Differences between Well-Differentiated Neuroendocrine Tumors and Ductal Adenocarcinomas of the Pancreas Assessed by Multi-Omics Profiling

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. Mutational Status
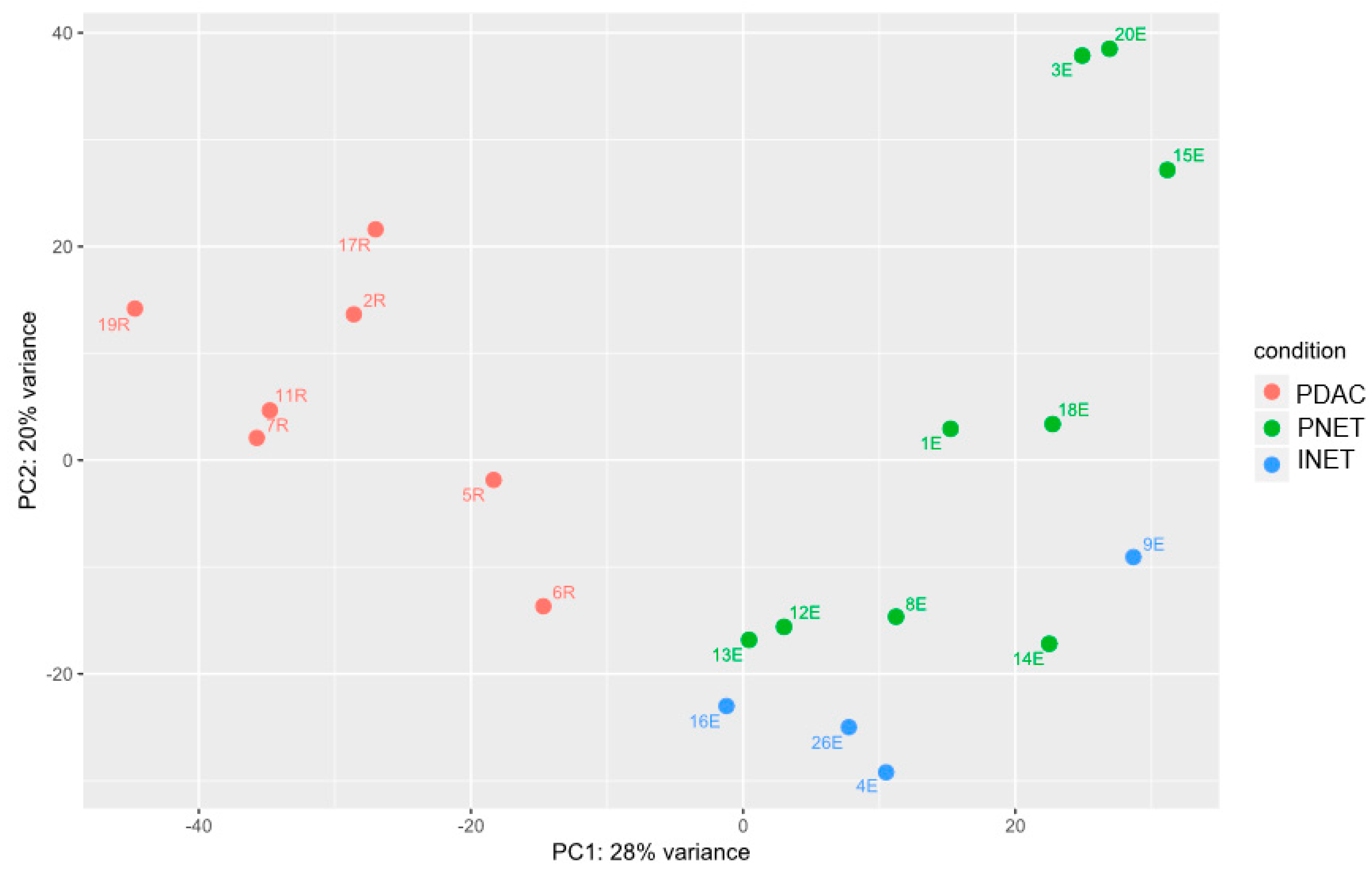
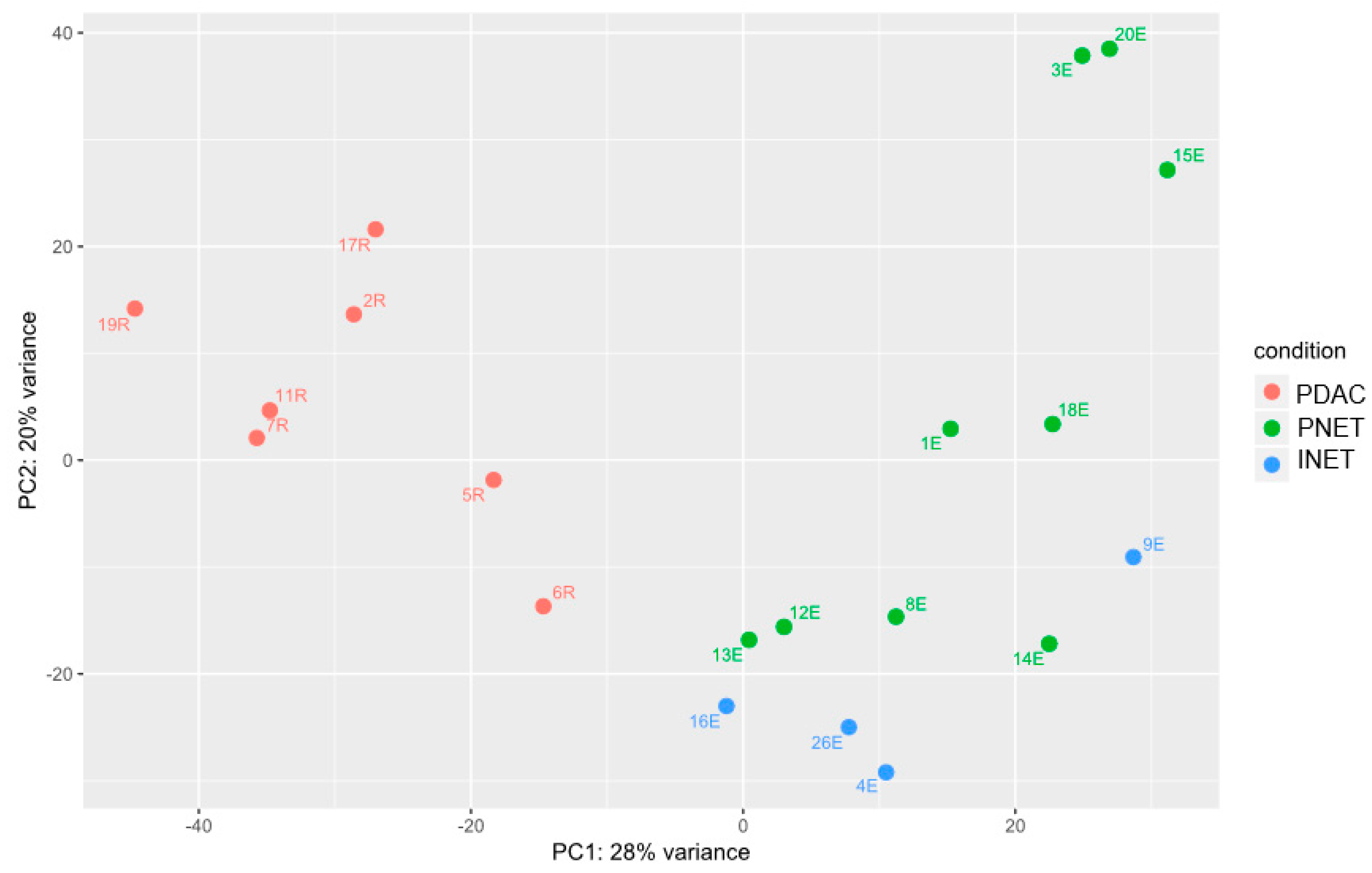
2.2. Gene Expression Analyses
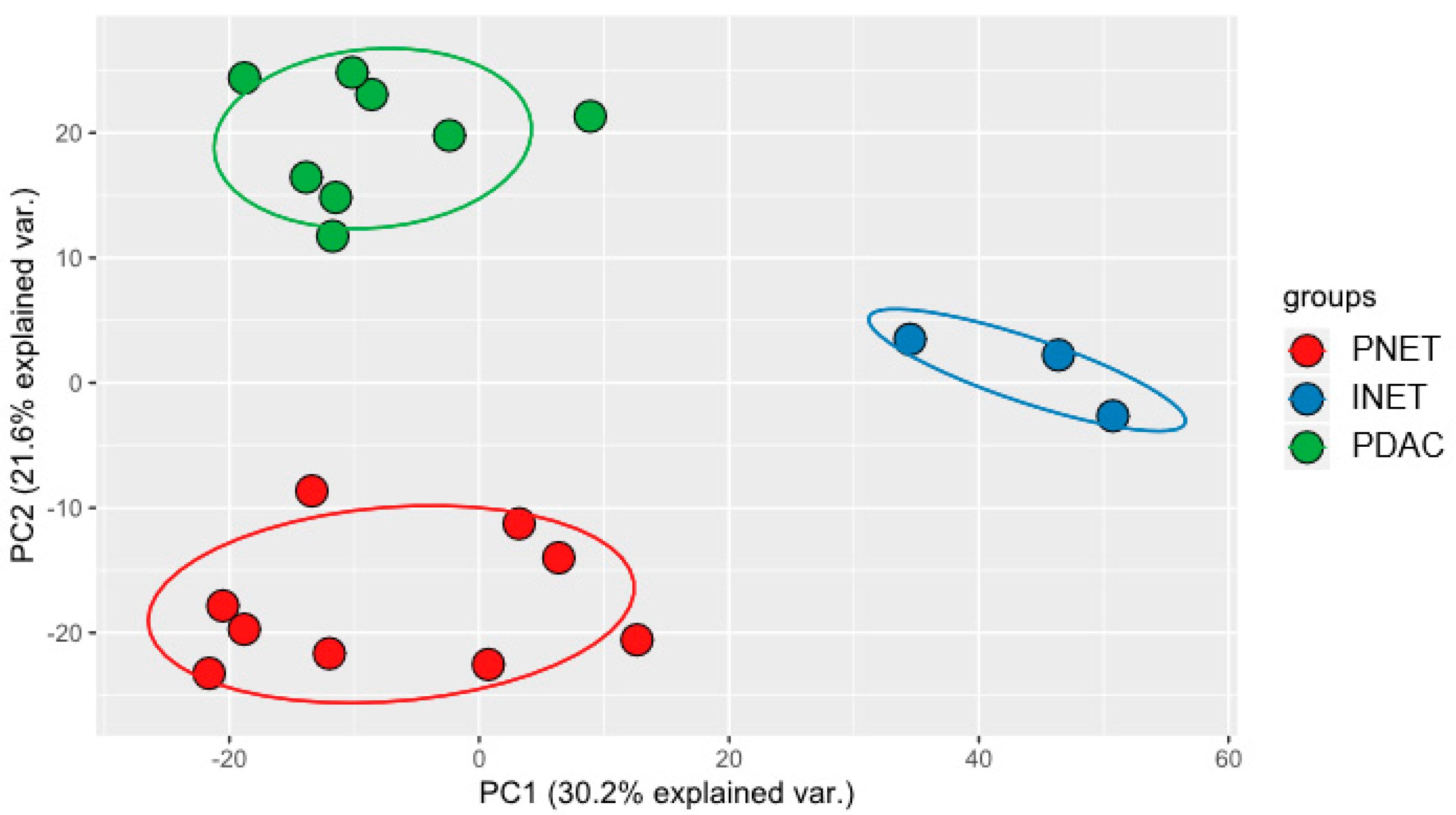
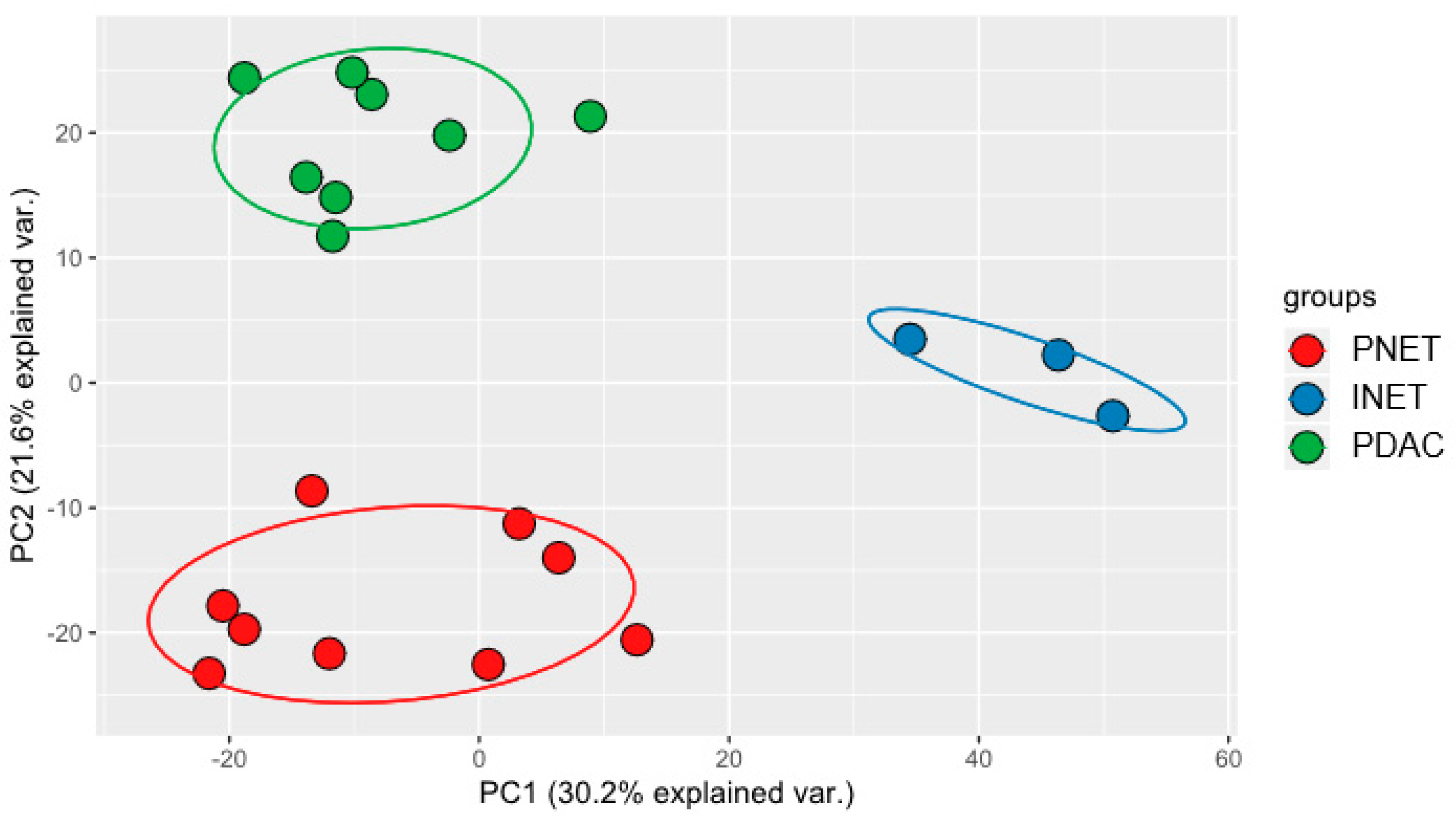
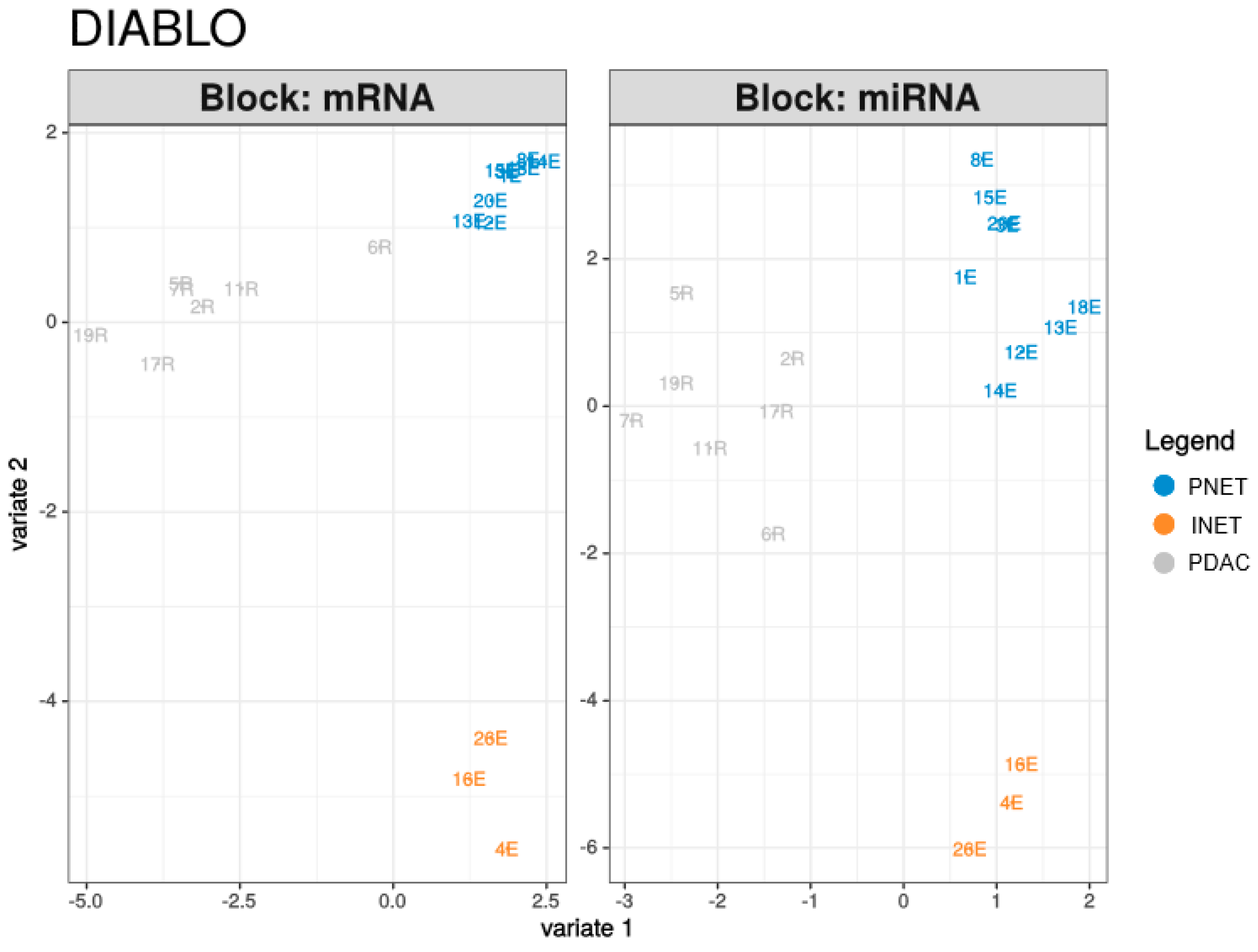
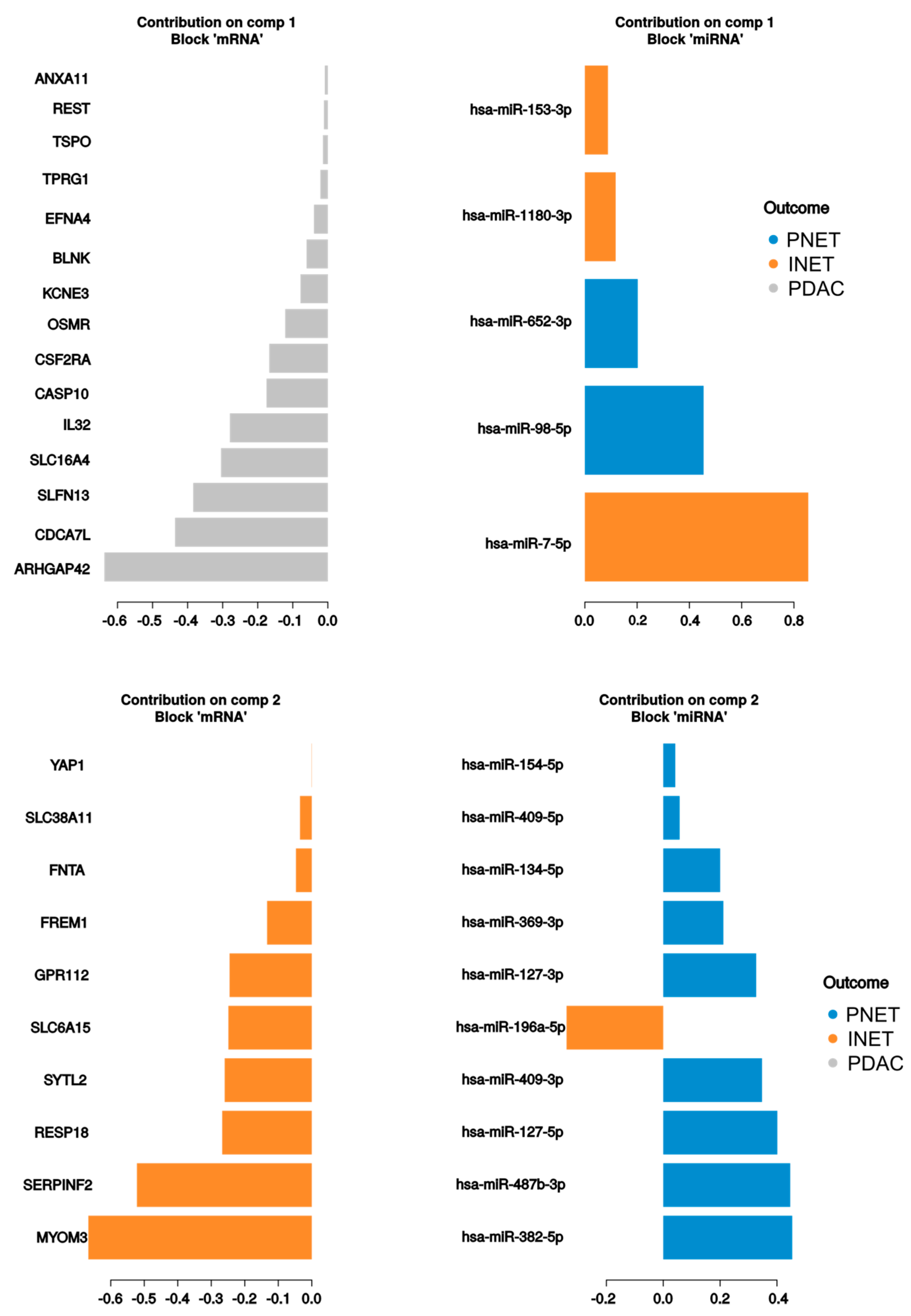
2.3. Integrated Analysis
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Nucleic Acid Isolation
4.3. Profiling of DNA Variants
4.4. Data Analysis and Variant Calling
4.5. Profiling of Transcriptome Panels
4.6. mRNA Data Analysis
4.7. miRNA Data Analysis
4.8. Integrated Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kos-Kudła, B.; Rosiek, V.; Borowska, M.; Bałdys-Waligórska, A.; Bednarczuk, T.; Blicharz-Dorniak, J.; Bolanowski, M.; Boratyn-Nowicka, A.; Cichocki, A.; Ćwikła, J.B.; et al. Pancreatic neuroendocrine neoplasms—Management guidelines (recommended by the Polish Network of Neuroendocrine Tumours). Endokrynol. Pol. 2017, 68, 169–197. [Google Scholar]
- Ro, C.; Chai, W.; Yu, V.E.; Yu, R. Pancreatic neuroendocrine tumors, biology, diagnosis, and treatment. Chin. J. Cancer 2013, 32, 312–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, L.M.; Steel, M.; Grewal, J.K.; Thibodeau, M.L.; Zhao, E.Y.; Loree, J.M.; Yang, K.C.; Gorski, S.M.; Mungall, A.J.; Mungall, K.L.; et al. Genomic characterization of a well-differentiated grade 3 pancreatic neuroendocrine tumor. Cold Spring Harb. Mol. Case Stud. 2019, 5, a003814. [Google Scholar] [CrossRef] [PubMed]
- Yoon, K.-A.; Woo, S.M.; Kim, Y.-H.; Kong, S.-Y.; Lee, M.K.; Han, S.-S.; Kim, T.H.; Lee, W.J.; Park, S.J. Comprehensive cancer panel sequencing defines genetic diversity and changes in the mutational characteristics of pancreatic cancer patients receiving neoadjuvant treatment. Gut Liver 2019, 13, 683–689. [Google Scholar] [CrossRef] [PubMed]
- Ilic, M.; Ilic, I. Epidemiology of pancreatic cancer. World J. Gastroenterol. 2016, 22, 9694–9705. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer, A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018, 24, 4846–4861. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of pancreatic cancer, global trends, etiology and risk factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Katabathina, V.S.; Rikhtehgar, O.Y.; Dasyam, A.K.; Manickam, R.; Prasad, S.R. Genetics of pancreatic neoplasms and role of screening. Magn. Reson. Imaging Clin. N. Am. 2018, 26, 375–389. [Google Scholar] [CrossRef]
- Kos-Kudła, B.; Blicharz-Dorniak, J.; Strzelczyk, J.; Bałdys-Waligórska, A.; Bednarczuk, T.; Bolanowski, M.; Kuśnierz, K.; Król, R.; Królicki, L.; Krzakowski, M.; et al. Diagnostic and therapeutic guidelines for gastro-entero-pancreatic neuroendocrine neoplasms (recommended by the Polish Network of Neuroendocrine Tumours). Endokrynol. Pol. 2017, 68, 418–443. [Google Scholar]
- Bosman, F.T.; Carneiro, F.; Hruban, R.H.; Theise, N.D. WHO Classification of Tumours of the Digestive System: WHO Classification of Tumours, Volume 3, 4th ed.; International Agency for Research on Cancer: Lyon, France, 2010; ISBN 978-92-832-2432-7. [Google Scholar]
- Haeberle, L.; Esposito, I. Pathology of pancreatic cancer. Transl. Gastroenterol. Hepatol. 2019, 4, 50. [Google Scholar] [CrossRef] [PubMed]
