Tau and Alpha Synuclein Synergistic Effect in Neurodegenerative Diseases: When the Periphery Is the Core


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Role of Tau in Physiological Condition in Axons
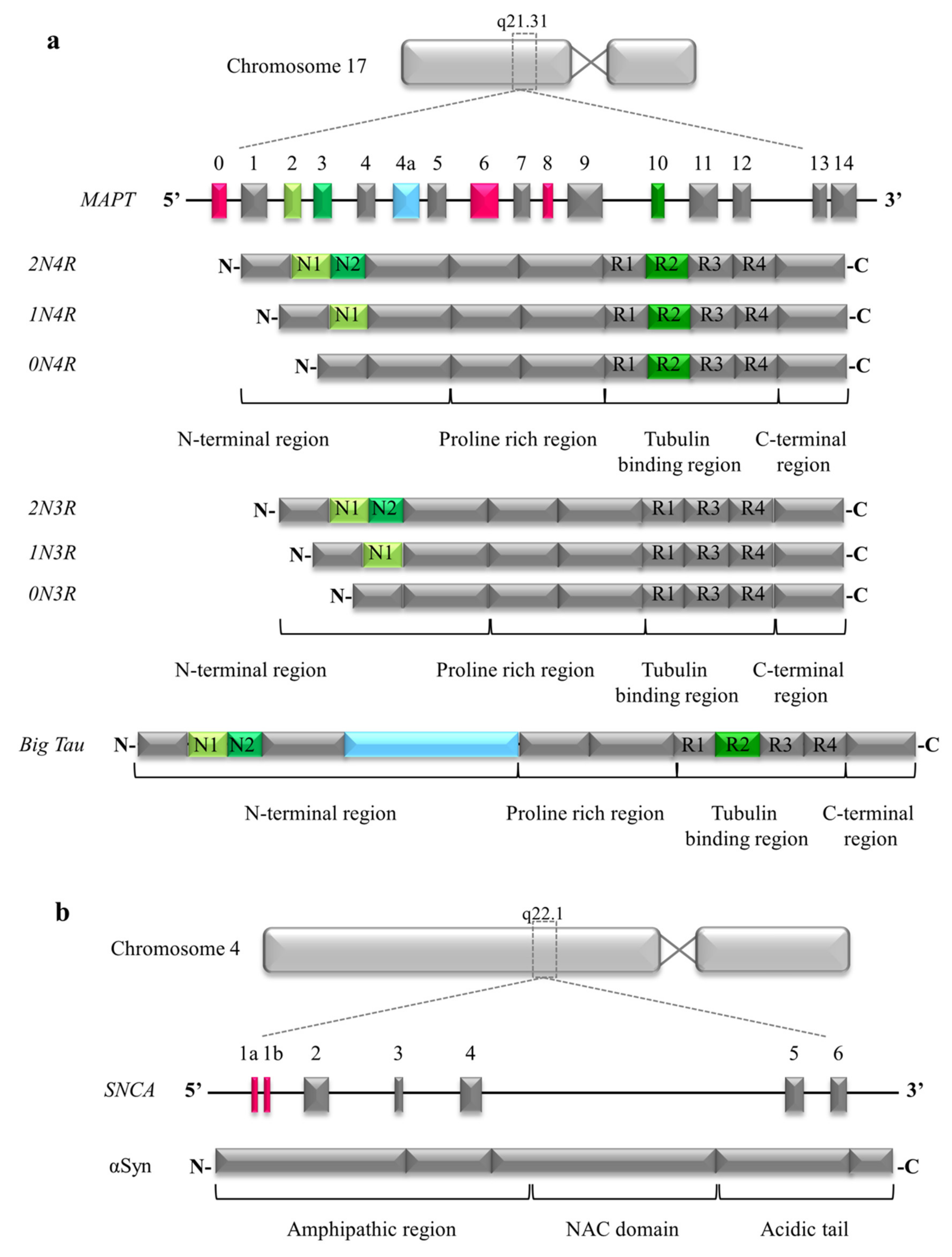
2.1. Structure of Tau
2.2. Big Tau in Peripheral Nervous System
2.3. Tau and Axonal Transport
2.4. Tau and Mitochondria
2.5. Tau at the Synaptic Terminals
3. The Role of Alpha Synuclein in Physiological Condition in Axons
3.1. Structure of Alpha Synuclein
3.2. Alpha Synuclein at the Synaptic Terminals
3.3. Alpha Synuclein and Mitochondria
3.4. Alpha Synuclein and Axonal Transport
4. The Role of Tau in Neurodegenerative Diseases
4.1. Tau Mutations Impair Microtubules Binding Affinity
4.2. Pathological Tau Hampers Axonal Transport
4.3. Neuroanatomical Stages of Tau Accumulation in Alzheimer’s Disease
5. The Role of Alpha Synuclein in Neurodegenerative Diseases
5.1. Axonal Transport Dysfunction Caused by Alpha Synuclein Aggregates May Be the Early Event in Neurodegeneration
5.2. Neuroanatomical Stages of Alpha Synuclein Accumulation in Parkinson’s Disease
6. Pathological Tau and the Peripheral Nervous System
6.1. Tau and Autonomic Nervous System
6.2. Tau and Somatosensory Nervous System
6.3. Tau and Olfactory Nervous System
7. Pathological Alpha Synuclein and the Peripheral Nervous System
7.1. Dual Hit Hypothesis
7.2. Alpha Synuclein in the Peripheral Nervous System as a Biomarker for Synucleinopathies
8. Tau and Alpha Synuclein Cross Talking in Causing Neurodegeneration
9. Tau and Alpha Synuclein Commonalities and Diversities
9.1. IDPs and Neurodegenerative Diseases
9.2. Aggregation, Propagation, and Prion Concept
9.3. Pathological Tau and Alpha Synuclein Target Different Neuronal Cells and Display Different Spreading Patterns
10. Conclusions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s Disease |
| PD | Parkinson’s Disease |
| αSyn | Alpha Synuclein |
| PNS | Peripheral Nervous System |
| CNS | Central Nervous System |
| IDP | Intrinsically Disordered Proteins |
| aa | amino acid |
| NAC | non-amyloid component |
| NFTs | NeuroFibrillary Tangles |
| NTs | Neuropil Threads |
| PSP | Progressive Sopranuclear Palsy |
| CBD | CorticoBasal Degeneration |
| PiD | Pick’s Disease |
| CTE | Chronic Traumatic Encephalopathy |
| AGD | Argyrophilic Grain Disease |
| PHFs | Paired Helical Filaments |
| DMV | Dorsal Motor Nucleus of the Vagal nerve |
| DLB | Dementia with Lewy Bodies |
| MSA | Multiple System Atrophy |
| LBs | Lewy Bodies |
| LNs | Lewy Neurites |
| RBD | Rem Behavior Disorder |
References
- Nussbaum, R.L. Alzheimer’s disease and Parkinson’s disease. N. Engl. J. Med. 2003, 348, 1356–1364. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M. NEURODEGENERATION. Alzheimer’s and Parkinson’s diseases: The prion concept in relation to assembled Abeta, tau, and alpha-synuclein. Science 2015, 349, 1255555. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Masuda-Suzukake, M.; Falcon, B. Like prions: The propagation of aggregated tau and alpha-synuclein in neurodegeneration. Brain 2017, 140, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Andreadis, A. Misregulation of tau alternative splicing in neurodegeneration and dementia. Prog. Mol. Subcell. Biol. 2006, 44, 89–107. [Google Scholar]
- Lee, G.; Cowan, N.; Kirschner, M. The primary structure and heterogeneity of tau protein from mouse brain. Science 1988, 239, 285–288. [Google Scholar] [CrossRef]
- Andreadis, A.; Broderick, J.A.; Kosik, K.S. Relative exon affinities and suboptimal splice site signals lead to non-equivalence of two cassette exons. Nucleic Acids Res. 1995, 23, 3585–3593. [Google Scholar] [CrossRef][Green Version]
- Schoenfeld, T.A.; Obar, R.A. Diverse distribution and function of fibrous microtubule-associated proteins in the nervous system. Int. Rev. Cytol. 1994, 151, 67–137. [Google Scholar]
- LoPresti, P.; Szuchet, S.; Papasozomenos, S.C.; Zinkowski, R.P.; Binder, L.I. Functional implications for the microtubule-associated protein tau: Localization in oligodendrocytes. Proc. Natl. Acad. Sci. USA 1995, 92, 10369–10373. [Google Scholar] [CrossRef]
- Papasozomenos, S.C.; Binder, L.I. Phosphorylation determines two distinct species of Tau in the central nervous system. Cell Motil. Cytoskelet. 1987, 8, 210–226. [Google Scholar] [CrossRef]
- Yi, S.; Liu, Q.; Wang, X.; Qian, T.; Wang, H.; Zha, G.; Yu, J.; Wang, P.; Gu, X.; Chu, D.; et al. Tau modulates Schwann cell proliferation, migration and differentiation following peripheral nerve injury. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Jakes, R.; Rutherford, D.; Crowther, R.A. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 1989, 3, 519–526. [Google Scholar] [CrossRef]
- Goedert, M.; Jakes, R. Expression of separate isoforms of human tau protein: Correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 1990, 9, 4225–4230. [Google Scholar] [CrossRef]
- Majounie, E.; Cross, W.; Newsway, V.; Dillman, A.; Vandrovcova, J.; Morris, C.M.; Nalls, M.A.; Ferrucci, L.; Owen, M.J.; O’Donovan, M.C.; et al. Variation in tau isoform expression in different brain regions and disease states. Neurobiol. Aging 2013, 34, 1922.e7–1922.e12. [Google Scholar] [CrossRef] [PubMed]
- Brandt, R.; Leger, J.; Lee, G. Interaction of tau with the neural plasma membrane mediated by tau’s amino-terminal projection domain. J. Cell Biol. 1995, 131, 1327–1340. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Newman, S.T.; Gard, D.L.; Band, H.; Panchamoorthy, G. Tau interacts with src-family non-receptor tyrosine kinases. J. Cell Sci. 1998, 111(Pt. 21), 3167–3177. [Google Scholar]
- Hwang, S.C.; Jhon, D.Y.; Bae, Y.S.; Kim, J.H.; Rhee, S.G. Activation of phospholipase C-gamma by the concerted action of tau proteins and arachidonic acid. J. Biol. Chem. 1996, 271, 18342–18349. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G. Ordered Assembly of Tau Protein and Neurodegeneration. Adv. Exp. Med. Biol. 2019, 1184, 3–21. [Google Scholar]
- Jeganathan, S.; von Bergen, M.; Brutlach, H.; Steinhoff, H.J.; Mandelkow, E. Global hairpin folding of tau in solution. Biochemistry 2006, 45, 2283–2293. [Google Scholar] [CrossRef]
- Correas, I.; Padilla, R.; Avila, J. The tubulin-binding sequence of brain microtubule-associated proteins, tau and MAP-2, is also involved in actin binding. Biochem. J. 1990, 269, 61–64. [Google Scholar] [CrossRef]
- Jensen, P.H.; Hager, H.; Nielsen, M.S.; Hojrup, P.; Gliemann, J.; Jakes, R. Alpha-synuclein binds to Tau and stimulates the protein kinase A-catalyzed tau phosphorylation of serine residues 262 and 356. J. Biol. Chem. 1999, 274, 25481–25489. [Google Scholar] [CrossRef]
- Mukrasch, M.D.; Bibow, S.; Korukottu, J.; Jeganathan, S.; Biernat, J.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol. 2009, 7, e1000034. [Google Scholar] [CrossRef]
- Melkova, K.; Zapletal, V.; Narasimhan, S.; Jansen, S.; Hritz, J.; Skrabana, R.; Zweckstetter, M.; Ringkjobing Jensen, M.; Blackledge, M.; Zidek, L. Structure and Functions of Microtubule Associated Proteins Tau and MAP2c: Similarities and Differences. Biomolecules 2019, 9, 105. [Google Scholar] [CrossRef]
- Himmler, A.; Drechsel, D.; Kirschner, M.W.; Martin, D.W., Jr. Tau consists of a set of proteins with repeated C-terminal microtubule-binding domains and variable N-terminal domains. Mol. Cell. Biol. 1989, 9, 1381–1388. [Google Scholar] [CrossRef] [PubMed]
- Drubin, D.G.; Caput, D.; Kirschner, M.W. Studies on the expression of the microtubule-associated protein, tau, during mouse brain development, with newly isolated complementary DNA probes. J. Cell Biol. 1984, 98, 1090–1097. [Google Scholar] [CrossRef]
- Fischer, I.; Baas, P.W. Resurrecting the Mysteries of Big Tau. Trends Neurosci. 2020. [Google Scholar] [CrossRef]
- Peng, I.; Binder, L.I.; Black, M.M. Cultured neurons contain a variety of microtubule-associated proteins. Brain Res. 1985, 361, 200–211. [Google Scholar] [CrossRef]
- Swanson, J.J.; Kuehl-Kovarik, M.C.; Wilson, M.C.; Elmquist, J.K.; Jacobson, C.D. Characterization and ontogeny of synapse-associated proteins in the developing facial and hypoglossal motor nuclei of the Brazilian opossum. J. Comp. Neurol. 1996, 368, 270–284. [Google Scholar] [CrossRef]
- Taleghany, N.; Oblinger, M.M. Regional distribution and biochemical characteristics of high molecular weight tau in the nervous system. J. Neurosci. Res. 1992, 33, 257–265. [Google Scholar] [CrossRef]
- Black, M.M.; Slaughter, T.; Moshiach, S.; Obrocka, M.; Fischer, I. Tau is enriched on dynamic microtubules in the distal region of growing axons. J. Neurosci. 1996, 16, 3601–3619. [Google Scholar] [CrossRef]
