Advances in Mast Cell Activation by IL-1 and IL-33 in Sjögren’s Syndrome: Promising Inhibitory Effect of IL-37

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
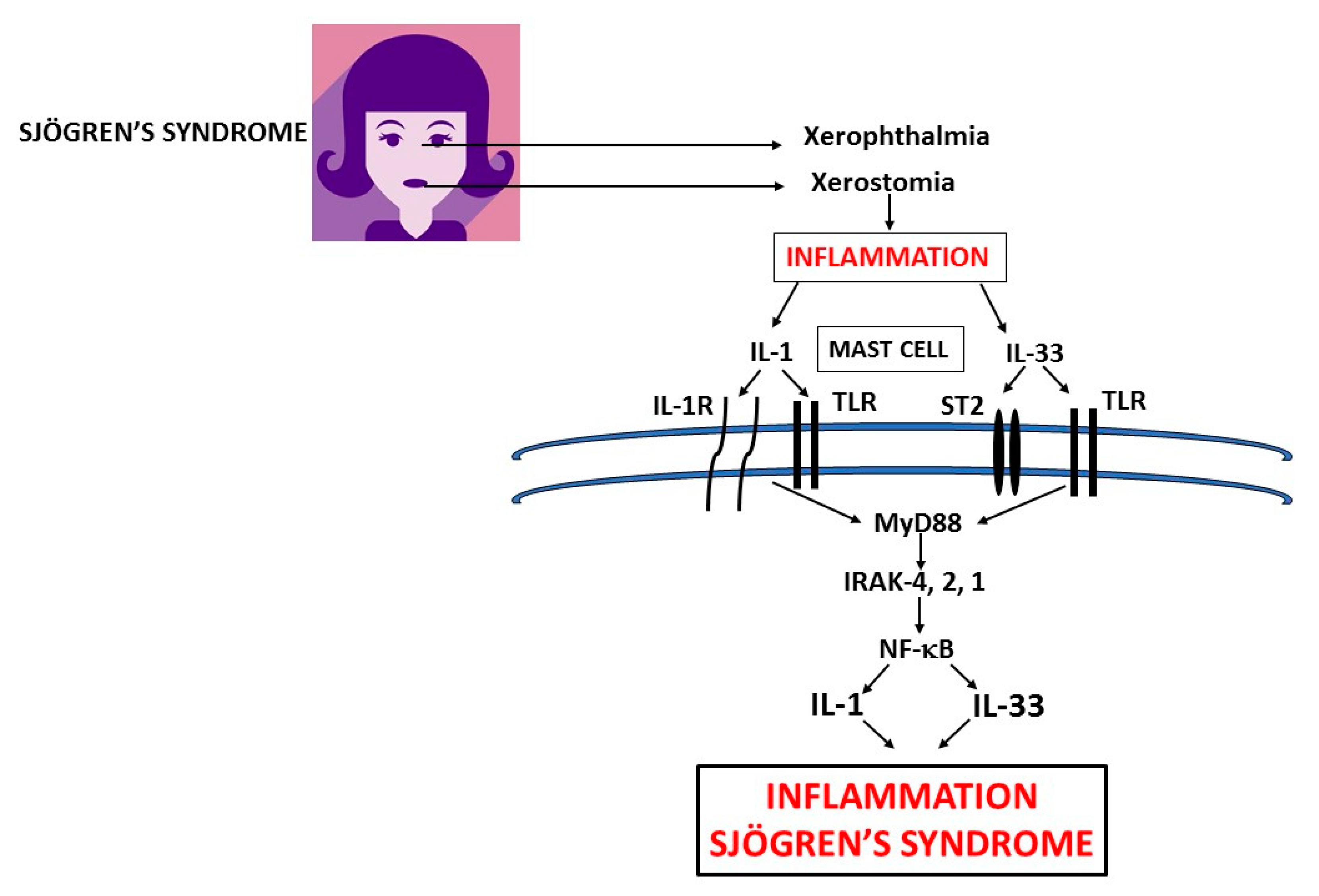
2. Mast Cells (MCs)
3. IL-1
4. IL-33
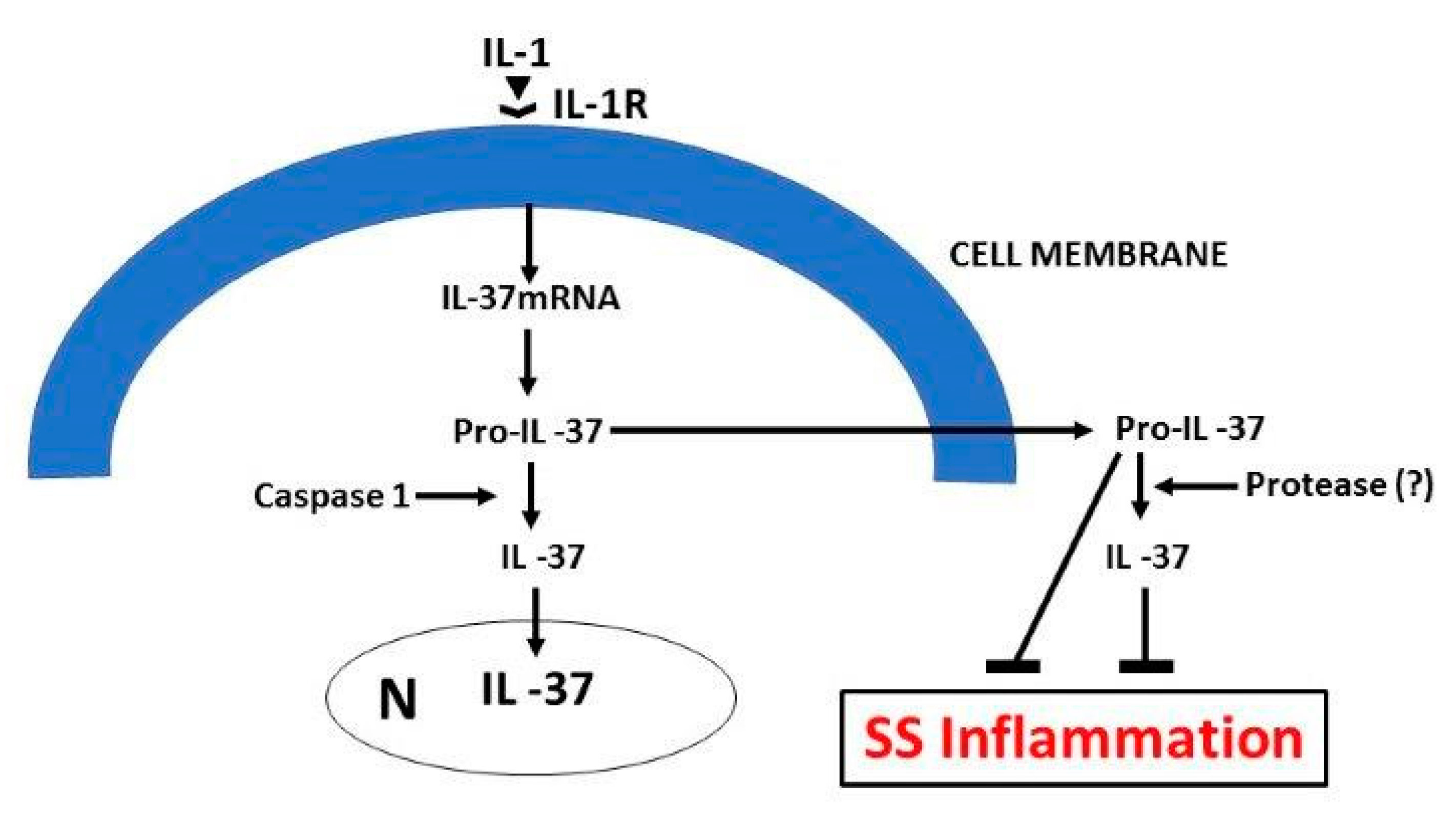
5. IL-37
Funding
Conflicts of Interest
References
- Theoharides, T.C.; Petra, A.I.; Taracanova, A.; Panagiotidou, S.; Conti, P. Targeting IL-33 in Autoimmunity and Inflammation. J. Pharmacol. Exp. Ther. 2015, 354, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Kamradt, T.; Mitchison, N.A. Tolerance and autoimmunity. N Engl. J. Med. 2001, 344, 655–664. [Google Scholar] [CrossRef]
- Peck, A.; Nguyen, C.Q. What can Sjögren’s syndrome-like disease in mice contribute to human Sjögren’s syndrome? Clin. Immunol. 2017, 182, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Fox, R.I.; Chilton, T.; Scott, S.; Benton, L.; Howell, F.V.; Vaughan, J.H. Potential role of Epstein-Barr virus in Sjögren’s syndrome. Rheum. Dis. Clin. North Am. 1987, 13, 275–292. [Google Scholar] [PubMed]
- Kiripolsky, J.; McCabe, L.G.; Kramer, J. Innate immunity in Sjögren’s syndrome. Clin. Immunol. 2017, 182, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Mariette, X.; Criswell, L.A. Primary Sjögren’s Syndrome. N. Engl. J. Med. 2018, 379, 97. [Google Scholar] [CrossRef]
- Moutsopoulos, H.M.; Mann, D.L.; Johnson, A.H.; Chused, T.M. Genetic Differences between Primary and Secondary Sicca Syndrome. N. Engl. J. Med. 1979, 301, 761–763. [Google Scholar] [CrossRef] [PubMed]
- Quartuccio, L.; Baldini, C.; Bartoloni, E.; Priori, R.; Carubbi, F.; Corazza, L.; Alunno, A.; Colafrancesco, S.; Luciano, N.; Giacomelli, R.; et al. Anti-SSA/SSB-negative Sjögren’s syndrome shows a lower prevalence of lymphoproliferative manifestations, and a lower risk of lymphoma evolution. Autoimmun. Rev. 2015, 14, 1019–1022. [Google Scholar] [CrossRef]
- Beckman, K.A.; Luchs, J.; Milner, M.S.; Ambrus, J.L. The Potential Role for Early Biomarker Testing as Part of a Modern, Multidisciplinary Approach to Sjögren’s Syndrome Diagnosis. Adv. Ther. 2017, 34, 799–812. [Google Scholar] [CrossRef] [PubMed]
- Kramer, J. Early events in Sjögren’s Syndrome pathogenesis: The importance of innate immunity in disease initiation. Cytokine 2014, 67, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Chused, T.M.; Kassan, S.S.; Opelz, G.; Moutsopoulos, H.M.; Terasaki, P.I. Sjögren’s syndrome association with HLA-Dw3. N. Engl. J. Med. 1977, 296, 895–897. [Google Scholar] [CrossRef]
- Conti, P.; Younes, A. Coronavirus COV-19/SARS-CoV-2 affects women less than men: Clinical response to viral infection. J. Biol. Regul. Homeost. Agents 2020, 34. [Google Scholar]