- Schlitter, A.M.; Segler, A.; Steiger, K.; Michalski, C.W.; Jäger, C.; Konukiewitz, B.; Pfarr, N.; Endris, V.; Bettstetter, M.; Kong, B.; et al. Molecular, morphological and survival analysis of 177 resected pancreatic ductal adenocarcinomas (PDACs), Identification of prognostic subtypes. Sci. Rep. 2017, 7, 41064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, I.; Penzel, R.; Chaib-Harrireche, M.; Barcena, U.; Bergmann, F.; Riedl, S.; Pfarr, N.; Endris, V.; Bettstetter, M.; Kong, B.; et al. Tenascin C and annexin II expression in the process of pancreatic carcinogenesis. J. Pathol. 2006, 208, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, M.; Hruban, R.H.; Wood, L.D. New developments in the molecular mechanisms of pancreatic tumorigenesis. Adv. Anat. Pathol. 2018, 25, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.-C.; Mansour, J.; Mollaee, M.; Wagner, K.-U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef] [PubMed]
- Waddell, N.; Pajic, M.; Patch, A.-M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.-C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.-M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef]
- Hosoda, W.; Wood, L.D. Molecular genetics of pancreatic neoplasms. Surg. Pathol. Clin. 2016, 9, 685–703. [Google Scholar] [CrossRef]
- Jiao, Y.; Shi, C.; Edil, B.H.; de Wilde, R.F.; Klimstra, D.S.; Maitra, A.; Schulick, R.D.; Tang, L.H.; Wolfgang, C.L.; Choti, M.A.; et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 2011, 331, 1199–1203. [Google Scholar] [CrossRef] [Green Version]
- Scarpa, A.; Chang, D.K.; Nones, K.; Corbo, V.; Patch, A.-M.; Bailey, P.; Lawlor, R.T.; Johns, A.L.; Miller, D.K.; Mafficini, A.; et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature 2017, 543, 65–71. [Google Scholar] [CrossRef]
- Wong, H.-L.; Yang, K.C.; Shen, Y.; Zhao, E.Y.; Loree, J.M.; Kennecke, H.F.; Kalloger, S.E.; Karasinska, J.M.; Lim, H.J.; Mungall, A.J.; et al. Molecular characterization of metastatic pancreatic neuroendocrine tumors (PNETs) using whole-genome and transcriptome sequencing. Cold Spring Harb. Mol. Case Stud. 2018, 4. [Google Scholar] [CrossRef]
- Zhang, C.; Wu, Y.; Zhuang, H.; Li, D.; Lin, Y.; Yin, Z.; Lu, X.; Hou, B.; Jian, Z. Establishment and validation of an AJCC stage- and histologic grade-based nomogram for pancreatic neuroendocrine tumors after surgical resection. Cancer Manag. Res. 2019, 11, 7345–7352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.R.; LaBauve, E.; Pomo, J.M.; Chiu, V.K.; Hanson, J.A.; Gullapalli, R.R. Site-specific genomic alterations in a well-differentiated pancreatic neuroendocrine tumor with high-grade progression. Pancreas 2018, 47, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Camilli, M.; Papadimitriou, K.; Nogueira, A.; Incorvaia, L.; Galvano, A.; D’Antonio, F.; Ferri, J.; Santini, D.; Silvestris, N.; Russo, A.; et al. Molecular profiling of pancreatic neuroendocrine tumors (pNETS) and the clinical potential. Expert Rev. Gastroenterol. Hepatol. 2018, 12, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Vortmeyer, A.O.; Huang, S.; Lubensky, I.; Zhuang, Z. Non-islet origin of pancreatic islet cell tumors. J. Clin. Endocrinol. Metab. 2004, 89, 1934–1938. [Google Scholar] [CrossRef] [Green Version]
- Obazee, O.; Capurso, G.; Tavano, F.; Archibugi, L.; De Bonis, A.; Greenhalf, W.; Key, T.; Pasquali, C.; Milanetto, A.C.; Hackert, T.; et al. Common genetic variants associated with pancreatic adenocarcinoma may also modify risk of pancreatic neuroendocrine neoplasms. Carcinogenesis 2018, 39, 360–367. [Google Scholar] [CrossRef]
- Haugvik, S.-P.; Hedenström, P.; Korsæth, E.; Valente, R.; Hayes, A.; Siuka, D.; Maisonneuve, P.; Gladhaug, I.P.; Lindkvist, B.; Capurso, G. Diabetes, smoking, alcohol use, and family history of cancer as risk factors for pancreatic neuroendocrine tumors, A systematic review and meta-analysis. Neuroendocrinology 2015, 101, 133–142. [Google Scholar] [CrossRef]