- Nothias, F.; Boyne, L.; Murray, M.; Tessler, A.; Fischer, I. The expression and distribution of tau proteins and messenger RNA in rat dorsal root ganglion neurons during development and regeneration. Neuroscience 1995, 66, 707–719. [Google Scholar] [CrossRef]
- Couchie, D.; Mavilia, C.; Georgieff, I.S.; Liem, R.K.; Shelanski, M.L.; Nunez, J. Primary structure of high molecular weight tau present in the peripheral nervous system. Proc. Natl. Acad. Sci. USA 1992, 89, 4378–4381. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Kanai, Y.; Cowan, N.J.; Hirokawa, N. Projection domains of MAP2 and tau determine spacings between microtubules in dendrites and axons. Nature 1992, 360, 674–677. [Google Scholar] [CrossRef] [PubMed]
- Boyne, L.J.; Tessler, A.; Murray, M.; Fischer, I. Distribution of Big tau in the central nervous system of the adult and developing rat. J. Comp. Neurol. 1995, 358, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Y.; Wang, C.; Destin, G.; Szaro, B.G. Microtubule-associated protein tau promotes neuronal class II -tubulin microtubule formation and axon elongation in embryonic Xenopus laevis. Eur. J. Neurosci. 2015, 41, 1263–1275. [Google Scholar] [CrossRef]
- Avila, J. Tau aggregation into fibrillar polymers: Taupathies. FEBS Lett. 2000, 476, 89–92. [Google Scholar] [CrossRef]
- Dujardin, S.; Hyman, B.T. Tau Prion-Like Propagation: State of the Art and Current Challenges. Adv. Exp. Med. Biol. 2019, 1184, 305–325. [Google Scholar]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef]
- Binder, L.I.; Frankfurter, A.; Rebhun, L.I. The distribution of tau in the mammalian central nervous system. J. Cell Biol. 1985, 101, 1371–1378. [Google Scholar] [CrossRef]
- Chen, Q.; Zhou, Z.; Zhang, L.; Wang, Y.; Zhang, Y.W.; Zhong, M.; Xu, S.C.; Chen, C.H.; Li, L.; Yu, Z.P. Tau protein is involved in morphological plasticity in hippocampal neurons in response to BDNF. Neurochem. Int. 2012, 60, 233–242. [Google Scholar] [CrossRef]
- Hoover, B.R.; Reed, M.N.; Su, J.; Penrod, R.D.; Kotilinek, L.A.; Grant, M.K.; Pitstick, R.; Carlson, G.A.; Lanier, L.M.; Yuan, L.L.; et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 2010, 68, 1067–1081. [Google Scholar] [CrossRef]
- Kimura, T.; Whitcomb, D.J.; Jo, J.; Regan, P.; Piers, T.; Heo, S.; Brown, C.; Hashikawa, T.; Murayama, M.; Seok, H.; et al. Microtubule-associated protein tau is essential for long-term depression in the hippocampus. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130144. [Google Scholar] [CrossRef] [PubMed]
- Maas, T.; Eidenmuller, J.; Brandt, R. Interaction of tau with the neural membrane cortex is regulated by phosphorylation at sites that are modified in paired helical filaments. J. Biol. Chem. 2000, 275, 15733–15740. [Google Scholar] [CrossRef] [PubMed]
- Sultan, A.; Nesslany, F.; Violet, M.; Begard, S.; Loyens, A.; Talahari, S.; Mansuroglu, Z.; Marzin, D.; Sergeant, N.; Humez, S.; et al. Nuclear tau, a key player in neuronal DNA protection. J. Biol. Chem. 2011, 286, 4566–4575. [Google Scholar] [CrossRef]
- Qiang, L.; Sun, X.; Austin, T.O.; Muralidharan, H.; Jean, D.C.; Liu, M.; Yu, W.; Baas, P.W. Tau does not stabilize axonal microtubules but rather enables them to have long labile domains. Curr. Biol. 2018, 28, 2181–2189.e2184. [Google Scholar] [CrossRef] [PubMed]
- Seitz, A.; Kojima, H.; Oiwa, K.; Mandelkow, E.M.; Song, Y.H.; Mandelkow, E. Single-molecule investigation of the interference between kinesin, tau and MAP2c. EMBO J. 2002, 21, 4896–4905. [Google Scholar] [CrossRef]
- Vershinin, M.; Carter, B.C.; Razafsky, D.S.; King, S.J.; Gross, S.P. Multiple-motor based transport and its regulation by Tau. Proc. Natl. Acad. Sci. USA 2007, 104, 87–92. [Google Scholar] [CrossRef]
- Dixit, R.; Ross, J.L.; Goldman, Y.E.; Holzbaur, E.L. Differential regulation of dynein and kinesin motor proteins by tau. Science 2008, 319, 1086–1089. [Google Scholar] [CrossRef]
- Kanaan, N.M.; Morfini, G.A.; LaPointe, N.E.; Pigino, G.F.; Patterson, K.R.; Song, Y.; Andreadis, A.; Fu, Y.; Brady, S.T.; Binder, L.I. Pathogenic forms of tau inhibit kinesin-dependent axonal transport through a mechanism involving activation of axonal phosphotransferases. J. Neurosci. 2011, 31, 9858–9868. [Google Scholar] [CrossRef]
- Magnani, E.; Fan, J.; Gasparini, L.; Golding, M.; Williams, M.; Schiavo, G.; Goedert, M.; Amos, L.A.; Spillantini, M.G. Interaction of tau protein with the dynactin complex. EMBO J. 2007, 26, 4546–4554. [Google Scholar] [CrossRef]
- Marx, A.; Pless, J.; Mandelkow, E.M.; Mandelkow, E. On the rigidity of the cytoskeleton: Are MAPs crosslinkers or spacers of microtubules? Cell. Mol. Biol. 2000, 46, 949–965. [Google Scholar]
- Mephon-Gaspard, A.; Boca, M.; Pioche-Durieu, C.; Desforges, B.; Burgo, A.; Hamon, L.; Pietrement, O.; Pastre, D. Role of tau in the spatial organization of axonal microtubules: Keeping parallel microtubules evenly distributed despite macromolecular crowding. Cell. Mol. Life Sci. 2016, 73, 3745–3760. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.; Li, Y.; Brautigan, D.L.; Gundersen, G.G. Protein phosphatase 1 is targeted to microtubules by the microtubule-associated protein Tau. J. Biol. Chem. 1998, 273, 21901–21908. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Qureshi, H.Y.; Cafferty, P.W.; Sobue, K.; Agarwal-Mawal, A.; Neufield, K.D.; Paudel, H.K. Glycogen synthase kinase-3,beta is complexed with tau protein in brain microtubules. J. Biol. Chem. 2002, 277, 11933–11940. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Hawkes, C.; Qureshi, H.Y.; Kar, S.; Paudel, H.K. Cyclin-dependent protein kinase 5 primes microtubule-associated protein tau site-specifically for glycogen synthase kinase 3 beta. Biochemistry 2006, 45, 3134–3145. [Google Scholar] [CrossRef]
- Qureshi, H.Y.; Paudel, H.K. Parkinsonian neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and alpha-synuclein mutations promote Tau protein phosphorylation at Ser262 and destabilize microtubule cytoskeleton in vitro. J. Biol. Chem. 2011, 286, 5055–5068. [Google Scholar] [CrossRef]
- Cieri, D.; Vicario, M.; Vallese, F.; D’Orsi, B.; Berto, P.; Grinzato, A.; Catoni, C.; De Stefani, D.; Rizzuto, R.; Brini, M.; et al. Tau localises within mitochondrial sub-compartments and its caspase cleavage affects ER-mitochondria interactions and cellular Ca2+ handling. BBA Mol. Basis Dis. 2018, 1864, 3247–3256. [Google Scholar] [CrossRef]
- Stoothoff, W.; Jones, P.B.; Spires-Jones, T.L.; Joyner, D.; Chhabra, E.; Bercury, K.; Fan, Z.; Xie, H.; Bacskai, B.; Edd, J.; et al. Differential effect of three-repeat and four-repeat tau on mitochondrial axonal transport. J. Neurochem. 2009, 111, 417–427. [Google Scholar] [CrossRef]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Clos, A.L.; Jackson, G.R.; Kayed, R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol. Neurodegener. 2011, 6, 39. [Google Scholar] [CrossRef]
- Briston, T.; Hicks, A.R. Mitochondrial dysfunction and neurodegenerative proteinopathies: Mechanisms and prospects for therapeutic intervention. Biochem. Soc. Trans. 2018, 46, 829–842. [Google Scholar] [CrossRef]
- Kandimalla, R.; Reddy, P.H. Multiple faces of dynamin-related protein 1 and its role in Alzheimer’s disease pathogenesis. Biochim. Biophys. Acta 2016, 1862, 814–828. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Lee, H.G.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 2009, 29, 9090–9103. [Google Scholar] [CrossRef] [PubMed]
- Stefanova, N.A.; Muraleva, N.A.; Korbolina, E.E.; Kiseleva, E.; Maksimova, K.Y.; Kolosova, N.G. Amyloid accumulation is a late event in sporadic Alzheimer’s disease-like pathology in nontransgenic rats. Oncotarget 2015, 6, 1396–1413. [Google Scholar] [CrossRef]
- Rai, S.N.; Singh, C.; Singh, A.; Singh, M.P.; Singh, B.K. Mitochondrial Dysfunction: A Potential Therapeutic Target to Treat Alzheimer’s Disease. Mol. Neurobiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Melov, S.; Adlard, P.A.; Morten, K.; Johnson, F.; Golden, T.R.; Hinerfeld, D.; Schilling, B.; Mavros, C.; Masters, C.L.; Volitakis, I.; et al. Mitochondrial Oxidative Stress Causes Hyperphosphorylation of Tau. PLoS ONE 2007, 2, e536. [Google Scholar] [CrossRef] [PubMed]
- Shahpasand, K.; Uemura, I.; Saito, T.; Asano, T.; Hata, K.; Shibata, K.; Toyoshima, Y.; Hasegawa, M.; Hisanaga, S. Regulation of mitochondrial transport and inter-microtubule spacing by tau phosphorylation at the sites hyperphosphorylated in Alzheimer’s disease. J. Neurosci. 2012, 32, 2430–2441. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Li, X.C.; Wang, Z.H.; Luo, Y.; Zhang, X.N.; Liu, X.P.; Feng, Q.; Wang, Q.; Yue, Z.Y.; Chen, Z.; et al. Tau accumulation impairs mitophagy via increasing mitochondrial membrane potential and reducing mitochondrial Parkin. Oncotarget 2016, 7, 17356–17368. [Google Scholar] [CrossRef] [PubMed]
