scholar] [CrossRef]
- Fukuchi, S.; Amemiya, T.; Sakamoto, S.; Nobe, Y.; Hosoda, K.; Kado, Y.; Murakami, S.D.; Koike, R.; Hiroaki, H.; Ota, M. IDEAL in 2014 illustrates interaction networks composed of intrinsically disordered proteins and their binding partners. Nucleic Acids Res. 2014, 42, D320–D325. [Google Scholar] [CrossRef]
- Piovesan, D.; Tabaro, F.; Mičetić, I.; Necci, M.; Quaglia, F.; Oldfield, C.J.; Aspromonte, M.C.; Davey, N.E.; Davidović, R.; Dosztányi, Z.; et al. DisProt 7.0: A major update of the database of disordered proteins. Nucleic Acids Res. 2017, 45, D1123–D1124. [Google Scholar] [CrossRef]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef]
- Mohan, A.; Uversky, V.N.; Radivojac, P. Influence of sequence changes and environment on intrinsically disordered proteins. PLoS Comput. Biol. 2009, 5, e1000497. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- The Gene Ontology Consortium The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [CrossRef] [PubMed]
- Dunker, A.K.; Brown, C.J.; Lawson, J.D.; Iakoucheva, L.M.; Obradović, Z. Intrinsic Disorder and Protein Function. Biochemistry 2002, 41, 6573–6582. [Google Scholar] [CrossRef] [PubMed]
- Deiana, A.; Forcelloni, S.; Porrello, A.; Giansanti, A. Intrinsically disordered proteins and structured proteins with intrinsically disordered regions have different functional roles in the cell. PLoS ONE 2019, 14. [Google Scholar] [CrossRef]
- Necci, M.; Piovesan, D.; Tosatto, S.C.E. Large-scale analysis of intrinsic disorder flavors and associated functions in the protein sequence universe. Protein Sci. Publ. Protein Soc. 2016, 25, 2164–2174. [Google Scholar] [CrossRef]
- Vucetic, S.; Xie, H.; Iakoucheva, L.M.; Oldfield, C.J.; Dunker, A.K.; Obradovic, Z.; Uversky, V.N. Functional Anthology of Intrinsic Disorder. II. Cellular Components, Domains, Technical Terms, Developmental Processes and Coding Sequence Diversities Correlated with Long Disordered Regions. J. Proteome Res. 2007, 6, 1899–1916. [Google Scholar] [CrossRef]
- Walsh, I.; Martin, A.J.M.; Di Domenico, T.; Tosatto, S.C.E. ESpritz: Accurate and fast prediction of protein disorder. Bioinformatics 2012, 28, 503–509. [Google Scholar] [CrossRef]
- Felli, I.C.; Pierattelli, R. Intrinsically Disordered Proteins Studied by NMR Spectroscopy; Springer: Cham, Switzerland, 2015; ISBN 978-3-319-20164-1. [Google Scholar]
- Walsh, I.; Giollo, M.; Di Domenico, T.; Ferrari, C.; Zimmermann, O.; Tosatto, S.C.E. Comprehensive large-scale assessment of intrinsic protein disorder. Bioinformatics 2015, 31, 201–208. [Google Scholar] [CrossRef]
- Fichó, E.; Reményi, I.; Simon, I.; Mészáros, B. MFIB: A repository of protein complexes with mutual folding induced by binding. Bioinformatics 2017, 33, 3682–3684. [Google Scholar] [CrossRef] [PubMed]
- Magyar, C.; Mentes, A.; Fichó, E.; Cserző, M.; Simon, I. Physical Background of the Disordered Nature of “Mutual Synergetic Folding” Proteins. Int. J. Mol. Sci. 2018, 19, 3340. [Google Scholar] [CrossRef]
- Mészáros, B.; Dobson, L.; Fichó, E.; Tusnády, G.E.; Dosztányi, Z.; Simon, I. Sequential, Structural and Functional Properties of Protein Complexes Are Defined by How Folding and Binding Intertwine. J. Mol. Biol. 2019, 431, 4408–4428. [Google Scholar] [CrossRef]
- Mentes, A.; Magyar, C.; Fichó, E.; Simon, I. Analysis of Heterodimeric “Mutual Synergistic Folding”-Complexes. Int. J. Mol. Sci. 2019, 20, 5136. [Google Scholar] [CrossRef]
- Fuxreiter, M. Fuzziness in Protein Interactions-A Historical Perspective. J. Mol. Biol. 2018, 430, 2278–2287. [Google Scholar] [CrossRef]
- Miskei, M.; Horvath, A.; Vendruscolo, M.; Fuxreiter, M. Sequence-Based Prediction of Fuzzy Protein Interactions. J. Mol. Biol. 2020, 432, 2289–2303. [Google Scholar] [CrossRef] [PubMed]
- Schad, E.; Fichó, E.; Pancsa, R.; Simon, I.; Dosztányi, Z.; Mészáros, B. DIBS: A repository of disordered binding sites mediating interactions with ordered proteins. Bioinformatics 2018, 34, 535–537. [Google Scholar] [CrossRef] [PubMed]