- Batukbhai, B.D.O.; De Jesus-Acosta, A. The molecular and clinical landscape of pancreatic neuroendocrine tumors. Pancreas 2019, 48, 9–21. [Google Scholar] [CrossRef]
- Gebauer, N.; Schmidt-Werthern, C.; Bernard, V.; Feller, A.C.; Keck, T.; Begum, N.; Rades, D.; Lehnert, H.; Brabant, G.; Thorns, C. Genomic landscape of pancreatic neuroendocrine tumors. World J. Gastroenterol. 2014, 20, 17498–17506. [Google Scholar] [CrossRef]
- Ehehalt, F.; Franke, E.; Pilarsky, C.; Grützmann, R. Molecular pathogenesis of pancreatic neuroendocrine tumors. Cancers 2010, 2, 1901–1910. [Google Scholar] [CrossRef] [Green Version]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Basturk, O.; Tang, L.; Hruban, R.H.; Adsay, V.; Yang, Z.; Krasinskas, A.M.; Vakiani, E.; La Rosa, S.; Jang, K.-T.; Frankel, W.L.; et al. Poorly differentiated neuroendocrine carcinomas of the pancreas, a clinicopathologic analysis of 44 Cases. Am. J. Surg. Pathol. 2014, 38, 437–447. [Google Scholar] [CrossRef]
- Hackeng, W.M.; Hruban, R.H.; Offerhaus, G.J.A.; Brosens, L.A.A. Surgical and molecular pathology of pancreatic neoplasms. Diagn. Pathol. 2016, 11, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hijioka, S.; Hosoda, W.; Matsuo, K.; Ueno, M.; Furukawa, M.; Yoshitomi, H.; Kobayashi, N.; Ikeda, M.; Ito, T.; Nakamori, S.; et al. Rb loss and KRAS mutation are predictors of the response to platinum-based chemotherapy in pancreatic neuroendocrine neoplasm with grade 3, a japanese multicenter pancreatic NEN-G3 study. Clin. Cancer Res. 2017, 23, 4625–4632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konukiewitz, B.; Schlitter, A.M.; Jesinghaus, M.; Pfister, D.; Steiger, K.; Segler, A.; Agaimy, A.; Sipos, B.; Zamboni, G.; Weichert, W.; et al. Somatostatin receptor expression related to TP53 and RB1 alterations in pancreatic and extrapancreatic neuroendocrine neoplasms with a Ki67-index above 20%. Mod Pathol. 2017, 30, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Yachida, S.; Vakiani, E.; White, C.M.; Zhong, Y.; Saunders, T.; Morgan, R.; de Wilde, R.F.; Maitra, A.; Hicks, J.; Demarzo, A.M.; et al. Small Cell and Large Cell Neuroendocrine Carcinomas of the Pancreas are Genetically Similar and Distinct From Well-differentiated Pancreatic Neuroendocrine Tumors. Am. J. Surg. Pathol. 2012, 36, 173–184. [Google Scholar] [CrossRef]
- Xiao, Y.; Yang, Y.; Wang, Y.; Li, X.; Wang, W. Five novel genes related to the pathogenesis and progression of pancreatic neuroendocrine tumors by bioinformatics analysis with RT-qPCR verification. Front. Neurosci. 2019, 13, 937. [Google Scholar] [CrossRef]
- Missiaglia, E.; Dalai, I.; Barbi, S.; Beghelli, S.; Falconi, M.; della Peruta, M.; Piemonti, L.; Capurso, G.; Di Florio, A.; delle Fave, G.; et al. Pancreatic endocrine tumors, expression profiling evidences a role for AKT-mTOR pathway. J. Clin. Oncol. 2010, 28, 245–255. [Google Scholar] [CrossRef] [Green Version]
- Sadanandam, A.; Wullschleger, S.; Lyssiotis, C.A.; Grotzinger, C.; Barbi, S.; Bersani, S.; Körner, J.; Wafy, I.; Mafficini, A.; Lawlor, R.T.; et al. A cross-species analysis in pancreatic neuroendocrine tumors reveals molecular subtypes with distinctive clinical, Metastatic, Developmental, and Metabolic Characteristics. Cancer Discov. 2015, 5, 1296–1313. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.-D.; Liu, Z.-W.; Han, M.-M.; Zhu, Z.-M.; Tu, Y.-L.; Dou, C.-Q.; Jin, X.; Cai, S.-W.; Du, N. Microarray based analysis of gene expression patterns in pancreatic neuroendocrine tumors. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 3367–3374. [Google Scholar]