- Decker, J.M.; Kruger, L.; Sydow, A.; Zhao, S.; Frotscher, M.; Mandelkow, E.; Mandelkow, E.M. Pro-aggregant Tau impairs mossy fiber plasticity due to structural changes and Ca(++) dysregulation. Acta Neuropathol. Commun. 2015, 3, 1–18. [Google Scholar] [CrossRef]
- Liu, C.; Song, X.; Nisbet, R.; Gotz, J. Co-immunoprecipitation with Tau Isoform-specific Antibodies Reveals Distinct Protein Interactions and Highlights a Putative Role for 2N Tau in Disease. J. Biol. Chem. 2016, 291, 8173–8188. [Google Scholar] [CrossRef]
- Wang, P.; Joberty, G.; Buist, A.; Vanoosthuyse, A.; Stancu, I.C.; Vasconcelos, B.; Pierrot, N.; Faelth-Savitski, M.; Kienlen-Campard, P.; Octave, J.N.; et al. Tau interactome mapping based identification of Otub1 as Tau deubiquitinase involved in accumulation of pathological Tau forms in vitro and in vivo. Acta Neuropathol. 2017, 133, 731–749. [Google Scholar] [CrossRef]
- McInnes, J.; Wierda, K.; Snellinx, A.; Bounti, L.; Wang, Y.C.; Stancu, I.C.; Apostolo, N.; Gevaert, K.; Dewachter, I.; Spires-Jones, T.L.; et al. Synaptogyrin-3 Mediates Presynaptic Dysfunction Induced by Tau. Neuron 2018, 97, 823–835.e828. [Google Scholar] [CrossRef]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wolfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Mondragon-Rodriguez, S.; Trillaud-Doppia, E.; Dudilot, A.; Bourgeois, C.; Lauzon, M.; Leclerc, N.; Boehm, J. Interaction of endogenous tau protein with synaptic proteins is regulated by N-methyl-D-aspartate receptor-dependent tau phosphorylation. J. Biol. Chem. 2012, 287, 32040–32053. [Google Scholar] [CrossRef] [PubMed]
- Malmqvist, T.; Anthony, K.; Gallo, J.M. Tau mRNA is present in axonal RNA granules and is associated with elongation factor 1A. Brain Res. 2014, 1584, 22–27. [Google Scholar] [CrossRef]
- Liu, L.; Drouet, V.; Wu, J.W.; Witter, M.P.; Small, S.A.; Clelland, C.; Duff, K. Trans-synaptic spread of tau pathology in vivo. PLoS ONE 2012, 7, e31302. [Google Scholar] [CrossRef] [PubMed]
- Pooler, A.M.; Phillips, E.C.; Lau, D.H.; Noble, W.; Hanger, D.P. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 2013, 14, 389–394. [Google Scholar] [CrossRef]
- Yamada, K.; Holth, J.K.; Liao, F.; Stewart, F.R.; Mahan, T.E.; Jiang, H.; Cirrito, J.R.; Patel, T.K.; Hochgrafe, K.; Mandelkow, E.M.; et al. Neuronal activity regulates extracellular tau in vivo. J. Exp. Med. 2014, 211, 387–393. [Google Scholar] [CrossRef]
- He, H.J.; Wang, X.S.; Pan, R.; Wang, D.L.; Liu, M.N.; He, R.Q. The proline-rich domain of tau plays a role in interactions with actin. BMC Cell Biol. 2009, 10, 81. [Google Scholar] [CrossRef]
- Yu, J.Z.; Rasenick, M.M. Tau associates with actin in differentiating PC12 cells. FASEB J. 2006, 20, 1452–1461. [Google Scholar] [CrossRef]
- Maroteaux, L.; Scheller, R.H. The rat-brain synucleins; family of proteins transiently associated with neuronal membrane. Mol. Brain Res. 1991, 11, 335–343. [Google Scholar] [CrossRef]
- Yuan, J.W.; Zhao, Y.W. Evolutionary aspects of the synuclein super-family and sub-families based on large-scale phylogenetic and group-discrimination analysis. Biochem. Biophys. Res. Commun. 2013, 441, 308–317. [Google Scholar] [CrossRef]
- Davidson, W.S.; Jonas, A.; Clayton, D.F.; George, J.M. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 1998, 273, 9443–9449. [Google Scholar] [CrossRef] [PubMed]
- Trexler, A.J.; Rhoades, E. N-terminal acetylation is critical for forming a-helical oligomer of a-synuclein. Protein Sci. 2012, 21, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Longhena, F.; Faustini, G.; Spillantini, M.G.; Bellucci, A. Living in promiscuity: The multiple partners of alpha-synuclein at the synapse in physiology and pathology. Int. J. Mol. Sci. 2019, 20, 141. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Fukushima, H.; Masliah, E.; Xia, Y.; Iwai, A.; Yoshimoto, M.; Otero, D.A.C.; Kondo, J.; Ihara, Y.; Saitoh, T. Molecular-cloning of cDNA-encoding an unrecognized component of amyloid in alzheimer-disease. Proc. Natl. Acad. Sci. USA 1993, 90, 11282–11286. [Google Scholar] [CrossRef] [PubMed]
- Bendor, J.T.; Logan, T.P.; Edwards, R.H. The function of alpha-synuclein. Neuron 2013, 79, 1044–1066. [Google Scholar] [CrossRef]
- Sevcsik, E.; Trexler, A.J.; Dunn, J.M.; Rhoades, E. Allostery in a Disordered Protein: Oxidative Modifications to alpha-Synuclein Act Distally To Regulate Membrane Binding. J. Am. Chem. Soc. 2011, 133, 7152–7158. [Google Scholar] [CrossRef]
- Snead, D.; Eliezer, D. Intrinsically disordered proteins in synaptic vesicle trafficking and release. J. Biol. Chem. 2019, 294, 3325–3342. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of alpha-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013, 14, 38–48. [Google Scholar] [CrossRef]
- George, J.M.; Jin, H.; Woods, W.S.; Clayton, D.F. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron 1995, 15, 361–372. [Google Scholar] [CrossRef]
- Iwai, A.; Masliah, E.; Yoshimoto, M.; Ge, N.F.; Flanagan, L.; Desilva, H.A.R.; Kittel, A.; Saitoh, T. The Precursor Protein of Non-a-Beta Component of Alzheimers-Disease Amyloid Is a Presynaptic Protein of the Central-Nervous-System. Neuron 1995, 14, 467–475. [Google Scholar] [CrossRef]
- Hijaz, B.A.; Volpicelli-Daley, L.A. Initiation and propagation of alpha-synuclein aggregation in the nervous system. Mol. Neurodegener. 2020, 15, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Tofaris, G.K.; Spillantini, M.G. Physiological and pathological properties of alpha-synuclein. Cell. Mol. Life Sci. 2007, 64, 2194–2201. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.; Roy, S. Alpha-Synuclein inhibits intersynaptic vesicle mobility and maintains recycling-pool homeostasis. J. Neurosci. 2012, 32, 10129–10135. [Google Scholar] [CrossRef] [PubMed]
- Farrer, M.; Destee, A.; Levecque, C.; Singleton, A.; Engelender, S.; Becquet, E.; Mouroux, V.; Richard, F.; Defebvre, L.; Crook, R.; et al. Genetic analysis of synphilin-1 in familial Parkinson’s disease. Neurobiol. Dis. 2001, 8, 317–323. [Google Scholar] [CrossRef][Green Version]
- Zaltieri, M.; Grigoletto, J.; Longhena, F.; Navarria, L.; Favero, G.; Castrezzati, S.; Colivicchi, M.A.; Della Corte, L.; Rezzani, R.; Pizzi, M.; et al. Alpha-synuclein and synapsin III cooperatively regulate synaptic function in dopamine neurons. J. Cell Sci. 2015, 128, 2231–2243. [Google Scholar] [CrossRef]
- Faustini, G.; Longhena, F.; Varanita, T.; Bubacco, L.; Pizzi, M.; Missale, C.; Benfenati, F.; Bjorklund, A.; Span, P.; Bellucci, A. Synapsin III deficiency hampers alpha-synuclein aggregation, striatal synaptic damage and nigral cell loss in an AAV-based mouse model of Parkinson’s disease. Acta Neuropathol. 2018, 136, 621–639. [Google Scholar] [CrossRef]
- Burre, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Sudhof, T.C. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef]
- Dunn, A.R.; Stout, K.A.; Ozawa, M.; Lohr, K.M.; Hoffman, C.A.; Bernstein, A.I.; Li, Y.; Wang, M.; Sgobio, C.; Sastry, N.; et al. Synaptic vesicle glycoprotein 2C (SV2C) modulates dopamine release and is disrupted in Parkinson disease. Proc. Natl. Acad. Sci. USA 2017, 114, E2253–E2262. [Google Scholar] [CrossRef] [PubMed]
- Gorenberg, E.L.; Chandra, S.S. The Role of Co-chaperones in Synaptic Proteostasis and Neurodegenerative Disease. Front. Neurosci. 2017, 11, 248. [Google Scholar] [CrossRef]
- Gitler, A.D.; Bevis, B.J.; Shorter, J.; Strathearn, K.E.; Hamamichi, S.; Su, L.J.; Caldwell, K.A.; Caldwell, G.A.; Rochet, J.C.; McCaffery, J.M.; et al. The Parkinson’s disease protein alpha-synuclein disrupts cellular Rab homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 145–150. [Google Scholar] [CrossRef]
- Shimizu, H.; Kawamura, S.; Ozaki, K. An essential role of Rab5 in uniformity of synaptic vesicle size. J. Cell Sci. 2003, 116, 3583–3590. [Google Scholar] [CrossRef]
- Masaracchia, C.; Hnida, M.; Gerhardt, E.; Lopes da Fonseca, T.; Villar-Pique, A.; Branco, T.; Stahlberg, M.A.; Dean, C.; Fernandez, C.O.; Milosevic, I.; et al. Membrane binding, internalization, and sorting of alpha-synuclein in the cell. Acta Neuropathol. Commun. 2018, 6, 1–17. [Google Scholar] [CrossRef]
- Shi, M.M.; Shi, C.H.; Xu, Y.M. Rab GTPases: The Key Players in the Molecular Pathway of Parkinson’s Disease. Front. Cell. Neurosci. 2017, 11, 81. [Google Scholar] [CrossRef]