- Conti, P.; Gallenga, C.E.; Tetè, G.; Caraffa, A.; Ronconi, G.; Younes, A.; Toniato, E.; Ross, R.; Kritas, S.K. How to reduce the likelihood of coronavirus-19 (CoV-19 or SARS-CoV-2) infection and lung inflammation mediated by IL-1. J. Biol. Regul. Homeost. Agents 2020, 34. [Google Scholar]
- Maitland, N.; Flint, S.; Scully, C.; Crean, S.J. Detection of cytomegalovirus and Epstein-Barr virus in labial salivary glands in Sjogren’s syndrome and non-specific sialadenitis. J. Oral Pathol. Med. 1995, 24, 293–298. [Google Scholar] [CrossRef]
- Psianou, K.; Panagoulias, I.; Papanastasiou, A.D.; de Lastic, A.-L.; Rodi, M.; Spantidea, P.I.; Degn, S.E.; Georgiou, P.; Mouzaki, A. Clinical and immunological parameters of Sjögren’s syndrome. Autoimmun. Rev. 2018, 17, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Cohen, P.L. The T cell in Sjogren’s syndrome: Force majeure, not spectateur. J. Autoimmun. 2012, 39, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Talotta, R.; Sarzi-Puttini, P.; Atzeni, F. Microbial Agents as Putative Inducers of B Cell Lymphoma in Sjögren’s Syndrome through an Impaired Epigenetic Control: The State-of-The-Art. J. Immunol. Res. 2019, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Nocturne, G.; Mariette, X. B cells in the pathogenesis of primary Sjögren syndrome. Nat. Rev. Rheumatol. 2018, 14, 133–145. [Google Scholar] [CrossRef]
- Voulgarelis, M.; Tzioufas, A.G. Pathogenetic mechanisms in the initiation and perpetuation of Sjögren’s syndrome. Nat. Rev. Rheumatol. 2010, 6, 529–537. [Google Scholar] [CrossRef]
- Verstappen, G.M.; Corneth, O.B.; Bootsma, H.; Kroese, F.G.M. Th17 cells in primary Sjögren’s syndrome: Pathogenicity and plasticity. J. Autoimmun. 2018, 87, 16–25. [Google Scholar] [CrossRef]
- Talal, N.; Flescher, E. Rheumatoid arthritis: An editorial perspective based on cytokine imbalance. J. Autoimmun. 1988, 1, 309–317. [Google Scholar] [CrossRef]
- Popov, Y.; Salomon-Escoto, K. Gastrointestinal and Hepatic Disease in Sjogren Syndrome. Rheum. Dis. Clin. North Am. 2018, 44, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, C.; la Barbera, L.; Pizzo, M.L.; Ciccia, F.; Sireci, G.; Guggino, G. Invariant NKT Cells and Rheumatic Disease: Focus on Primary Sjogren Syndrome. Int. J. Mol. Sci. 2019, 20, 5435. [Google Scholar] [CrossRef] [PubMed]
- Atzeni, F.; Doria, A.; Carrabba, M.; Turiel, M.; Sarzi-Puttini, P. Potential target of infliximab in autoimmune and inflammatory diseases. Autoimmun. Rev. 2007, 6, 529–536. [Google Scholar] [CrossRef]
- Hebbar, M.; Lassalle, P.; Janin, A.; Vanhée, D.; Bisiau, S.; Hatron, P.Y.; Tonnel, A.B.; Gosselin, B. E-selectin expression in salivary endothelial cells and sera from patients with systemic sclerosis. Role of resident mast cell-derived tumor necrosis factor alpha. Arthritis Rheum. 1995, 38, 406–412. [Google Scholar] [CrossRef]