- Klieser, E.; Urbas, R.; Swierczynski, S.; Stättner, S.; Primavesi, F.; Jäger, T.; Mayr, C.; Kiesslich, T.; Fazio, P.D.; Helm, K.; et al. HDAC-linked “proliferative” miRNA expression pattern in pancreatic neuroendocrine tumors. Int. J. Mol. Sci. 2018, 19, 2781. [Google Scholar] [CrossRef] [Green Version]
- Croce, C.M. Causes and consequences of microRNA dysregulation in cancer. Nat. Rev. Genet. 2009, 10, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, Z.; Li, B.; Zhang, Z.; Luo, H.; Wang, Y.; Lu, Z.; Wu, X. Epigenetic silencing of miRNA-9 is correlated with promoter-proximal CpG island hypermethylation in gastric cancer in vitro and in vivo. Int. J. Oncol. 2014, 45, 2576–2586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiaschetti, G.; Abela, L.; Nonoguchi, N.; Dubuc, A.M.; Remke, M.; Boro, A.; Grunder, E.; Siler, U.; Ohgaki, H.; Taylor, M.D.; et al. Epigenetic silencing of miRNA-9 is associated with HES1 oncogenic activity and poor prognosis of medulloblastoma. Br. J. Cancer 2014, 110, 636–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayr, C.; Beyreis, M.; Wagner, A.; Pichler, M.; Neureiter, D.; Kiesslich, T. Deregulated MicroRNAs in Biliary Tract Cancer, Functional Targets and Potential Biomarkers. BioMed Res. Int. 2016, 2016, 1–15. [Google Scholar] [CrossRef]
- Holubekova, V.; Mendelova, A.; Jasek, K.; Mersakova, S.; Zubor, P.; Lasabova, Z. Epigenetic regulation by DNA methylation and miRNA molecules in cancer. Future Oncol. 2017, 13, 2217–2222. [Google Scholar] [CrossRef] [Green Version]
- Kwon, H.; Song, K.; Han, C.; Zhang, J.; Lu, L.; Chen, W.; Wu, T. Epigenetic Silencing of miRNA-34a in Human Cholangiocarcinoma via EZH2 and DNA Methylation. Am. J. Pathol. 2017, 187, 2288–2299. [Google Scholar] [CrossRef] [Green Version]
- Jiao, L.R.; Frampton, A.E.; Jacob, J.; Pellegrino, L.; Krell, J.; Giamas, G.; Tsim, N.; Vlavianos, P.; Cohen, P.; Ahmad, R.; et al. MicroRNAs Targeting Oncogenes Are Down-Regulated in Pancreatic Malignant Transformation from Benign Tumors. Tjwa M, editor. PLoS ONE 2012, 7, e32068. [Google Scholar] [CrossRef] [Green Version]
- Papaconstantinou, I.G.; Manta, A.; Gazouli, M.; Lyberopoulou, A.; Lykoudis, P.M.; Polymeneas, G.; Voros, D. Expression of MicroRNAs in Patients With Pancreatic Cancer and Its Prognostic Significance. Pancreas 2013, 42, 67–71. [Google Scholar] [CrossRef]
- Zimmermann, N.; Knief, J.; Kacprowski, T.; Lazar-Karsten, P.; Keck, T.; Billmann, F.; Schmid, S.; Luley, K.; Lehnert, H.; Brabant, G.; et al. MicroRNA analysis of gastroenteropancreatic neuroendocrine tumors and metastases. Oncotarget 2018, 9, 28379–28390. [Google Scholar] [CrossRef] [Green Version]
- Fellenberg, J.; Lehner, B.; Saehr, H.; Schenker, A.; Kunz, P. Tumor Suppressor Function of miR-127-3p and miR-376a-3p in Osteosarcoma Cells. Cancers (Basel) 2019, 11, 2019. [Google Scholar] [CrossRef] [Green Version]
- Herr, I.; Sähr, H.; Zhao, Z.; Yin, L.; Omlor, G.; Lehner, B.; Fellenberg, J. MiR-127 and miR-376a act as tumor suppressors by in vivo targeting of COA1 and PDIA6 in giant cell tumor of bone. Cancer Lett. 2017, 409, 49–55. [Google Scholar] [CrossRef]
- Fan, J.; Du, W.; Zhang, H.; Wang, Y.; Li, K.; Meng, Y.; Wang, J. Transcriptional downregulation of miR-127-3p by CTCF promotes prostate cancer bone metastasis by targeting PSMB5. FEBS Lett. 2020, 594, 466–476. [Google Scholar] [CrossRef]