- Guo, J.T.; Chen, A.Q.; Kong, Q.; Zhu, H.; Ma, C.M.; Qin, C. Inhibition of vesicular monoamine transporter-2 activity in alpha-synuclein stably transfected SH-SY5Y cells. Cell. Mol. Neurobiol. 2008, 28, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Fukae, J.; Mori, H.; Mizuno, Y.; Hattori, N. Positive immunoreactivity for vesicular monoamine transporter 2 in Lewy bodies and Lewy neurites in substantia nigra. Neurosci. Lett. 2006, 396, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Lee, F.J.; Liu, F.; Pristupa, Z.B.; Niznik, H.B. Direct binding and functional coupling of alpha-synuclein to the dopamine transporters accelerate dopamine-induced apoptosis. FASEB J. 2001, 15, 916–926. [Google Scholar] [PubMed]
- Buddhala, C.; Loftin, S.K.; Kuley, B.M.; Cairns, N.J.; Campbell, M.C.; Perlmutter, J.S.; Kotzbauer, P.T. Dopaminergic, serotonergic, and noradrenergic deficits in Parkinson disease. Ann. Clin. Transl. Neurol. 2015, 2, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Calo, L.; Wegrzynowicz, M.; Santivanez-Perez, J.; Grazia Spillantini, M. Synaptic failure and alpha-synuclein. Mov. Disord. 2016, 31, 169–177. [Google Scholar] [CrossRef]
- Schulz-Schaeffer, W.J. The synaptic pathology of alpha-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol. 2010, 120, 131–143. [Google Scholar] [CrossRef]
- Perez, R.G. Editorial: The Protein Alpha-Synuclein: Its Normal Role (in Neurons) and Its Role in Disease. Front. Neurosci. Switz. 2020, 14, 116. [Google Scholar] [CrossRef]
- Devi, L.; Anandatheerthavarada, H.K. Mitochondrial trafficking of APP and alpha synuclein: Relevance to mitochondrial dysfunction in Alzheimer’s and Parkinson’s diseases. BBA Mol. Basis Dis. 2010, 1802, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Ulmer, T.S.; Bax, A.; Cole, N.B.; Nussbaum, R.L. Structure and dynamics of micelle-bound human alpha-synuclein. J. Biol. Chem. 2005, 280, 9595–9603. [Google Scholar] [CrossRef] [PubMed]
- Ellis, C.E.; Murphy, E.J.; Mitchell, D.C.; Golovko, M.Y.; Scaglia, F.; Barcelo-Coblijn, G.C.; Nussbaum, R.L. Mitochondrial lipid abnormality and electron transport chain impairment in mice lacking alpha-synuclein. Mol. Cell. Biol. 2005, 25, 10190–10201. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef] [PubMed]
- Ludtmann, M.H.; Angelova, P.R.; Ninkina, N.N.; Gandhi, S.; Buchman, V.L.; Abramov, A.Y. Monomeric Alpha-Synuclein Exerts a Physiological Role on Brain ATP Synthase. J. Neurosci. 2016, 36, 10510–10521. [Google Scholar] [CrossRef] [PubMed]
- Kamp, F.; Exner, N.; Lutz, A.K.; Wender, N.; Hegermann, J.; Brunner, B.; Nuscher, B.; Bartels, T.; Giese, A.; Beyer, K.; et al. Inhibition of mitochondrial fusion by alpha-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J. 2010, 29, 3571–3589. [Google Scholar] [CrossRef]
- Vicario, M.; Cieri, D.; Brini, M.; Cali, T. The Close Encounter Between Alpha-Synuclein and Mitochondria. Front. Neurosci. Switz. 2018, 12, 388. [Google Scholar] [CrossRef]
- O’Donnell, K.C.; Lulla, A.; Stahl, M.C.; Wheat, N.D.; Bronstein, J.M.; Sagasti, A. Axon degeneration and PGC-1 alpha-mediated protection in a zebrafish model of alpha-synuclein toxicity. Dis. Models Mech. 2014, 7, 571–582. [Google Scholar] [CrossRef]
- Xie, W.; Chung, K.K. Alpha-synuclein impairs normal dynamics of mitochondria in cell and animal models of Parkinson’s disease. J. Neurochem. 2012, 122, 404–414. [Google Scholar] [CrossRef]
- Bender, A.; Desplats, P.; Spencer, B.; Rockenstein, E.; Adame, A.; Elstner, M.; Laub, C.; Mueller, S.; Koob, A.O.; Mante, M.; et al. TOM40 mediates mitochondrial dysfunction induced by alpha-synuclein accumulation in Parkinson’s disease. PLoS ONE 2013, 8, e62277. [Google Scholar] [CrossRef]
- Boassa, D.; Berlanga, M.L.; Yang, M.A.; Terada, M.; Hu, J.R.; Bushong, E.A.; Hwang, M.; Masliah, E.; George, J.M.; Ellisman, M.H. Mapping the subcellular distribution of alpha-synuclein in neurons using genetically encoded probes for correlated light and electron microscopy: Implications for Parkinson’s disease pathogenesis. J. Neurosci. 2013, 33, 2605–2615. [Google Scholar] [CrossRef] [PubMed]
- Volpicelli-Daley, L.A. Effects of alpha-synuclein on axonal transport. Neurobiol. Dis. 2017, 105, 321–327. [Google Scholar] [CrossRef]
- Jensen, P.H.; Li, J.Y.; Dahlstrom, A.; Dotti, C.G. Axonal transport of synucleins is mediated by all rate components. Eur. J. Neurosci. 1999, 11, 3369–3376. [Google Scholar] [CrossRef] [PubMed]
- Alim, M.A.; Hossain, M.S.; Arima, K.; Takeda, K.; Izumiyama, Y.; Nakamura, M.; Kaji, H.; Shinoda, T.; Hisanaga, S.; Ueda, K. Tubulin seeds alpha-synuclein fibril formation. J. Biol. Chem. 2002, 277, 2112–2117. [Google Scholar] [CrossRef]
- Toba, S.; Jin, M.Y.; Yamada, M.; Kumamoto, K.; Matsumoto, S.; Yasunaga, T.; Fukunaga, Y.; Miyazawa, A.; Fujita, S.; Itoh, K.; et al. Alpha-synuclein facilitates to form short unconventional microtubules that have a unique function in the axonal transport. Sci. Rep. UK 2017, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Prots, I.; Veber, V.; Brey, S.; Campioni, S.; Buder, K.; Riek, R.; Bohm, K.J.; Winner, B. Alpha-Synuclein Oligomers Impair Neuronal Microtubule-Kinesin Interplay. J. Biol. Chem. 2013, 288, 21742–21754. [Google Scholar] [CrossRef]
- Plotegher, N.; Kumar, D.; Tessari, I.; Brucale, M.; Munari, F.; Tosatto, L.; Belluzzi, E.; Greggio, E.; Bisaglia, M.; Capaldi, S.; et al. The chaperone-like protein 14-3-3eta interacts with human alpha-synuclein aggregation intermediates rerouting the amyloidogenic pathway and reducing alpha-synuclein cellular toxicity. Hum. Mol. Genet. 2014, 23, 5615–5629. [Google Scholar] [CrossRef]
- McFarland, M.A.; Ellis, C.E.; Markey, S.P.; Nussbaum, R.L. Proteomics Analysis Identifies Phosphorylation-dependent alpha-Synuclein Protein Interactions. Mol. Cell. Proteom. 2008, 7, 2123–2137. [Google Scholar] [CrossRef]
- Wang, B.; Underwood, R.; Kamath, A.; Britain, C.; McFerrin, M.B.; McLean, X.J.; Volpicelli-Daley, L.A.; Whitaker, R.H.; Placzek, W.J.; Becker, K.; et al. 14-3-3 Proteins Reduce Cell-to-Cell Transfer and Propagation of Pathogenic alpha-Synuclein. J. Neurosci. 2018, 38, 8211–8232. [Google Scholar] [CrossRef]
- Esposito, A.; Dohm, C.P.; Kermer, P.; Bahr, M.; Wouters, F.S. Alpha-synuclein and its disease-related mutants interact differentially with the microtubule protein tau and associate with the actin cytoskeleton. Neurobiol. Dis. 2007, 26, 521–531. [Google Scholar] [CrossRef]
- Combs, B.; Mueller, R.L.; Morfini, G.; Brady, S.T.; Kanaan, N.M. Tau and axonal transport misregulation in tauopathies. Adv. Exp. Med. Biol. 2019, 1184, 81–95. [Google Scholar] [PubMed]
- Kovacs, G.G. Tauopathies. Handb. Clin. Neurol. 2017, 145, 355–368. [Google Scholar] [PubMed]
- Kanaan, N.M.; Pigino, G.F.; Brady, S.T.; Lazarov, O.; Binder, L.I.; Morfini, G.A. Axonal degeneration in Alzheimer’s disease: When signaling abnormalities meet the axonal transport system. Exp. Neurol. 2013, 246, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Kneynsberg, A.; Combs, B.; Christensen, K.; Morfini, G.; Kanaan, N.M. Axonal degeneration in tauopathies: Disease relevance and underlying mechanisms. Front. Neurosci. 2017, 11, 572. [Google Scholar] [CrossRef]
- Narasimhan, S.; Guo, J.L.; Changolkar, L.; Stieber, A.; McBride, J.D.; Silva, L.V.; He, Z.H.; Zhang, B.; Gathagan, R.J.; Trojanowski, J.Q.; et al. Pathological tau strains from human brains recapitulate the diversity of tauopathies in nontransgenic mouse brain. J. Neurosci. 2017, 37, 11406–11423. [Google Scholar] [CrossRef]
- Guo, J.L.; Narasimhan, S.; Changolkar, L.; He, Z.; Stieber, A.; Zhang, B.; Gathagan, R.J.; Iba, M.; McBride, J.D.; Trojanowski, J.Q.; et al. Unique pathological tau conformers from Alzheimer’s brains transmit tau pathology in nontransgenic mice. J. Exp. Med. 2016, 213, 2635–2654. [Google Scholar] [CrossRef]
- Barghorn, S.; Zheng-Fischhofer, Q.; Ackmann, M.; Biernat, J.; von Bergen, M.; Mandelkow, E.M.; Mandelkow, E. Structure, microtubule interactions, and paired helical filament aggregation by tau mutants of frontotemporal dementias. Biochemistry 2000, 39, 11714–11721. [Google Scholar] [CrossRef]
- Hong, M.; Zhukareva, V.; Vogelsberg-Ragaglia, V.; Wszolek, Z.; Reed, L.; Miller, B.I.; Geschwind, D.H.; Bird, T.D.; McKeel, D.; Goate, A.; et al. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science 1998, 282, 1914–1917. [Google Scholar] [CrossRef]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21. [Google Scholar] [CrossRef]
- Goode, B.L.; Chau, M.; Denis, P.E.; Feinstein, S.C. Structural and functional differences between 3-repeat and 4-repeat tau isoforms. Implications for normal tau function and the onset of neurodegenetative disease. J. Biol. Chem. 2000, 275, 38182–38189. [Google Scholar] [CrossRef]