- Mészáros, B.; Erdős, G.; Szabó, B.; Schád, É.; Tantos, Á.; Abukhairan, R.; Horváth, T.; Murvai, N.; Kovács, O.P.; Kovács, M.; et al. PhaSePro: The database of proteins driving liquid–liquid phase separation. Nucleic Acids Res. 2020, 48, D360–D367. [Google Scholar] [CrossRef] [PubMed]
- UniProt Consortium UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [CrossRef] [PubMed]
- Piovesan, D.; Tabaro, F.; Paladin, L.; Necci, M.; Micetic, I.; Camilloni, C.; Davey, N.; Dosztányi, Z.; Mészáros, B.; Monzon, A.M.; et al. MobiDB 3.0: More annotations for intrinsic disorder, conformational diversity and interactions in proteins. Nucleic Acids Res. 2018, 46, D471–D476. [Google Scholar] [CrossRef]
- Necci, M.; Piovesan, D.; Dosztányi, Z.; Tosatto, S.C.E. MobiDB-lite: Fast and highly specific consensus prediction of intrinsic disorder in proteins. Bioinformatics 2017, 33, 1402–1404. [Google Scholar] [CrossRef] [PubMed]
- Dosztányi, Z.; Csizmok, V.; Tompa, P.; Simon, I. IUPred: Web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 2005, 21, 3433–3434. [Google Scholar] [CrossRef] [PubMed]
- Linding, R.; Jensen, L.J.; Diella, F.; Bork, P.; Gibson, T.J.; Russell, R.B. Protein disorder prediction: Implications for structural proteomics. Structure 2003, 11, 1453–1459. [Google Scholar] [CrossRef] [PubMed]
- Linding, R.; Russell, R.B.; Neduva, V.; Gibson, T.J. GlobPlot: Exploring protein sequences for globularity and disorder. Nucleic Acids Res. 2003, 31, 3701–3708. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Proteins | Median Protein Length | Short Disordered Regions (SDR) | Long Disordered Regions (LDR) | Missing Residues (Disordered) | Observed Residues (Structure) | Unknown Residues |
|---|---|---|---|---|---|---|---|
| Proteins with LDRs | 3133 | 473.0 | 3742 | 3553 | 270,656 (14.1%) | 1,140,513 (59.4%) | 509,174 (26.5%) |
| Proteins with SDRs | 15,968 | 358.5 | 25,087 | 0 | 303,611 (3.9%) | 4,724,722 (60.4%) | 2,787,457 (35.7%) |
| Structured proteins | 18,294 | 274.0 | 0 | 0 | 28,338 (0.4%) | 4,242,966 (62.8%) | 2,485,372 (36.8%) |
| Total | 37,395 | 441.0 | 28,829 | 3533 | 602,605 (3.7%) | 10,108,201 (61,3%) | 5,782,003 (35.1%) |
| MCC | F1 Score | Accuracy | Precision | Specificity | Recall | |
|---|---|---|---|---|---|---|
| Espritz-X | 0.456 | 0.498 | 0.672 | 0.738 | 0.968 | 0.376 |
| IUPred-short | 0.411 | 0.473 | 0.662 | 0.656 | 0.954 | 0.370 |
| MobiDB-Lite | 0.389 | 0.350 | 0.606 | 0.872 | 0.992 | 0.219 |
| VSL2b | 0.384 | 0.516 | 0.720 | 0.432 | 0.800 | 0.640 |
| IUPred-long | 0.375 | 0.456 | 0.655 | 0.590 | 0.939 | 0.372 |
| DisEMBL-465 | 0.364 | 0.416 | 0.633 | 0.644 | 0.960 | 0.307 |
| Espritz-N | 0.361 | 0.485 | 0.683 | 0.478 | 0.872 | 0.493 |
| Espritz-D | 0.257 | 0.214 | 0.558 | 0.746 | 0.990 | 0.125 |
| DisEMBL-HL | 0.209 | 0.388 | 0.622 | 0.316 | 0.742 | 0.503 |
| GlobPlot | 0.192 | 0.329 | 0.588 | 0.368 | 0.879 | 0.297 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monzon, A.M.; Necci, M.; Quaglia, F.; Walsh, I.; Zanotti, G.; Piovesan, D.; Tosatto, S.C.E. Experimentally Determined Long Intrinsically Disordered Protein Regions Are Now Abundant in the Protein Data Bank. Int. J. Mol. Sci. 2020, 21, 4496. https://doi.org/10.3390/ijms21124496
Monzon AM, Necci M, Quaglia F, Walsh I, Zanotti G, Piovesan D, Tosatto SCE. Experimentally Determined Long Intrinsically Disordered Protein Regions Are Now Abundant in the Protein Data Bank. International Journal of Molecular Sciences. 2020; 21(12):4496. https://doi.org/10.3390/ijms21124496
Chicago/Turabian StyleMonzon, Alexander Miguel, Marco Necci, Federica Quaglia, Ian Walsh, Giuseppe Zanotti, Damiano Piovesan, and Silvio C. E. Tosatto. 2020. "Experimentally Determined Long Intrinsically Disordered Protein Regions Are Now Abundant in the Protein Data Bank" International Journal of Molecular Sciences 21, no. 12: 4496. https://doi.org/10.3390/ijms21124496
APA StyleMonzon, A. M., Necci, M., Quaglia, F., Walsh, I., Zanotti, G., Piovesan, D., & Tosatto, S. C. E. (2020). Experimentally Determined Long Intrinsically Disordered Protein Regions Are Now Abundant in the Protein Data Bank. International Journal of Molecular Sciences, 21(12), 4496. https://doi.org/10.3390/ijms21124496