- Bi, L.; Yang, Q.; Yuan, J.; Miao, Q.; Duan, L.; Li, F.; Wang, S. MicroRNA-127-3p acts as a tumor suppressor in epithelial ovarian cancer by regulating the BAG5 gene. Oncol Rep. 2016, 36, 2563–2570. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Jin, C.; Liu, J.; Hua, D.; Zhou, F.; Lou, X.; Zhao, N.; Lan, Q.; Huang, Q.; Yoon, J.-G.; et al. Next generation sequencing analysis of miRNAs, MiR-127-3p inhibits glioblastoma proliferation and activates TGF-β signaling by targeting SKI. OMICS 2014, 18, 196–206. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Zhang, Y.; Huang, Z.; Gu, H.; Zhou, K.; Yin, X.; Xu, J. MiR-409-3p Inhibits Cell Proliferation and Invasion of Osteosarcoma by Targeting Zinc-Finger E-Box-Binding Homeobox-1. Front. Pharmacol. 2019, 10, 137. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Dai, J. miR-409-3p suppresses the proliferation invasion migration of tongue squamous cell carcinoma via targeting, R.D.X. Oncol Lett. 2018, 16, 543–551. [Google Scholar]
- Zhang, G.; Liu, Z.; Xu, H.; Yang, Q. miR-409-3p suppresses breast cancer cell growth and invasion by targeting Akt1. Biochem. Biophys. Res. Commun. 2016, 469, 189–195. [Google Scholar] [CrossRef]
- Josson, S.; Gururajan, M.; Hu, P.; Shao, C.; Chu, G.-Y.; Zhau, H.E.; Liu, C.; Lao, K.; Lu, C.L.; Lu, Y.T.; et al. miR-409-3p/-5p promotes tumorigenesis, epithelial-to-mesenchymal transition, and bone metastasis of human prostate cancer. Clin. Cancer Res. 2014, 20, 4636–4646. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.; Zhang, A.; Liu, J.; Wu, K.; Chen, Z.; Wang, Q. Down-regulation of MAP2K1 by miR-539 inhibits hepatocarcinoma progression. Biochem. Biophys. Res. Commun. 2018, 504, 784–791. [Google Scholar] [CrossRef]
- Lv, L.; Wang, Y.; Zhang, Q.; Zang, H.; Wang, X. miR-539 induces cell cycle arrest in nasopharyngeal carcinoma by targeting cyclin-dependent kinase 4. Cell Biochem. Funct. 2015, 33, 534–540. [Google Scholar] [CrossRef]
- Yu, H.; Gao, G.; Cai, J.; Song, H.; Ma, Z.; Jin, X.; Ji, W.; Pan, B. MiR-539 functions as a tumor suppressor in pancreatic cancer by targeting TWIST1. Exp. Mol. Pathol. 2019, 108, 143–149. [Google Scholar] [CrossRef]
- Wen, D.; Li, S.; Jiang, W.; Zhu, J.; Liu, J.; Zhao, S. miR-539 inhibits human colorectal cancer progression by targeting RUNX2. Biomed. Pharmacother. 2017, 95, 1314–1320. [Google Scholar] [CrossRef]
- Sun, X.; Dongol, S.; Qiu, C.; Xu, Y.; Sun, C.; Zhang, Z.; Yang, X.; Zhang, Q.; Kong, B. miR-652 Promotes Tumor Proliferation and Metastasis by Targeting RORA in Endometrial Cancer. Mol. Cancer Res. 2018, 16, 1927–1939. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zou, X. MiR-652 serves as a prognostic biomarker in gastric cancer and promotes tumor proliferation, migration, and invasion via targeting RORA. Cancer Biomark. 2019, 26, 323–331. [Google Scholar] [CrossRef]
- Xia, Z.; Yang, C.; Yang, X.; Wu, S.; Feng, Z.; Qu, L.; Chen, X.; Liu, L.; Ma, Y. miR-652 Promotes Proliferation and Migration of Uveal Melanoma Cells by Targeting HOXA9. Med. Sci. Monit. 2019, 25, 8722–8732. [Google Scholar] [CrossRef]
- Yang, W.; Zhou, C.; Luo, M.; Shi, X.; Li, Y.; Sun, Z.; Zhou, F.; Chen, Z.; He, J. MiR-652-3p is upregulated in non-small cell lung cancer and promotes proliferation and metastasis by directly targeting Lgl1. Oncotarget 2016, 7, 16703–16715. [Google Scholar] [CrossRef] [Green Version]
- Tang, L.; Peng, Y.-Z.; Li, C.-G.; Jiang, H.-W.; Mei, H.; Hu, Y. Prognostic and clinicopathological significance of MiR-155 in hematologic malignancies. a systematic review and meta-analysis. J. Cancer 2019, 10, 654–664. [Google Scholar] [CrossRef] [Green Version]