- Lacovich, V.; Espindola, S.L.; Alloatti, M.; Pozo Devoto, V.; Cromberg, L.E.; Carna, M.E.; Forte, G.; Gallo, J.M.; Bruno, L.; Stokin, G.B.; et al. Tau isoforms imbalance impairs the axonal transport of the amyloid precursor protein in human neurons. J. Neurosci. 2017, 37, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Bouge, A.L.; Parmentier, M.L. Tau excess impairs mitosis and kinesin-5 function, leading to aneuploidy and cell death. Dis. Model. Mech. 2016, 9, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Malmanche, N.; Dourlen, P.; Gistelinck, M.; Demiautte, F.; Link, N.; Dupont, C.; Vanden Broeck, L.; Werkmeister, E.; Amouyel, P.; Bongiovanni, A.; et al. Developmental expression of 4-repeat-tau induces neuronal aneuploidy in drosophila tauopathy models. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.; Conconi, D.; Panzeri, E.; Paoletta, L.; Piccoli, E.; Ferretti, M.G.; Mangieri, M.; Ruggerone, M.; Dalpra, L.; Tagliavini, F. Mutations in MAPT give rise to aneuploidy in animal models of tauopathy. Neurogenetics 2014, 15, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.; Conconi, D.; Panzeri, E.; Redaelli, S.; Piccoli, E.; Paoletta, L.; Dalpra, L.; Tagliavini, F. Mutations in MAPT gene cause chromosome instability and introduce copy number variations widely in the genome. J. Alzheimers Dis. 2013, 33, 969–982. [Google Scholar] [CrossRef]
- Galas, M.C.; Bonnefoy, E.; Buee, L.; Lefebvre, B. Emerging Connections Between Tau and Nucleic Acids. Adv. Exp. Med. Biol. 2019, 1184, 135–143. [Google Scholar]
- Hanger, D.P.; Anderton, B.H.; Noble, W. Tau phosphorylation: The therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 2009, 15, 112–119. [Google Scholar] [CrossRef]
- Petrucelli, L.; Dickson, D.; Kehoe, K.; Taylor, J.; Snyder, H.; Grover, A.; De Lucia, M.; McGowan, E.; Lewis, J.; Prihar, G.; et al. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum. Mol. Genet. 2004, 13, 703–714. [Google Scholar] [CrossRef]
- Funk, K.E.; Thomas, S.N.; Schafer, K.N.; Cooper, G.L.; Liao, Z.; Clark, D.J.; Yang, A.J.; Kuret, J. Lysine methylation is an endogenous post-translational modification of tau protein in human brain and a modulator of aggregation propensity. Biochem. J. 2014, 462, 77–88. [Google Scholar] [CrossRef]
- Watanabe, A.; Hong, W.K.; Dohmae, N.; Takio, K.; Morishima-Kawashima, M.; Ihara, Y. Molecular aging of tau: Disulfide-independent aggregation and non-enzymatic degradation in vitro and in vivo. J. Neurochem. 2004, 90, 1302–1311. [Google Scholar] [CrossRef]
- Mukrasch, M.D.; von Bergen, M.; Biernat, J.; Fischer, D.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. The “jaws” of the tau-microtubule interaction. J. Biol. Chem. 2007, 282, 12230–12239. [Google Scholar] [CrossRef] [PubMed]
- Peterson, D.W.; Zhou, H.; Dahlquist, F.W.; Lew, J. A soluble oligomer of tau associated with fiber formation analyzed by NMR. Biochemistry 2008, 47, 7393–7404. [Google Scholar] [CrossRef]
- Berriman, J.; Serpell, L.C.; Oberg, K.A.; Fink, A.L.; Goedert, M.; Crowther, R.A. Tau filaments from human brain and from in vitro assembly of recombinant protein show cross-beta structure. Proc. Natl. Acad. Sci. USA 2003, 100, 9034–9038. [Google Scholar] [CrossRef] [PubMed]
- Gerson, J.E.; Kayed, R. Formation and propagation of tau oligomeric seeds. Front. Neurol. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Margittai, M.; Langen, R. Template-assisted filament growth by parallel stacking of tau. Proc. Natl. Acad. Sci. USA 2004, 101, 10278–10283. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M. Tau filaments in neurodegenerative diseases. FEBS Lett. 2018, 592, 2383–2391. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Derisbourg, M.; Leghay, C.; Chiappetta, G.; Fernandez-Gomez, F.J.; Laurent, C.; Demeyer, D.; Carrier, S.; Buee-Scherrer, V.; Blum, D.; Vinh, J.; et al. Role of the Tau N-terminal region in microtubule stabilization revealed by new endogenous truncated forms. Sci. Rep. UK 2015, 5, 9659. [Google Scholar] [CrossRef]
- Wang, Y.P.; Biernat, J.; Pickhardt, M.; Mandelkow, E.; Mandelkow, E.M. Stepwise proteolysis liberates tau fragments that nucleate the Alzheimer-like aggregation of full-length tau in a neuronal cell model. Proc. Natl. Acad. Sci. USA 2007, 104, 10252–10257. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Jakes, R. Mutations causing neurodegenerative tauopathies. Biochim. Biophys. Acta 2005, 1739, 240–250. [Google Scholar] [CrossRef]
- Li, B.; Chohan, M.O.; Grundke-Iqbal, I.; Iqbal, K. Disruption of microtubule network by Alzheimer abnormally hyperphosphorylated tau. Acta Neuropathol. 2007, 113, 501–511. [Google Scholar] [CrossRef]
- Ebneth, A.; Godemann, R.; Stamer, K.; Illenberger, S.; Trinczek, B.; Mandelkow, E. Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: Implications for Alzheimer’s disease. J. Cell Biol. 1998, 143, 777–794. [Google Scholar] [CrossRef] [PubMed]
- Combs, B.; Hamel, C.; Kanaan, N.M. Pathological conformations involving the amino terminus of tau occur early in Alzheimer’s disease and are differentially detected by monoclonal antibodies. Neurobiol. Dis. 2016, 94, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Combs, B.; Kanaan, N.M. Exposure of the amino terminus of tau is a pathological event in multiple tauopathies. Am. J. Pathol. 2017, 187, 1222–1229. [Google Scholar] [CrossRef]
- LaPointe, N.E.; Morfini, G.; Pigino, G.; Gaisina, I.N.; Kozikowski, A.P.; Binder, L.I.; Brady, S.T. The amino terminus of tau inhibits kinesin-dependent axonal transport: Implications for filament toxicity. J. Neurosci. Res. 2009, 87, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Gunawardena, S.; Goldstein, L.S. Cargo-carrying motor vehicles on the neuronal highway: Transport pathways and neurodegenerative disease. J. Neurobiol. 2004, 58, 258–271. [Google Scholar] [CrossRef]
- Mondragon-Rodriguez, S.; Perry, G.; Zhu, X.; Moreira, P.I.; Acevedo-Aquino, M.C.; Williams, S. Phosphorylation of tau protein as the link between oxidative stress, mitochondrial dysfunction, and connectivity failure: Implications for Alzheimer’s disease. Oxid. Med. Cell. Longev. 2013, 6. [Google Scholar] [CrossRef]
- Berrocal, M.; Corbacho, I.; Sepulveda, M.R.; Gutierrez-Merino, C.; Mata, A.M. Phospholipids and calmodulin modulate the inhibition of PMCA activity by tau. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1028–1035. [Google Scholar] [CrossRef]
- Eckert, A.; Nisbet, R.; Grimm, A.; Gotz, J. March separate, strike together--role of phosphorylated TAU in mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Wang, Y.; Gao, D.; Ye, J.; Wang, X.; Fang, L.; Wu, D.; Pi, G.; Lu, C.; Zhou, X.W.; et al. Accumulation of human full-length tau induces degradation of nicotinic acetylcholine receptor alpha4 via activating calpain-2. Sci. Rep. 2016, 6, 27283. [Google Scholar] [CrossRef]
- Moreno, H.; Morfini, G.; Buitrago, L.; Ujlaki, G.; Choi, S.; Yu, E.; Moreira, J.E.; Avila, J.; Brady, S.T.; Pant, H.; et al. Tau pathology-mediated presynaptic dysfunction. Neuroscience 2016, 325, 30–38. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fa, M.; Puzzo, D.; Piacentini, R.; Staniszewski, A.; Zhang, H.; Baltrons, M.A.; Li Puma, D.D.; Chatterjee, I.; Li, J.; Saeed, F.; et al. Extracellular Tau Oligomers Produce An Immediate Impairment of LTP and Memory. Sci. Rep. 2016, 6, 19393. [Google Scholar] [CrossRef] [PubMed]
- Mietelska-Porowska, A.; Wasik, U.; Goras, M.; Filipek, A.; Niewiadomska, G. Tau protein modifications and interactions: Their role in function and dysfunction. Int. J. Mol. Sci. 2014, 15, 4671–4713. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K. Potential pathways of abnormal tau and alpha-synuclein dissemination in sporadic Alzheimer’s and Parkinson’s diseases. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Trecidi, K. Neuroanatomy and pathology of sporadic Alzheimer’s disease. Adv. Anat. Embryol. Cell Biol. 2015, 215, 1–162. [Google Scholar]
- Braak, H.; Del Tredici, K. The preclinical phase of the pathological process underlying sporadic Alzheimer’s disease. Brain 2015, 138, 2814–2833. [Google Scholar] [CrossRef]
- Kovacs, G.G.; Lukic, M.J.; Irwin, D.J.; Arzberger, T.; Respondek, G.; Lee, E.B.; Coughlin, D.; Giese, A.; Grossman, M.; Kurz, C.; et al. Distribution patterns of tau pathology in progressive supranuclear palsy. Acta Neuropathol. 2020. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Goedert, M. The alpha-synucleinopathies: Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy. Ann. N. Y. Acad. Sci. 2000, 920, 16–27. [Google Scholar] [CrossRef]