- Morita, H.; Arae, K.; Unno, H.; Miyauchi, K.; Toyama, S.; Nambu, A.; Oboki, K.; Ohno, T.; Motomura, K.; Matsuda, A.; et al. An Interleukin-33-Mast Cell-Interleukin-2 Axis Suppresses Papain-Induced Allergic Inflammation by Promoting Regulatory T Cell Numbers. Immunity 2015, 43, 175–186. [Google Scholar] [CrossRef]
- McCarthy, P.C.; Phair, I.R.; Greger, C.; Pardali, K.; McGuire, V.A.; Clark, A.R.; Arthur, J.S. CIL-33 regulates cytokine production and neutrophil recruitment via the p38 MAPK-activated kinases MK2/3. Immunol. Cell Biol. 2019, 97, 54–71. [Google Scholar] [CrossRef]
- Brown, M.A.; Weinberg, R.B. Mast Cells, and Innate Lymphoid Cells: Underappreciated Players in CNS Autoimmune Demyelinating Disease. Front. Immunol. 2018, 9, 514. [Google Scholar] [CrossRef]
- Jung, S.M.; Lee, J.; Baek, S.Y.; Lee, J.H.; Lee, J.; Park, K.S.; Kwok, S.K. The Interleukin 33/ST2 axis in patients with primary Sjögren syndrome: Expression in serum and salivary glands, and the clinical association. J. Rheumatol. 2015, 42, 264–271. [Google Scholar] [CrossRef]
- Ho, L.H.; Ohno, T.; Oboki, K.; Kajiwara, N.; Suto, H.; Iikura, M.; Nakae, S. IL-33 induces IL-13 production by mouse mast cells independently of IgE-FcepsilonRI signals. J. Leukoc. Biol. 2007, 82, 1481–1490. [Google Scholar] [CrossRef]
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The interleukin-1 family: Back to the future. Immunity 2013, 39, 1003–1018. [Google Scholar] [CrossRef]
- Dinarello, C.A.; Kaplanski, G. Indeed, IL-18 is more than an inducer of IFN-γ. J. Leukoc. Boil. 2018, 104, 237–238. [Google Scholar] [CrossRef]
- Boraschi, D.; Dinarello, C.A. IL-18 in autoimmunity: Review. Eur. Cytokine Netw. 2006, 17, 224–252. [Google Scholar]
- Conti, P.; Youinou, P.; Theoharides, T.C. Modulation of autoimmunity by the latest interleukins (with special emphasis on IL-32). Autoimmun. Rev. 2007, 6, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Aota, K.; Kani, K.; Yamanoi, T.; Nakashiro, K.-I.; Ishimaru, N.; Azuma, M. Distinct Regulation of CXCL10 Production by Cytokines in Human Salivary Gland Ductal and Acinar Cells. Inflammation 2018, 41, 1172–1181. [Google Scholar] [CrossRef] [PubMed]
- Margiotta, D.P.; Navarini, L.; Vadacca, M.; lo Vullo, M.; Pignataro, F.; Basta, F.; Afeltra, A. The IL33/ST2 axis in Sjogren syndrome in relation to disease activity. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 1295–1299. [Google Scholar] [PubMed]
- Nagashima, H.; Ogawa, J.; Chida, E.; Yamauchi, K. A case of Sjögren syndrome with nifedipine-induced pneumonia. Nihon Kokyuki Gakkai Zasshi 2009, 47, 476–486. [Google Scholar]