- Jurkovicova, D.; Magyerkova, M.; Kulcsar, L.; Krivjanska, M.; Krivjansky, V.; Gibadulinova, A.; Oveckova, I.; Chovanec, M. miR-155 as a diagnostic and prognostic marker in hematological and solid malignancies. Neoplasma 2014, 61, 241–251. [Google Scholar] [CrossRef]
- Lv, Y.; Yang, H.; Ma, X.; Wu, G. Strand-specific miR-28-3p and miR-28-5p have differential effects on nasopharyngeal cancer cells proliferation, apoptosis, migration and invasion. Cancer Cell Int. 2019, 19, 187. [Google Scholar] [CrossRef]
- Almeida, M.I.; Nicoloso, M.S.; Zeng, L.; Ivan, C.; Spizzo, R.; Gafà, R.; Xiao, L.; Zhang, X.; Vannini, I.; Fanini, F.; et al. Strand-specific miR-28-5p and miR-28-3p have distinct effects in colorectal cancer cells. Gastroenterology 2012, 142, 886–896.e9. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Wu, C.; Yang, Q.; Ding, M.; Zhong, J.; Zhang, C.-Y.; Ge, J.; Wang, J.; Zhang, C. miR-28-5p acts as a tumor suppressor in renal cell carcinoma for multiple antitumor effects by targeting, R.A.P.1.B. Oncotarget 2016, 7, 73888–73902. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Yao, Y.; Song, Q.; Li, S.; Hu, Z.; Yu, Y.; Hu, C.; Da, X.; Li, H.; Chen, Q.; et al. Up-regulation of miR-95-3p in hepatocellular carcinoma promotes tumorigenesis by targeting p21 expression. Sci. Rep. Nat. Publ. Group 2016, 6, 34034. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Chen, S.; Hang, W.; Huang, H.; Ma, H. MiR-95 induces proliferation and chemo- or radioresistance through directly targeting sorting nexin1 (SNX1) in non-small cell lung cancer. Biomed. Pharm. 2014, 68, 589–595. [Google Scholar] [CrossRef]
- Liu, X.; Ma, W.; Ma, J.; Xiao, L.; Hao, D. Upregulation of miR-95-3p inhibits growth of osteosarcoma by targeting HDGF. Pathol. Res. Pract. 2019, 215, 152492. [Google Scholar] [CrossRef]
- Thorns, C.; Schurmann, C.; Gebauer, N.; Wallaschofski, H.; Kümpers, C.; Bernard, V.; Feller, A.C.; Keck, T.; Habermann, J.K.; Begum, N.; et al. Global microRNA profiling of pancreatic neuroendocrine neoplasias. Anticancer Res. 2014, 34, 2249–2254. [Google Scholar]
- Pentheroudakis, G.; Pavlidis, N.; Fountzilas, G.; Krikelis, D.; Goussia, A.; Stoyianni, A.; Sanden, M.; St Cyr, B.; Yerushalmi, N.; Benjamin, H.; et al. Novel microRNA-based assay demonstrates 92% agreement with diagnosis based on clinicopathologic and management data in a cohort of patients with carcinoma of unknown primary. Mol. Cancer. 2013, 12, 57. [Google Scholar] [CrossRef] [Green Version]
- Meiri, E.; Mueller, W.C.; Rosenwald, S.; Zepeniuk, M.; Klinke, E.; Edmonston, T.B.; Werner, M.; Lass, U.; Barshack, I.; Feinmesser, M.; et al. A second-generation microRNA-based assay for diagnosing tumor tissue origin. Oncologist 2012, 17, 801–812. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, K.A.; Moore, A.R.; Parsons, B.N.; O’Hara, A.; Boyce, M.; Dockray, G.J.; Varro, A.; Pritchard, D.M. Gastrin-induced miR-222 promotes gastric tumor development by suppressing p27kip1. Oncotarget 2016, 7, 45462–45478. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.A.; Zubair, H.; Srivastava, S.K.; Singh, S.; Singh, A.P. Insights into the role of microRNAs in pancreatic Cancer pathogenesis, potential for diagnosis, prognosis, and therapy. Adv. Exp. Med. Biol. 2015, 889, 71–87. [Google Scholar]
- Karpathakis, A.; Dibra, H.; Thirlwell, C. Neuroendocrine tumours, cracking the epigenetic code. Endocrine-Relat. Cancer 2013, 20, R65–R82. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Kim, H.; Kim, H.W.; Lee, J.-C.; Paik, K.-H.; Kang, J.; Kim, J.; Yoon, Y.-S.; Han, H.-S.; Sohn, I.; et al. High expression of microRNA-196a indicates poor prognosis in resected pancreatic neuroendocrine tumor. Medicine 2015, 94, e2224. [Google Scholar] [CrossRef]