- Krismer, F.; Wenning, G.K. Multiple system atrophy: Insights into a rare and debilitating movement disorder. Nat. Rev. Neurol. 2017, 13, 232–243. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Appel-Cresswell, S.; Vilarino-Guell, C.; Encarnacion, M.; Sherman, H.; Yu, I.; Shah, B.; Weir, D.; Thompson, C.; Szu-Tu, C.; Trinh, J.; et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov. Disord. 2013, 28, 811–813. [Google Scholar] [CrossRef] [PubMed]
- Kruger, R.; Kuhn, W.; Muller, T.; Woitalla, D.; Graeber, M.; Kosel, S.; Przuntek, H.; Epplen, J.T.; Schols, L.; Riess, O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef] [PubMed]
- Lesage, S.; Anheim, M.; Letournel, F.; Bousset, L.; Honore, A.; Rozas, N.; Pieri, L.; Madiona, K.; Durr, A.; Melki, R.; et al. G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol. 2013, 73, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Pasanen, P.; Myllykangas, L.; Siitonen, M.; Raunio, A.; Kaakkola, S.; Lyytinen, J.; Tienari, P.J.; Poyhonen, M.; Paetau, A. Novel alpha-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol. Aging 2014, 35, 2180.e1–2180.e5. [Google Scholar] [CrossRef]
- Proukakis, C.; Dudzik, C.G.; Brier, T.; MacKay, D.S.; Cooper, J.M.; Millhauser, G.L.; Houlden, H.; Schapira, A.H. A novel alpha-synuclein missense mutation in Parkinson disease. Neurology 2013, 80, 1062–1064. [Google Scholar] [CrossRef]
- Zarranz, J.J.; Alegre, J.; Gomez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atares, B.; et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef]
- Chartier-Harlin, M.C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. Alpha-synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841. [Google Scholar] [CrossRef]
- Balestrino, R.; Schapira, A.H.V. Glucocerebrosidase and Parkinson Disease: Molecular, clinical, and therapeutic implications. Neuroscientist 2018, 24, 540–559. [Google Scholar] [CrossRef] [PubMed]
- Kalinderi, K.; Bostantjopoulou, S.; Fidani, L. The genetic background of Parkinson’s disease: Current progress and future prospects. Acta Neurol. Scand. 2016, 134, 314–326. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Q.; Tan, L.; Yu, J.T. The role of the LRRK2 gene in Parkinsonism. Mol. Neurodegener. 2014, 9, 47. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Zheng, W.; Wang, X.; Chen, Z. Proteostasis of alpha-Synuclein and Its Role in the Pathogenesis of Parkinson’s Disease. Front. Cell. Neurosci. 2020, 14, 45. [Google Scholar] [CrossRef] [PubMed]
- Rey, N.L.; Bousset, L.; George, S.; Madaj, Z.; Meyerdirk, L.; Schulz, E.; Steiner, J.A.; Melki, R.; Brundin, P. Alpha-Synuclein conformational strains spread, seed and target neuronal cells differentially after injection into the olfactory bulb. Acta Neuropathol. Commun. 2019, 7, 1–18. [Google Scholar] [CrossRef]
- Hunn, B.H.; Cragg, S.J.; Bolam, J.P.; Spillantini, M.G.; Wade-Martins, R. Impaired intracellular trafficking defines early Parkinson’s disease. Trends Neurosci. 2015, 38, 178–188. [Google Scholar] [CrossRef]
- Tagliaferro, P.; Burke, R.E. Retrograde axonal degeneration in Parkinson disease. J. Parkinsons. Dis. 2016, 6, 1–15. [Google Scholar] [CrossRef]
- Griffin, J.W.; George, E.B.; Chaudhry, V. Wallerian degeneration in peripheral nerve disease. Baillieres Clin. Neurol. 1996, 5, 65–75. [Google Scholar]
- Zhai, Q.; Wang, J.; Kim, A.; Liu, Q.; Watts, R.; Hoopfer, E.; Mitchison, T.; Luo, L.; He, Z. Involvement of the ubiquitin-proteasome system in the early stages of wallerian degeneration. Neuron 2003, 39, 217–225. [Google Scholar] [CrossRef]
- Xia, Q.; Liao, L.; Cheng, D.; Duong, D.M.; Gearing, M.; Lah, J.J.; Levey, A.I.; Peng, J. Proteomic identification of novel proteins associated with Lewy bodies. Front. Biosci. 2008, 13, 3850–3856. [Google Scholar] [CrossRef]
- Cartelli, D.; Cappelletti, G. Microtubule Destabilization Paves the Way to Parkinson’s Disease. Mol. Neurobiol. 2017, 54, 6762–6774. [Google Scholar] [CrossRef]
- Godena, V.K.; Brookes-Hocking, N.; Moller, A.; Shaw, G.; Oswald, M.; Sancho, R.M.; Miller, C.C.J.; Whitworth, A.J.; De Vos, K.J. Increasing microtubule acetylation rescues axonal transport and locomotor deficits caused by LRRK2 Roc-COR domain mutations. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Jiang, H.; Yang, F.; Nakaso, K.; Feng, J. Parkin protects dopaminergic neurons against microtubule-depolymerizing toxins by attenuating microtubule-associated protein kinase activation. J. Biol. Chem. 2009, 284, 4009–4017. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Jiang, H.; Hu, Z.; Fan, K.; Wang, J.; Janoschka, S.; Wang, X.; Ge, S.; Feng, J. Parkin mutations reduce the complexity of neuronal processes in iPSC-derived human neurons. Stem Cells 2015, 33, 68–78. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cartelli, D.; Aliverti, A.; Barbiroli, A.; Santambrogio, C.; Ragg, E.M.; Casagrande, F.V.M.; Cantele, F.; Beltramone, S.; Marangon, J.; De Gregorio, C.; et al. Alpha-Synuclein is a novel microtubule dynamase. Sci. Rep. UK 2016, 6, 1–13. [Google Scholar]
- Braak, H.; Rub, U.; Gai, W.P.; Del Tredici, K. Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural. Transm. 2003, 110, 517–536. [Google Scholar] [CrossRef]
- Orimo, S.; Uchihara, T.; Nakamura, A.; Mori, F.; Kakita, A.; Wakabayashi, K.; Takahashi, H. Axonal alpha-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain 2008, 131, 642–650. [Google Scholar] [CrossRef]
- Uchihara, T.; Giasson, B.I. Propagation of alpha-synuclein pathology: Hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol. 2016, 131, 49–73. [Google Scholar] [CrossRef]
- Luna, E.; Decker, S.C.; Riddle, D.M.; Caputo, A.; Zhang, B.; Cole, T.; Caswell, C.; Xie, S.X.; Lee, V.M.Y.; Luk, K.C. Differential alpha-synuclein expression contributes to selective vulnerability of hippocampal neuron subpopulations to fibril-induced toxicity. Acta Neuropathol. 2018, 135, 855–875. [Google Scholar] [CrossRef]
- Peng, C.; Trojanowski, J.Q.; Lee, V.M. Protein transmission in neurodegenerative disease. Nat. Rev. Neurol. 2020, 16, 199–212. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rub, U.; de Vos, R.A.I.; Steur, E.N.H.J.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Del Tredici, K.; Braak, H. Spinal cord lesions in sporadic Parkinson’s disease. Acta Neuropathol. 2012, 124, 643–664. [Google Scholar] [CrossRef] [PubMed]
- Arendt, T.; Bruckner, M.K.; Bigl, V.; Marcova, L. Dendritic reorganisation in the basal forebrain under degenerative conditions and its defects in Alzheimer’s disease. II. Ageing, Korsakoff’s disease, Parkinson’s disease, and Alzheimer’s disease. J. Comp. Neurol. 1995, 351, 189–222. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.E.; Hyman, B.T.; Flory, J.; Damasio, A.R.; Van Hoesen, G.W. The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer’s disease. Cereb. Cortex 1991, 1, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Holzer, M.; Holzapfel, H.P.; Zedlick, D.; Bruckner, M.K.; Arendt, T. Abnormally phosphorylated tau protein in Alzheimer’s disease: Heterogeneity of individual regional distribution and relationship to clinical severity. Neuroscience 1994, 63, 499–516. [Google Scholar] [CrossRef]
- Ishii, T. Distribution of Alzheimer’s neurofibrillary changes in the brain stem and hypothalamus of senile dementia. Acta Neuropathol. 1966, 6, 181–187. [Google Scholar] [CrossRef]
- Rodriguez-Leyva, I.; Chi-Ahumada, E.G.; Carrizales, J.; Rodriguez-Violante, M.; Velazquez-Osuna, S.; Medina-Mier, V.; Martel-Gallegos, M.G.; Zarazua, S.; Enriquez-Macias, L.; Castro, A.; et al. Parkinson disease and progressive supranuclear palsy: Protein expression in skin. Ann. Clin. Transl. Neurol. 2016, 3, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Dugger, B.N.; Hoffman, B.R.; Scroggins, A.; Serrano, G.E.; Adler, C.H.; Shill, H.A.; Belden, C.M.; Sabbagh, M.N.; Caviness, J.N.; Driver Dunckley, E.; et al. Tau immunoreactivity in peripheral tissues of human aging and select tauopathies. Neurosci. Lett. 2019, 696, 132–139. [Google Scholar] [CrossRef]
- Gu, Y.; Oyama, F.; Ihara, Y. Tau is widely expressed in rat tissues. J. Neurochem. 1996, 67, 1235–1244. [Google Scholar] [CrossRef]
- Lionnet, A.; Wade, M.A.; Corbille, A.G.; Prigent, A.; Paillusson, S.; Tasselli, M.; Gonzales, J.; Durieu, E.; Rolli-Derkinderen, M.; Coron, E.; et al. Characterisation of tau in the human and rodent enteric nervous system under physiological conditions and in tauopathy. Acta Neuropathol. Commun. 2018, 6. [Google Scholar] [CrossRef]
- Murofushi, H.; Suzuki, M.; Sakai, H.; Kobayashi, S. Immunohistochemical localization of microtubule-associated proteins in the nervous system of the small intestine of guinea pig. Cell Tissue Res. 1989, 255, 315–322. [Google Scholar] [CrossRef]