- Vivino, F.B.; Bunya, V.Y.; Massaro-Giordano, G.; Johr, C.R.; Giattino, S.L.; Schorpion, A.; Shafer, B.; Peck, A.; Sivils, K.L.; Rasmussen, A.; et al. Sjogren’s syndrome: An update on disease pathogenesis, clinical manifestations and treatment. Clin. Immunol. 2019, 203, 81–121. [Google Scholar] [CrossRef]
- Cornec, D.; Devauchelle-Pensec, V.; Tobón, G.J.; Pers, J.-O.; Jousse-Joulin, S.; Saraux, A. B cells in Sjögren’s syndrome: From pathophysiology to diagnosis and treatment. J. Autoimmun. 2012, 39, 161–167. [Google Scholar] [CrossRef]
- Hansen, A.; Dörner, T. Current therapeutic options in Sjögren’s syndrome. Z Rheumatol. 2010, 69, 19–24. [Google Scholar] [CrossRef]
- Conti, P.; Ronconi, G.; Caraffa, A.; Gallenga, C.E.; Ross, R.; Frydas, I.; Kritas, S.K. Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by Coronavirus-19 (COVI-19 or SARS-CoV-2): anti-inflammatory strategies. J. Biol. Regul. Homeost. Agents 2020, 34. [Google Scholar] [CrossRef]
- Li, B.; Smith, T.J. Regulation of IL-1 receptor antagonist by TSH in fibrocytes and orbital fibroblasts. J. Clin. Endocrinol. Metab. 2014, 99, E625–E633. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. The IL-1 family of cytokines and receptors in rheumatic diseases. Nat. Rev. Rheumatol. 2019, 15, 612–632. [Google Scholar] [CrossRef]
- Cavalli, G.; Hayashi, M.; Jin, Y.; Yorgov, D.; Santorico, S.A.; Holcomb, C.; Rastrou, M.; Erlich, H.; Tengesdal, I.W.; Dagna, L.; et al. MHC class II super-enhancer increases surface expression of HLA-DR and HLA-DQ and affects cytokine production in autoimmune vitiligo. Proc. Natl. Acad. Sci. USA 2016, 113, 1363–1368. [Google Scholar] [CrossRef]
- Galli, S.J.; Kalesnikoff, J.; Grimbaldeston, M.A.; Piliponsky, A.M.; Williams, C.M.; Tsai, M. Mast cells as “tunable” effector and immunoregulatory cells: Recent advances. Annu. Rev. Immunol. 2005, 23, 749–786. [Google Scholar] [CrossRef]
- Kritas, S.K.; Ronconi, G.; Caraffa, A.; Gallenga, C.E.; Ross, R.; Conti, P. Mast cells contribute to coronavirus-induced inflammation: New anti-inflammatory strategy. J. Biol. Regu.l Homeost. Agents 2020, 34. [Google Scholar]
- Franza, L.; Carusi, V.; Altamura, S.; Caraffa, A.; E Gallenga, C.; Kritas, S.K.; Ronconi, G.; Conti, P.; Pandolfi, F. Interrelationship between inflammatory cytokines (IL-1, IL-6, IL-33, IL-37) and acquired immunity. J. Biol. Regul. Homeost. Agents 2019, 33, 1321–1326. [Google Scholar] [PubMed]
- Mukai, K.; Tsai, M.; Starkl, P.; Marichal, T.; Galli, S.J. IgE and mast cells in host defense against parasites and venoms. Semin. Immunopathol. 2016, 38, 581–603. [Google Scholar] [CrossRef]