- Garzon, R.; Marcucci, G.; Croce, C.M. Targeting microRNAs in cancer, rationale, strategies and challenges. Nat. Rev. Drug Discov. 2010, 9, 775–789. [Google Scholar] [CrossRef] [Green Version]
- Paziewska, A.; Mikula, M.; Dabrowska, M.; Kulecka, M.; Goryca, K.; Antoniewicz, A.; Dobruch, J.; Borowka, A.; Rutkowski, P.; Ostrowski, J. Candidate diagnostic miRNAs that can detect cancer in prostate biopsy. Prostate 2018, 78, 178–185. [Google Scholar] [CrossRef]
- Iorio, M.V.; Croce, C.M. MicroRNA dysregulation in cancer, diagnostics, monitoring and therapeutics. A comprehensive review. EMBO Mol. Med. 2012, 4, 143–159. [Google Scholar] [CrossRef]
- Ambrozkiewicz, F.; Karczmarski, J.; Kulecka, M.; Paziewska, A.; Cybulska, M.; Szymanski, M.; Dobruch, J.; Antoniewicz, A.; Mikula, M.; Ostrowski, J. Challenges in cancer biomarker discovery exemplified by the Identification of diagnostic microRNAs in prostate tissues. Biomed. Res. Int. 2020, 9086829. [Google Scholar]
- Iontorrent/TMAP. Ion Torrent. 2019. Available online: https://github.com/iontorrent/TMAP (accessed on 19 November 2019).
- Roberson, D. davidroberson/tvc [Internet]. 2019. Available online: https://github.com/davidroberson/tvc. (accessed on 19 November 2019).
- Douville, C.; Carter, H.; Kim, R.; Niknafs, N.; Diekhans, M.; Stenson, P.D.; Cooper, D.N.; Ryan, M.; Karchin, R. CRAVAT, Cancer-related analysis of variants toolkit. Bioinformatics 2013, 29, 647–648. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pagès, F.; Trajanoski, Z.; Galon, J.; et al. ClueGO, a cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools, A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR, a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Shannon, C.P.; Gautier, B.; Rohart, F.; Vacher, M.; Tebbutt, S.J.; Lê Cao, K.A. DIABLO, an integrative approach for identifying key molecular drivers from multi-omics assays. Bioinformatics 2019, 35, 3055–3062. [Google Scholar] [CrossRef]

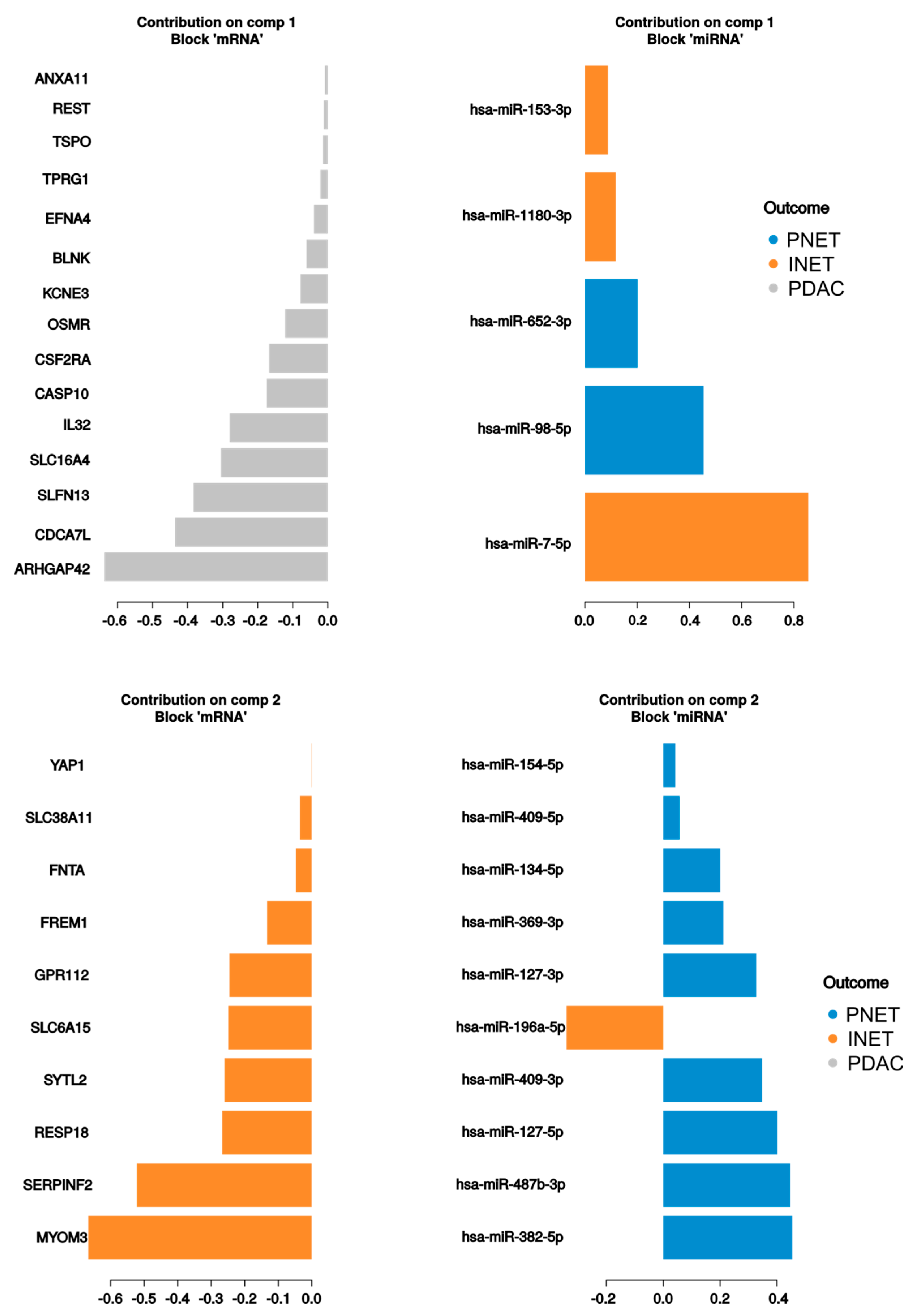

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sex | Age | Tumor Location | Tumor Size (cm) | Tumor Type/Grade | Vascular Invasion | Nerve Invasion | Surgery | TNM Stage |
|---|---|---|---|---|---|---|---|---|
| M | 70 | Tail | 1.9 | PNET G2 | + | resection | T4N0M0 | |
| F | 35 | Tail | 3.5 | PNET G2 | + | + | resection | T2N1M0 |
| F | 27 | Head | 3.5 | PNET G2 | + | + | resection | T2N1M0 |
| F | 27 | Tail | 2.3 | PNET G2 | + | resection | T2N0M0 | |
| F | 54 | Tail | 4.2 | PNET G1 | + | + | resection | T3N1M1 |
| F | 69 | Head | 4.2 | PNET G2 | + | resection | T3N1M0 | |
| F | 44 | Tail | 3.2 | PNET G1 | + | resection | T2N0M0 | |
| F | 24 | Head | 1.3 | PNET G2 | + | resection | T1N1M0 | |
| F | 60 | Body | Unknown | PNET G3 | laparotomy | Unknown | ||
| M | 67 | Duodenum | 3 | INET G1 | resection | T2N0M0 | ||
| F | 66 | Small intestine | 2 | INET G1 | resection | T4N1M0 | ||
| M | 59 | Small intestine | 2.7 | INET G1 | resection | T3N0M1 | ||
| M | 38 | Small intestine | 1.7 | INET G1 | resection | T1N0M0 | ||
| M | 45 | Head | 2.8 | PDAC G2 | + | + | resection | T2N2M0 |
| F | 72 | Body | Unknown | PDAC G? | + | laparotomy | T4N1M0 | |
| F | 51 | Head | 2.5 | PDAC G2 | + | resection | T2N2M0 | |
| F | 67 | Head | 3.3 | PDAC G3 | + | + | resection | T4N2M0 |
| F | 69 | Head | 3 | PDAC G2 | + | + | resection | T2N2M0 |
| M | 38 | Tail | 1.7 | PDAC G? | resection | T1N0M1 | ||
| F | 70 | Head | Unknown | PDAC G? | + | laparotomy | T3N1M1 | |