- Dugger, B.N.; Whiteside, C.M.; Maarouf, C.L.; Walker, D.G.; Beach, T.G.; Sue, L.I.; Garcia, A.; Dunckley, T.; Meechoovet, B.; Reiman, E.M.; et al. The presence of select tau species in human peripheral tissues and their relation to Alzheimer’s disease. J. Alzheimers Dis. 2016, 54, 1249. [Google Scholar] [CrossRef] [PubMed]
- Bohl, J.; Ulbricht, D.; Steinmetz, H. Neurofibrillary tangles in peripheral autonomic ganglion cells. Alzheimers. Dis. Biol. Diagn. Ther. 1997, 4, 281–287. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Furuta, A.; Takahashi, H.; Ikuta, F. Occurrence of neurofibrillary tangles in the celiac ganglia. Acta Neuropathol. 1989, 78, 448. [Google Scholar] [CrossRef] [PubMed]
- Holzer, M.; Holzapfel, H.P.; Krohn, K.; Gertz, H.J.; Arendt, T. Alterations in content and phosphorylation state of cytoskeletal proteins in the sciatic nerve during ageing and in Alzheimer’s disease. J. Neural. Transm. (Vienna) 1999, 106, 743–755. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, C.H.; Doty, R.L. The Neurology of Olfaction; Cambridge University Press: Cambridge, UK, 2009. [Google Scholar]
- Viereck, C.; Tucker, R.P.; Matus, A. The adult rat olfactory system expresses microtubule-associated proteins found in the developing brain. J. Neurosci. 1989, 9, 3547–3557. [Google Scholar] [CrossRef] [PubMed]
- Attems, J.; Walker, L.; Jellinger, K.A. Olfactory bulb involvement in neurodegenerative diseases. Acta Neuropathol. 2014, 127, 459–475. [Google Scholar] [CrossRef] [PubMed]
- Bathini, P.; Mottas, A.; Jaquet, M.; Brai, E.; Alberi, L. Progressive signaling changes in the olfactory nerve of patients with Alzheimer’s disease. Neurobiol. Aging 2019, 76, 80–95. [Google Scholar] [CrossRef]
- Murphy, C.; Gilmore, M.M.; Seery, C.S.; Salmon, D.P.; Lasker, B.R. Olfactory thresholds are associated with degree of dementia in Alzheimer’s disease. Neurobiol. Aging 1990, 11, 465–469. [Google Scholar] [CrossRef]
- Wilson, R.S.; Arnold, S.E.; Schneider, J.A.; Tang, Y.; Bennett, D.A. The relationship between cerebral Alzheimer’s disease pathology and odour identification in old age. J. Neurol. Neurosurg. Psychiatry 2007, 78, 30–35. [Google Scholar] [CrossRef]
- Talamo, B.R.; Feng, W.H.; Perez-Cruet, M.; Adelman, L.; Kosik, K.; Lee, M.Y.; Cork, L.C.; Kauer, J.S. Pathologic changes in olfactory neurons in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 1991, 640, 1–7. [Google Scholar] [CrossRef]
- Talamo, B.R.; Rudel, R.; Kosik, K.S.; Lee, V.M.; Neff, S.; Adelman, L.; Kauer, J.S. Pathological changes in olfactory neurons in patients with Alzheimer’s disease. Nature 1989, 337, 736–739. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.E.; Lee, E.B.; Moberg, P.J.; Stutzbach, L.; Kazi, H.; Han, L.Y.; Lee, V.M.; Trojanowski, J.Q. Olfactory epithelium amyloid-beta and paired helical filament-tau pathology in Alzheimer disease. Ann. Neurol. 2010, 67, 462–469. [Google Scholar] [CrossRef]
- Trojanowski, J.Q.; Newman, P.D.; Hill, W.D.; Lee, V.M.Y. Human Olfactory Epithelium in Normal Aging, Alzheimers-Disease, and Other Neurodegenerative Disorders. J. Comp. Neurol. 1991, 310, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, M.; Ishizuka, Y.; Seki, K. Pathology of olfactory mucosa in patients with Alzheimers-disease. Ann. Otol. Rhinol. Laryngol. 1994, 103, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, C.H.; Del Tredici, K.; Braak, H. Parkinson’s disease: A dual-hit hypothesis. Neuropathol. Appl. Neurobiol. 2007, 33, 599–614. [Google Scholar] [CrossRef]
- Hilton, D.; Stephens, M.; Kirk, L.; Edwards, P.; Potter, R.; Zajicek, J.; Broughton, E.; Hagan, H.; Carroll, C. Accumulation of alpha-synuclein in the bowel of patients in the pre-clinical phase of Parkinson’s disease. Acta Neuropathol. 2014, 127, 235–241. [Google Scholar] [CrossRef]
- Liu, B.; Fang, F.; Pedersen, N.L.; Tillander, A.; Ludvigsson, J.F.; Ekbom, A.; Svenningsson, P.; Chen, H.; Wirdefeldt, K. Vagotomy and Parkinson disease: A Swedish register-based matched-cohort study. Neurology 2017, 88, 1996–2002. [Google Scholar] [CrossRef]
- Van Den Berge, N.; Ferreira, N.; Gram, H.; Mikkelsen, T.W.; Alstrup, A.K.O.; Casadei, N.; Tsung-Pin, P.; Riess, O.; Nyengaard, J.R.; Tamguney, G.; et al. Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol. 2019, 138, 535–550. [Google Scholar] [CrossRef]
- Borghammer, P.; Van Den Berge, N. Brain-First versus Gut-First Parkinson’s Disease: A Hypothesis. J. Parkinsons. Dis. 2019, 9, S281–S295. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Mori, F.; Tanji, K.; Orimo, S.; Takahashi, H. Involvement of the peripheral nervous system in synucleinopathies, tauopathies and other neurodegenerative proteinopathies of the brain. Acta Neuropathol. 2010, 120, 1–12. [Google Scholar] [CrossRef]
- Nishie, M.; Mori, F.; Fujiwara, H.; Hasegawa, M.; Yoshimoto, M.; Iwatsubo, T.; Takahashi, H.; Wakabayashi, K. Accumulation of phosphorylated alpha-synuclein in the brain and peripheral ganglia of patients with multiple system atrophy. Acta Neuropathol. 2004, 107, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Mori, F.; Inenaga, C.; Yoshimoto, M.; Umezu, H.; Tanaka, R.; Takahashi, H.; Wakabayashi, K. Alpha-synuclein immunoreactivity in normal and neoplastic Schwann cells. Acta Neuropathol. 2002, 103, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Melli, G.; Vacchi, E.; Biemmi, V.; Galati, S.; Staedler, C.; Ambrosini, R.; Kaelin-Lang, A. Cervical skin denervation associates with alpha-synuclein aggregates in Parkinson disease. Ann. Clin. Transl. Neurol. 2018, 5, 1394–1407. [Google Scholar] [CrossRef] [PubMed]
- Kanda, T.; Tsukagoshi, H.; Oda, M.; Miyamoto, K.; Tanabe, H. Changes of unmyelinated nerve fibers in sural nerve in amyotrophic lateral sclerosis, Parkinson’s disease and multiple system atrophy. Acta Neuropathol. 1996, 91, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Tsukita, K.; Sakamaki-Tsukita, H.; Tanaka, K.; Suenaga, T.; Takahashi, R. Value of in vivo alpha-synuclein deposits in Parkinson’s disease: A systematic review and meta-analysis. Mov. Disord. 2019, 34, 1452–1463. [Google Scholar] [CrossRef]
- Donadio, V.; Doppler, K.; Incensi, A.; Kuzkina, A.; Janzen, A.; Mayer, G.; Volkmann, J.; Rizzo, G.; Antelmi, E.; Plazzi, G.; et al. Abnormal alpha-synuclein deposits in skin nerves: Intra- and inter-laboratory reproducibility. Eur. J. Neurol. 2019, 26, 1245–1251. [Google Scholar] [CrossRef]
- Antelmi, E.; Donadio, V.; Incensi, A.; Plazzi, G.; Liguori, R. Skin nerve phosphorylated alpha-synuclein deposits in idiopathic REM sleep behavior disorder. Neurology 2017, 88, 2128–2131. [Google Scholar] [CrossRef]
- Doppler, K.; Jentschke, H.M.; Schulmeyer, L.; Vadasz, D.; Janzen, A.; Luster, M.; Hoffken, H.; Mayer, G.; Brumberg, J.; Booij, J.; et al. Dermal phospho-alpha-synuclein deposits confirm REM sleep behaviour disorder as prodromal Parkinson’s disease. Acta Neuropathol. 2017, 133, 535–545. [Google Scholar] [CrossRef]
- Vacchi, E.; Pinton, S.; Kaelin-Lang, A.; Melli, G. Targeting Alpha Synuclein Aggregates in Cutaneous Peripheral Nerve Fibers by Free-floating Immunofluorescence Assay. J. Vis. Exp. 2019, e59558. [Google Scholar] [CrossRef]
- Mazzetti, S.; Basellini, M.J.; Ferri, V.; Cassani, E.; Cereda, E.; Paolini, M.; Calogero, A.M.; Bolliri, C.; De Leonardis, M.; Sacilotto, G.; et al. alpha-Synuclein oligomers in skin biopsy of idiopathic and monozygotic twin patients with Parkinson’s disease. Brain 2020, 143, 920–931. [Google Scholar] [CrossRef]
- Nolano, M.; Provitera, V.; Estraneo, A.; Selim, M.M.; Caporaso, G.; Stancanelli, A.; Saltalamacchia, A.M.; Lanzillo, B.; Santoro, L. Sensory deficit in Parkinson’s disease: Evidence of a cutaneous denervation. Brain 2008, 131, 1903–1911. [Google Scholar] [CrossRef] [PubMed]
- Nolano, M.; Provitera, V.; Stancanelli, A.; Saltalamacchia, A.M.; Caporaso, G.; Lullo, F.; Borreca, I.; Piscosquito, G.; Mozzillo, S.; Esposito, M.; et al. Small fiber pathology parallels disease progression in Parkinson disease: A longitudinal study. Acta Neuropathol. 2018, 136, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, R.L. Lewy bodies in Alzheimer’s disease: A neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathol. 2000, 10, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Bancher, C.; Braak, H.; Fischer, P.; Jellinger, K.A. Neuropathological staging of Alzheimer lesions and intellectual status in Alzheimer’s and Parkinson’s disease patients. Neurosci. Lett. 1993, 162, 179–182. [Google Scholar] [CrossRef]