- Galli, S.J.; Maurer, M.; Lantz, C.S. Mast cells as sentinels of innate immunity. Curr. Opin. Immunol. 1999, 11, 53–59. [Google Scholar] [CrossRef]
- Caraffa, A.; Gallenga, C.E.; Kritas, S.K.; Ronconi, G.; Conti, P. Impact of mast cells in systemic lupus erythematosus: Can inflammation be inhibited? J. Biol. Regul. Homeost. Agents 2019, 33, 669–673. [Google Scholar]
- Nakae, S.; Suto, H.; Berry, G.J.; Galli, S.J. Mast cell–derived TNF can promote Th17 cell–dependent neutrophil recruitment in ovalbumin-challenged OTII mice. Blood 2006, 109, 3640–3648. [Google Scholar] [CrossRef] [PubMed]
- Conti, P.; Lessiani, G.; Kritas, S.K.; Ronconi, G.; Caraffa, A.L.; Theoharides, T.C. Mast cells emerge as mediators of atherosclerosis: Special emphasis on IL-37 inhibition. Tissue Cell 2017, 49, 393–400. [Google Scholar] [CrossRef]
- Conti, P.; Lauritano, D.; Caraffa, A.; Gallenga, C.E.; Kritas, S.K.; Ronconi, G.; Martinotti, S. Microglia and mast cells generate proinflammatory cytokines in the brain and worsen inflammatory state: Suppressor effect of IL-37. Eur. J. Pharmacol. 2020, 875, 173035. [Google Scholar] [CrossRef] [PubMed]
- Galli, S.J.; Nakae, S.; Tsai, M. Mast cells in the development of adaptive immune responses. Nat. Immunol. 2005, 6, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.-L.; Gao, J.-M.; Li, P.-P.; Wang, X. IL-9 Contributes to Immunosuppression Mediated by Regulatory T Cells and Mast Cells in B-Cell Non-Hodgkin’s Lymphoma. J. Clin. Immunol. 2011, 31, 1084–1094. [Google Scholar] [CrossRef] [PubMed]
- Chavarría, A.; Alcocer-Varela, J. Is damage in central nervous system due to inflammation? Autoimmun. Rev. 2004, 3, 251–260. [Google Scholar] [CrossRef]
- Mukai, K.; Tsai, M.; Saito, H.; Galli, S.J. Mast cells as sources of cytokines, chemokines, and growth factors. Immunol. Rev. 2018, 282, 121–150. [Google Scholar] [CrossRef]
- Brown, M.A.; Hural, J. Functions of IL-4 and Control of Its Expression. Crit. Rev. Immunol. 2017, 37, 181–212. [Google Scholar] [CrossRef]
- Svenningsen, S.; Nair, P. Asthma Endotypes and an Overview of Targeted Therapy for Asthma. Front. Med. 2017, 4, 158. [Google Scholar] [CrossRef]
- Liu, X.; Xiao, Y.; Pan, Y.; He, L.; Zheng, S.G.; Wenru, S. The role of the IL-33/ST2 axis in autoimmune disorders: Friend or foe? Cytokine Growth Factor Rev. 2019, 50, 60–74. [Google Scholar] [CrossRef]
- Lauritano, D.; Ronconi, G.; Caraffa, A.; Gallenga, C.E.; Kritas, S.K.; di Emidio, P.; Martinotti, S.; Tetè, G.; Ross, R.; Conti, P. New aspect of allergic contact dermatitis, an inflammatory skin disorder mediated by mast cells: Can IL-38 help? Med. Hypotheses 2020, 139, 109687. [Google Scholar] [CrossRef] [PubMed]