| F | 71 | Head | Unknown | PDAC G? | + | laparotomy | T4N1M0 |
| Names | Total | Elements |
|---|---|---|
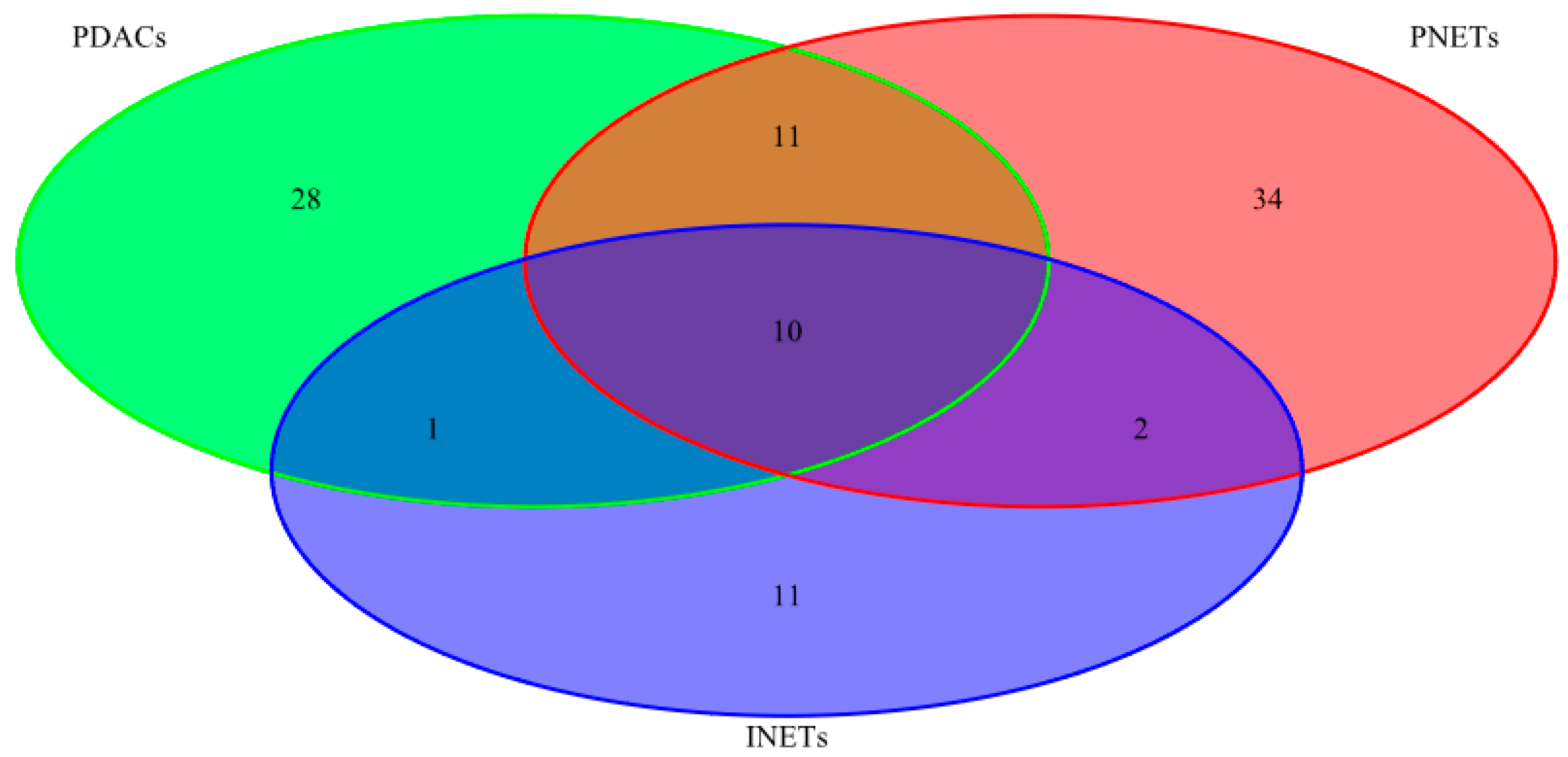
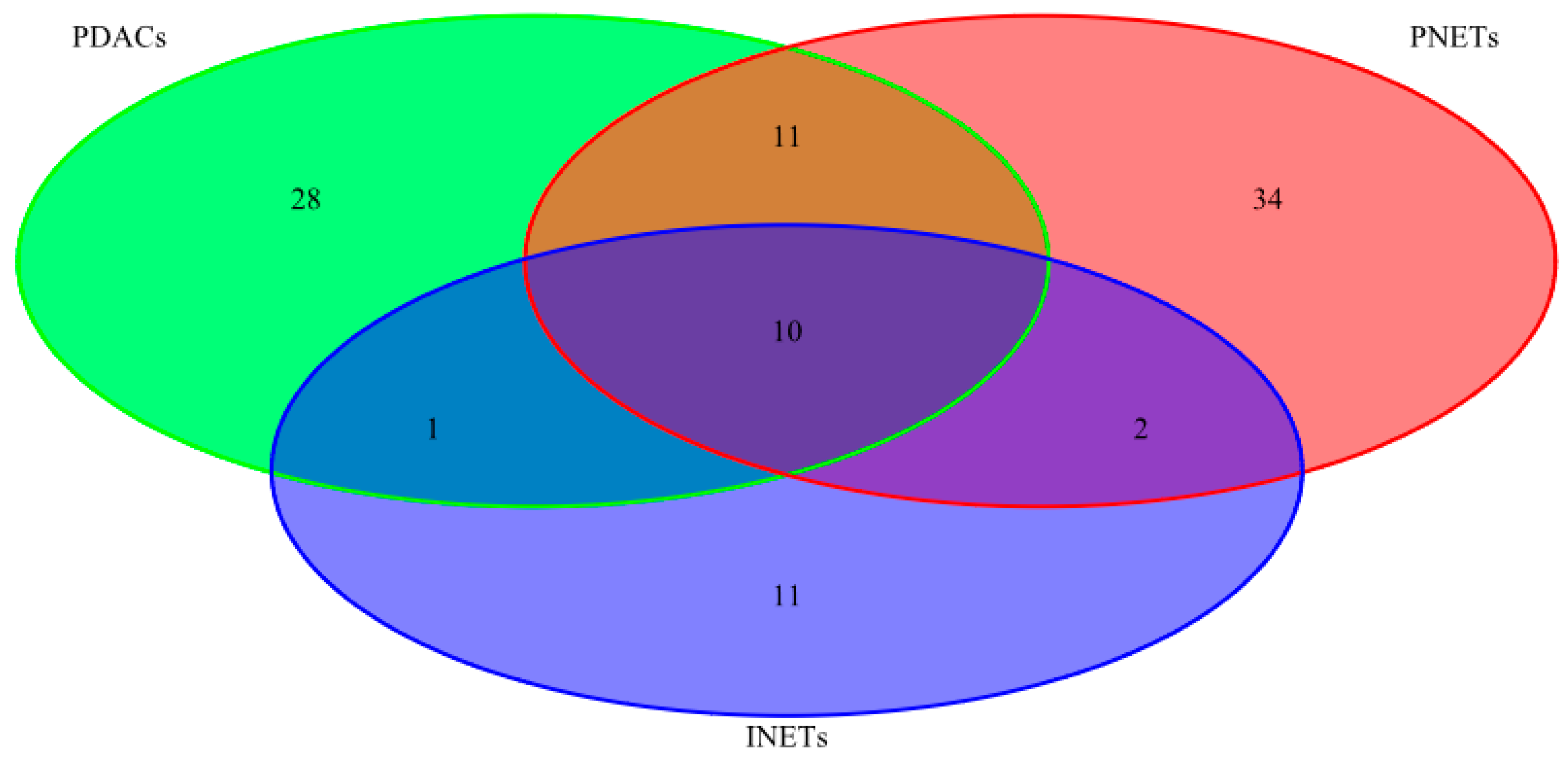
| INETs PDACs PNETs | 10 | CASC5 PAX7 ATRX NFKB2 HNF1A APC STK11 TSHR CREBBP RNF213 |
| PDACs PNETs | 11 | ARID2 MTOR COL1A1 MLL2 KAT6B BCL9 BLM FN1 NOTCH2 MSH6 ATM |
| INETs PDACs | 1 | ERBB4 |
| INETs PNETs | 2 | MLL3 LPHN3 |
| PDACs | 28 | EP400 CYP2D6 KRAS ABL2 BCL2 PTPRT TP53 RET KDM5C TET2 ING4 PER1 SMAD4 BCL6 PALB2 NPM1 NUP98 CDKN2A ERG BAP1 MET GATA1 PRDM1 ATR RNASEL FANCG PLEKHG5 KLF6 |
| PNETs | 34 | FGFR3 AURKB CDH1 FLI1 CKS1B CYP2C19 SMO UBR5 TNFAIP3 SOCS1 NTRK1 TNK2 MITF EPHA3 ARID1A MEN1 PIK3CG MLL VHL MAP2K1 MYD88 SF3B1 RPS6KA2 CSF1R BCR RUNX1T1 PRKDC TFE3 JAK3 AR PMS1 ESR1 DAXX NIN |
| INETs | 11 | FLT4 GDNF NOTCH1 SMARCA4 ZNF521 ITGB2 EP300 PTCH1 FLT3 BIRC3 TSC2 |
| Names | Total | Elements |
|---|---|---|
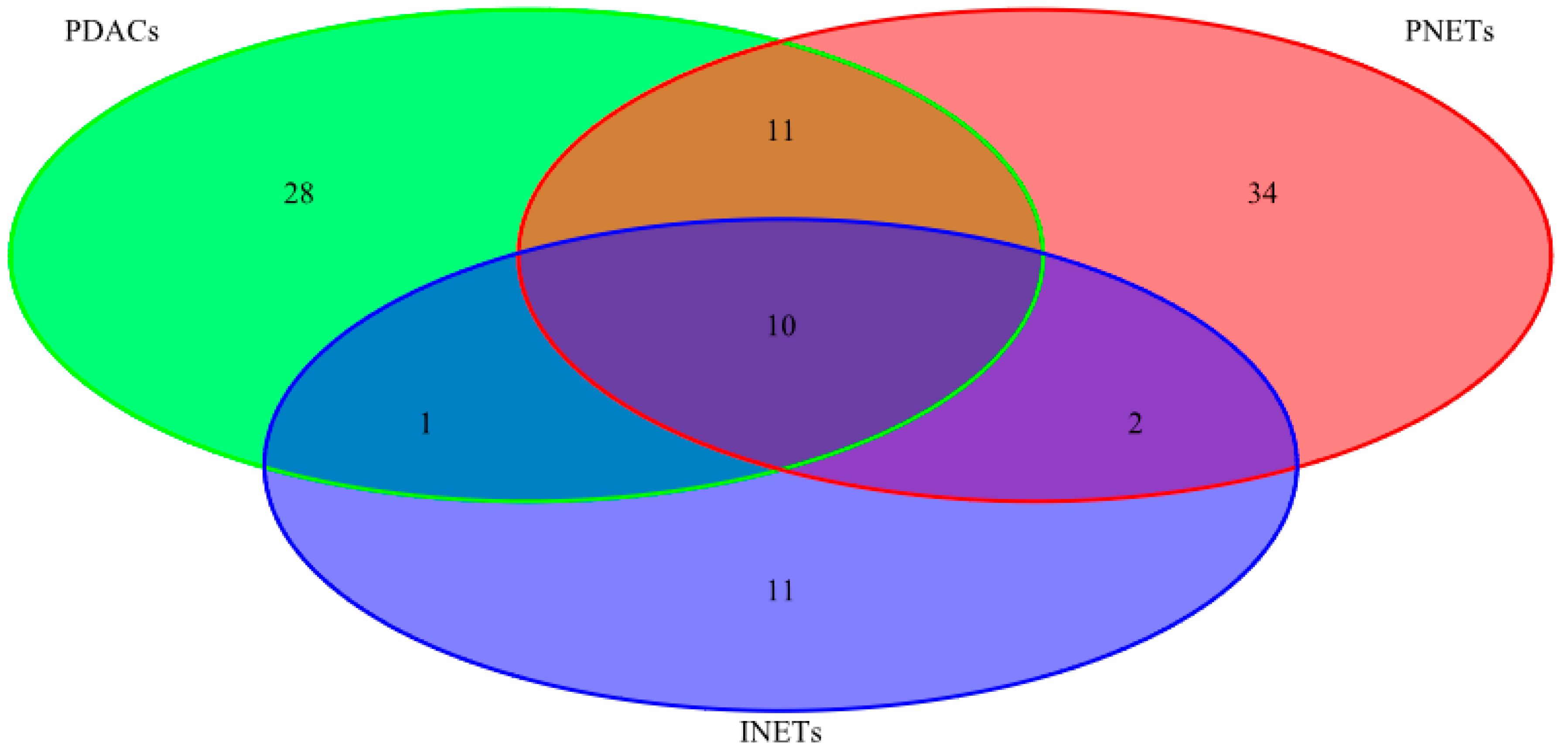
| INETs PDACs PNETs | 2 | CREBBP CASC5 |
| PDACs PNETs | 2 | HNF1A APC |
| INETs PDACs | 1 | RNF213 |
| PDACs | 2 | KRAS TP53 |
| PNETs | 4 | MLL2 KAT6B JAK3 MLL3 |
| INETs | 8 | FLT4 NFKB2 PAX7 FLT3 ZNF521 NOTCH1 EP300 STK11 |
| Group | AUC | p-Value | Component | Analysis |
|---|---|---|---|---|
| PNET | 0.9 | 0.003289 | 1 | mRNA |
| INET | 0.625 | 0.5023 | 1 | mRNA |
| PDAC | 1 | 0.000386 | 1 | mRNA |
| PNET | 1 | 0.000239 | 2 | mRNA |
| INET | 1 | 0.00729 | 2 | mRNA |
| PDAC | 1 | 0.000386 | 2 | mRNA |
| PNET | 1 | 0.000239 | 3 | mRNA |
| INET | 1 | 0.00729 | 3 | mRNA |
| PDAC | 1 | 0.000386 | 3 | mRNA |
| PNET | 0.8889 | 0.004267 | 1 | miRNA |
| INET | 0.6458 | 0.4338 | 1 | miRNA |
| PDAC | 1 | 0.000386 | 1 | miRNA |
| PNET | 1 | 0.000239 | 2 | miRNA |
| INET | 1 | 0.00729 | 2 | miRNA |
| PDAC | 1 | 0.000386 | 2 | miRNA |
| PNET | 1 | 0.000239 | 3 | miRNA |
| INET | 1 | 0.00729 | 3 | miRNA |
| PDAC | 1 | 0.000386 | 3 | miRNA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Starzyńska, T.; Karczmarski, J.; Paziewska, A.; Kulecka, M.; Kuśnierz, K.; Żeber-Lubecka, N.; Ambrożkiewicz, F.; Mikula, M.; Kos-Kudła, B.; Ostrowski, J. Differences between Well-Differentiated Neuroendocrine Tumors and Ductal Adenocarcinomas of the Pancreas Assessed by Multi-Omics Profiling. Int. J. Mol. Sci. 2020, 21, 4470. https://doi.org/10.3390/ijms21124470
Starzyńska T, Karczmarski J, Paziewska A, Kulecka M, Kuśnierz K, Żeber-Lubecka N, Ambrożkiewicz F, Mikula M, Kos-Kudła B, Ostrowski J. Differences between Well-Differentiated Neuroendocrine Tumors and Ductal Adenocarcinomas of the Pancreas Assessed by Multi-Omics Profiling. International Journal of Molecular Sciences. 2020; 21(12):4470. https://doi.org/10.3390/ijms21124470
Chicago/Turabian StyleStarzyńska, Teresa, Jakub Karczmarski, Agnieszka Paziewska, Maria Kulecka, Katarzyna Kuśnierz, Natalia Żeber-Lubecka, Filip Ambrożkiewicz, Michał Mikula, Beata Kos-Kudła, and Jerzy Ostrowski. 2020. "Differences between Well-Differentiated Neuroendocrine Tumors and Ductal Adenocarcinomas of the Pancreas Assessed by Multi-Omics Profiling" International Journal of Molecular Sciences 21, no. 12: 4470. https://doi.org/10.3390/ijms21124470
APA StyleStarzyńska, T., Karczmarski, J., Paziewska, A., Kulecka, M., Kuśnierz, K., Żeber-Lubecka, N., Ambrożkiewicz, F., Mikula, M., Kos-Kudła, B., & Ostrowski, J. (2020). Differences between Well-Differentiated Neuroendocrine Tumors and Ductal Adenocarcinomas of the Pancreas Assessed by Multi-Omics Profiling. International Journal of Molecular Sciences, 21(12), 4470. https://doi.org/10.3390/ijms21124470