- Kotzbauer, P.T.; Giasson, B.I.; Kravitz, A.V.; Golbe, L.I.; Mark, M.H.; Trojanowski, J.Q.; Lee, V.M. Fibrillization of alpha-synuclein and tau in familial Parkinson’s disease caused by the A53T alpha-synuclein mutation. Exp. Neurol. 2004, 187, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Arima, K.; Mizutani, T.; Alim, M.A.; Tonozuka-Uehara, H.; Izumiyama, Y.; Hirai, S.; Ueda, K. NACP/alpha-synuclein and tau constitute two distinctive subsets of filaments in the same neuronal inclusions in brains from a family of parkinsonism and dementia with Lewy bodies: Double-immunolabeling fluorescence and electron microscopic studies. Acta Neuropathol. 2000, 100, 115–121. [Google Scholar] [CrossRef]
- Ishizawa, T.; Mattila, P.; Davies, P.; Wang, D.; Dickson, D.W. Colocalization of tau and alpha-synuclein epitopes in Lewy bodies. J. Neuropathol. Exp. Neurol. 2003, 62, 389–397. [Google Scholar] [CrossRef]
- Baker, M.; Litvan, I.; Houlden, H.; Adamson, J.; Dickson, D.; Perez-Tur, J.; Hardy, J.; Lynch, T.; Bigio, E.; Hutton, M. Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum. Mol. Genet. 1999, 8, 711–715. [Google Scholar] [CrossRef]
- Vilarino-Guell, C.; Soto-Ortolaza, A.I.; Rajput, A.; Mash, D.C.; Papapetropoulos, S.; Pahwa, R.; Lyons, K.E.; Uitti, R.J.; Wszolek, Z.K.; Dickson, D.W.; et al. Mapt H1 Haplotype Is a Risk Factor for Essential Tremor and Multiple System Atrophy. Neurology 2011, 76, 670–672. [Google Scholar] [CrossRef]
- Peuralinna, T.; Oinas, M.; Polvikoski, T.; Paetau, A.; Sulkava, R.; Niinisto, L.; Kalimo, H.; Hernandez, D.; Hardy, J.; Singleton, A.; et al. Neurofibrillary tau pathology modulated by genetic variation of alpha-synuclein. Ann. Neurol. 2008, 64, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, F.; Suzuki, M.; Shimada, N.; Kagiya, G.; Ohta, E.; Tamura, K.; Maruyama, H.; Ichikawa, T. Stimulatory effect of alpha-synuclein on the tau-phosphorylation by GSK-3beta. FEBS J. 2011, 278, 4895–4904. [Google Scholar] [CrossRef] [PubMed]
- Badiola, N.; de Oliveira, R.M.; Herrera, F.; Guardia-Laguarta, C.; Goncalves, S.A.; Pera, M.; Suarez-Calvet, M.; Clarimon, J.; Outeiro, T.F.; Lleo, A. Tau enhances alpha-synuclein aggregation and toxicity in cellular models of synucleinopathy. PLoS ONE 2011, 6, e26609. [Google Scholar] [CrossRef] [PubMed]
- Ordonez, D.G.; Lee, M.K.; Feany, M.B. alpha-synuclein Induces Mitochondrial Dysfunction through Spectrin and the Actin Cytoskeleton. Neuron 2018, 97, 108–124.e106. [Google Scholar] [CrossRef] [PubMed]
- DuBoff, B.; Gotz, J.; Feany, M.B. Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron 2012, 75, 618–632. [Google Scholar] [CrossRef] [PubMed]
- Pech, U.; Verstreken, P. alpha-Synuclein and Tau: Mitochondrial Kill Switches. Neuron 2018, 97, 3–4. [Google Scholar] [CrossRef]
- Khandelwal, P.J.; Dumanis, S.B.; Herman, A.M.; Rebeck, G.W.; Moussa, C.E.H. Wild type and P301L mutant Tau promote neuro-inflammation and alpha-Synuclein accumulation in lentiviral gene delivery models. Mol. Cell. Neurosci. 2012, 49, 44–53. [Google Scholar] [CrossRef]
- Lewis, J.; McGowan, E.; Rockwood, J.; Melrose, H.; Nacharaju, P.; Van Slegtenhorst, M.; Gwinn-Hardy, K.; Paul Murphy, M.; Baker, M.; Yu, X.; et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat. Genet. 2000, 25, 402–405. [Google Scholar] [CrossRef]
- Ittner, L.M.; Fath, T.; Ke, Y.D.; Bi, M.; van Eersel, J.; Li, K.M.; Gunning, P.; Gotz, J. Parkinsonism and impaired axonal transport in a mouse model of frontotemporal dementia. Proc. Natl. Acad. Sci. USA 2008, 105, 15997–16002. [Google Scholar] [CrossRef]
- Emmer, K.L.; Waxman, E.A.; Covy, J.P.; Giasson, B.I. E46K human alpha-synuclein transgenic mice develop Lewy-like and tau pathology associated with age-dependent, detrimental motor impairment. J. Biol. Chem. 2011, 286, 35104–35118. [Google Scholar] [CrossRef]
- Wills, J.; Credle, J.; Haggerty, T.; Lee, J.H.; Oaks, A.W.; Sidhu, A. Tauopathic changes in the striatum of A53T alpha-synuclein mutant mouse model of Parkinson’s disease. PLoS ONE 2011, 6, e17953. [Google Scholar] [CrossRef]
- Jakes, R.; Spillantini, M.G.; Goedert, M. Identification of two distinct synucleins from human brain. FEBS Lett. 1994, 345, 27–32. [Google Scholar] [CrossRef]
- Cleveland, D.W.; Hwo, S.Y.; Kirschner, M.W. Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J. Mol. Biol. 1977, 116, 227–247. [Google Scholar] [CrossRef]
- Lee, V.M.Y.; Giasson, B.I.; Trojanowski, J.Q. More than just two peas in a pod: Common amyloidogenic properties of tau and alpha-synuclein in neurodegenerative diseases. Trends Neurosci. 2004, 27, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. Alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M. Alpha-synuclein and neurodegenerative diseases. Nat. Rev. Neurosci. 2001, 2, 492–501. [Google Scholar] [CrossRef]
- Lee, V.M.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef]
- Feng, S.T.; Wang, Z.Z.; Yuan, Y.H.; Sun, H.M.; Chen, N.H.; Zhang, Y. Update on the association between alpha-synuclein and tau with mitochondrial dysfunction: Implications for Parkinson’s disease. Eur. J. Neurosci. 2020. [Google Scholar] [CrossRef]
- Dunker, A.K.; Brown, C.J.; Lawson, J.D.; Iakoucheva, L.M.; Obradovic, Z. Intrinsic disorder and protein function. Biochemistry 2002, 41, 6573–6582. [Google Scholar] [CrossRef]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Uversky, V.N. Functional roles of transiently and intrinsically disordered regions within proteins. FEBS J. 2015, 282, 1182–1189. [Google Scholar] [CrossRef]
- De Luca, C.M.G.; Elia, A.E.; Portaleone, S.M.; Cazzaniga, F.A.; Rossi, M.; Bistaffa, E.; De Cecco, E.; Narkiewicz, J.; Salzano, G.; Carletta, O.; et al. Efficient RT-QuIC seeding activity for alpha-synuclein in olfactory mucosa samples of patients with Parkinson’s disease and multiple system atrophy. Transl. Neurodegener. 2019, 8, 24. [Google Scholar] [CrossRef]
- Saijo, E.; Metrick, M.A.; Koga, S.; Parchi, P.; Litvan, I.; Spina, S.; Boxer, A.; Rojas, J.C.; Galasko, D.; Kraus, A.; et al. 4-Repeat tau seeds and templating subtypes as brain and CSF biomarkers of frontotemporal lobar degeneration. Acta Neuropathol. 2020, 139, 63–77. [Google Scholar] [CrossRef]
- Shahnawaz, M.; Mukherjee, A.; Pritzkow, S.; Mendez, N.; Rabadia, P.; Liu, X.; Hu, B.; Schmeichel, A.; Singer, W.; Wu, G.; et al. Discriminating alpha-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature 2020, 578, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Andrews, H.; White, K.; Thomson, C.; Edgar, J.; Bates, D.; Griffiths, I.; Turnbull, D.; Nichols, P. Increased axonal mitochondrial activity as an adaptation to myelin deficiency in the Shiverer mouse. J. Neurosci. Res. 2006, 83, 1533–1539. [Google Scholar] [CrossRef]
- Bristow, E.A.; Griffiths, P.G.; Andrews, R.M.; Johnson, M.A.; Turnbull, D.M. The distribution of mitochondrial activity in relation to optic nerve structure. Arch. Ophthalmol. 2002, 120, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Monsma, P.C.; Li, Y.; Fenn, J.D.; Jung, P.; Brown, A. Local regulation of neurofilament transport by myelinating cells. J. Neurosci. 2014, 34, 2979–2988. [Google Scholar] [CrossRef] [PubMed]
- Uchida, A.; Colakoglu, G.; Wang, L.; Monsma, P.C.; Brown, A. Severing and end-to-end annealing of neurofilaments in neurons. Proc. Natl. Acad. Sci. USA 2013, 110, E2696–E2705. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, I.T.; Lin, K.J.; Huang, K.L.; Huang, C.C.; Chen, H.S.; Wey, S.P.; Yen, T.C.; Okamura, N.; Hsu, J.L. Biodistribution and radiation dosimetry for the tau tracer (18)F-THK-5351 in healthy human subjects. J. Nucl. Med. 2017, 58, 1498–1503. [Google Scholar] [CrossRef] [PubMed][Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vacchi, E.; Kaelin-Lang, A.; Melli, G. Tau and Alpha Synuclein Synergistic Effect in Neurodegenerative Diseases: When the Periphery Is the Core. Int. J. Mol. Sci. 2020, 21, 5030. https://doi.org/10.3390/ijms21145030
Vacchi E, Kaelin-Lang A, Melli G. Tau and Alpha Synuclein Synergistic Effect in Neurodegenerative Diseases: When the Periphery Is the Core. International Journal of Molecular Sciences. 2020; 21(14):5030. https://doi.org/10.3390/ijms21145030
Chicago/Turabian StyleVacchi, Elena, Alain Kaelin-Lang, and Giorgia Melli. 2020. "Tau and Alpha Synuclein Synergistic Effect in Neurodegenerative Diseases: When the Periphery Is the Core" International Journal of Molecular Sciences 21, no. 14: 5030. https://doi.org/10.3390/ijms21145030
APA StyleVacchi, E., Kaelin-Lang, A., & Melli, G. (2020). Tau and Alpha Synuclein Synergistic Effect in Neurodegenerative Diseases: When the Periphery Is the Core. International Journal of Molecular Sciences, 21(14), 5030. https://doi.org/10.3390/ijms21145030