- Shaik, Y.; Caraffa, A.; Ronconi, G.; Lessiani, G.; Conti, P. Impact of polyphenols on mast cells with special emphasis on the effect of quercetin and luteolin. Central Eur. J. Immunol. 2018, 43, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.I.; Lee, K.H.; Joo, Y.H.; Lee, J.M.; Jeon, J.; Jung, H.J.; Shin, M.; Cho, S.; Kim, T.H.; Park, S.; et al. Inflammasomes and autoimmune and rheumatic diseases: A comprehensive review. J. Autoimmun. 2019, 103, 102299. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Zhang, X.; Wang, M.; Chen, Z.; Yan, Y.; Gu, W.; Tan, J.; Jiang, W.; Ji, W. Exosomes from Thymic Stromal Lymphopoietin-Activated Dendritic Cells Promote Th2 Differentiation through the OX40 Ligand. Pathobiology 2018, 86, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Andonegui, G.; Wong, C.H.; Kubes, P. Role of endothelial TLR4 for neutrophil recruitment into central nervous system microvessels in systemic inflammation. J. Immunol. 2009, 183, 5244–5250. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A.; Conti, P.; Mier, J.W. Effects of human interleukin-1 on natural killer cell activity: Is fever a host defense mechanism for tumor killing? Yale J. Boil. Med. 1986, 59, 97–106. [Google Scholar]
- Madonna, S.; Girolomoni, G.; Dinarello, C.A.; Albanesi, C. The Significance of IL-36 Hyperactivation and IL-36R Targeting in Psoriasis. Int. J. Mol. Sci. 2019, 20, 3318. [Google Scholar] [CrossRef]
- Cavalli, G.; Dinarello, C.A. Suppression of inflammation and acquired immunity by IL-37. Immunol. Rev. 2017, 281, 179–190. [Google Scholar] [CrossRef]
- Levandowski, C.B.; Mailloux, C.M.; Ferrara, T.M.; Gowan, K.; Ben, S.; Jin, Y.; McFann, K.K.; Holland, P.J.; Fain, P.R.; Dinarello, C.A.; et al. NLRP1 haplotypes associated with vitiligo and autoimmunity increase interleukin-1β processing via the NLRP1 inflammasome. Proc. Natl. Acad. Sci. USA 2013, 110, 2952–2956. [Google Scholar] [CrossRef]
- Fox, R.I.; Kang, H.I.; Ando, D.; Abrams, J.; Pisa, E. Cytokine mRNA expression in salivary gland biopsies of Sjögren’s syndrome. J. Immunol. 1994, 152, 5532–5539. [Google Scholar]
- Moutsopoulos, H.M. Sjögren’s syndrome: Autoimmune epithelitis. Clin. Immunol. Immunopathol. 1994, 72, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Killedar, S.Y.; Eckenrode, S.E.; McIndoe, R.; She, J.-X.; Nguyen, C.Q.; Peck, A.B.; Cha, S. Early pathogenic events associated with Sjögren’s syndrome (SjS)-like disease of the nod mouse using microarray analysis. Lab. Investig. 2006, 86, 1243–1260. [Google Scholar] [CrossRef] [PubMed]
- Taracanova, A.; Alevizos, M.; Karagkouni, A.; Weng, Z.; Norwitz, E.; Conti, P.; Leeman, S.E.; Theoharides, T.C. SP and IL-33 together markedly enhance TNF synthesis and secretion from human mast cells mediated by the interaction of their receptors. Proc. Natl. Acad. Sci. USA 2017, 114, E4002–E4009. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Zhang, B.; Kempuraj, D.; Tagen, M.; Vasiadi, M.; Angelidou, A.; Alysandratos, K.-D.; Kalogeromitros, D.; Asadi, S.; Stavrianeas, N.; et al. IL-33 augments substance P-induced VEGF secretion from human mast cells and is increased in psoriatic skin. Proc. Natl. Acad. Sci. USA 2010, 107, 4448–4453. [Google Scholar] [CrossRef] [PubMed]
- Awada, A.; Nicaise, C.; Ena, S.; Schandéné, L.; Rasschaert, J.; Popescu, I.; Gangji, V.; Soyfoo, M.S. Potential involvement of the IL-33–ST2 axis in the pathogenesis of primary Sjögren’s syndrome. Ann. Rheum. Dis. 2014, 73, 1259–1263. [Google Scholar] [CrossRef] [PubMed]
- Kandere-Grzybowska, K.; Letourneau, R.; Kempuraj, D.; Donelan, J.; Poplawski, S.; Boucher, W.; Athanassiou, A.; Theoharides, T.C. IL-1 induces vesicular secretion of IL-6 without degranulation from human mast cells. J. Immunol. 2003, 171, 4830–4836. [Google Scholar] [CrossRef] [PubMed]
- Conti, P.; Kempuraj, D. Important role of mast cells in multiple sclerosis. Mult. Scler. Relat. Disord. 2016, 5, 77–80. [Google Scholar] [CrossRef]
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2017, 281, 8–27. [Google Scholar] [CrossRef]
- Conti, P.; Caraffa, A.; Mastrangelo, F.; Tettamanti, L.; Ronconi, G.; Frydas, I.; Kritas, S.K.; Theoharides, T.C. Critical role of inflammatory mast cell in fibrosis: Potential therapeutic effect of IL-37. Cell Prolif. 2018, 51, e12475. [Google Scholar] [CrossRef]
- Conti, P.; Ronconi, G.; Kritas, S.K.; Caraffa, A.; Theoharides, T.C. Activated Mast Cells Mediate Low-Grade Inflammation in Type 2 Diabetes: Interleukin-37 Could Be Beneficial. Can. J. Diabetes 2018, 42, 568–573. [Google Scholar] [CrossRef]
- Conti, P.; Caraffa, A.; Ronconi, G.; Kritas, S.K.; Mastrangelo, F.; Tettamanti, L.; Frydas, I.; Theoharides, T.C. Mast cells participate in allograft rejection: Can IL-37 play an inhibitory role? Inflamm. Res. 2018, 67, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Jiang, H.-R.; Kewin, P.; Li, Y.; Mu, R.; Fraser, A.R.; Pitman, N.; Kurowska-Stolarska, M.; McKenzie, A.N.J.; McInnes, I.; et al. IL-33 exacerbates antigen-induced arthritis by activating mast cells. Proc. Natl. Acad. Sci. USA 2008, 105, 10913–10918. [Google Scholar] [CrossRef] [PubMed]
- Rider, P.; Voronov, E.; Dinarello, C.A.; Apte, R.N.; Cohen, I. Alarmins: Feel the Stress. J. Immunol. 2017, 198, 1395–1402. [Google Scholar] [CrossRef] [PubMed]
- Martin, N.T.; Martin, M.U. Interleukin 33 is a guardian of barriers and a local alarmin. Nat. Immunol. 2016, 17, 122–131. [Google Scholar] [CrossRef]
- Eisenmesser, E.Z.; Gottschlich, A.; Redzic, J.S.; Paukovich, N.; Nix, J.C.; Azam, T.; Zhang, L.; Zhao, R.; Kieft, J.S.; The, E.; et al. Interleukin-37 monomer is the active form for reducing innate immunity. Proc. Natl. Acad. Sci. USA 2019, 116, 5514–5522. [Google Scholar] [CrossRef]
- Gallenga, C.E.; Pandolfi, F.; Caraffa, A.; Kritas, S.K.; Ronconi, G.; Toniato, E.; Martinotti, S.; Conti, P. Interleukin-1 family cytokines and mast cells: Activation and inhibition. J. Boil. Regul. Homeost. Agents 2019, 33, 1–6. [Google Scholar]
- Ronconi, G.; Teté, G.; Kritas, S.K.; Gallenga, C.E.; Caraffa, A.l.; Ross, R.; Conti, P. COVID-19 induced by SARS-CoV-2 causes Kawasaki-like disease in children: role of pro-inflammatory and anti-inflammatory cytokines. J. Biol. Regul. Homeost. Agents. 2020, 3. [Google Scholar] [CrossRef]
- Dinarello, C.A. Setting the cytokine trap for autoimmunity. Nat. Med. 2003, 9, 20–22. [Google Scholar] [CrossRef]
- Gugliandolo, A.; Caraffa, A.L.; Gallenga, C.E.; Kritas, S.K.; Ronconi, G.; Trubiani, O.; Conti, P.; di Emidio, P.; Mazzon, E. Mesenchymal stem cells and IL-37: A powerful combination. J. Biol. Regul. Homeost. Agents 2019, 33, 1019–1022. [Google Scholar]
- Caraffa, A.; Gallenga, C.E.; Kritas, S.K.; Ronconi, G.; di Emidio, P.; Conti, P. CAR-T cell therapy causes inflammation by IL-1 which activates inflammatory cytokine mast cells: Anti-inflammatory role of IL-37. J. Biol. Regul. Homeost. Agents 2020, 33, 1981–1985. [Google Scholar]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conti, P.; Stellin, L.; Caraffa, A.; Gallenga, C.E.; Ross, R.; Kritas, S.K.; Frydas, I.; Younes, A.; Di Emidio, P.; Ronconi, G. Advances in Mast Cell Activation by IL-1 and IL-33 in Sjögren’s Syndrome: Promising Inhibitory Effect of IL-37. Int. J. Mol. Sci. 2020, 21, 4297. https://doi.org/10.3390/ijms21124297
Conti P, Stellin L, Caraffa A, Gallenga CE, Ross R, Kritas SK, Frydas I, Younes A, Di Emidio P, Ronconi G. Advances in Mast Cell Activation by IL-1 and IL-33 in Sjögren’s Syndrome: Promising Inhibitory Effect of IL-37. International Journal of Molecular Sciences. 2020; 21(12):4297. https://doi.org/10.3390/ijms21124297
Chicago/Turabian StyleConti, Pio, Luisa Stellin, Alesssandro Caraffa, Carla E. Gallenga, Rhiannon Ross, Spyros K. Kritas, Ilias Frydas, Ali Younes, Paolo Di Emidio, and Gianpaolo Ronconi. 2020. "Advances in Mast Cell Activation by IL-1 and IL-33 in Sjögren’s Syndrome: Promising Inhibitory Effect of IL-37" International Journal of Molecular Sciences 21, no. 12: 4297. https://doi.org/10.3390/ijms21124297
APA StyleConti, P., Stellin, L., Caraffa, A., Gallenga, C. E., Ross, R., Kritas, S. K., Frydas, I., Younes, A., Di Emidio, P., & Ronconi, G. (2020). Advances in Mast Cell Activation by IL-1 and IL-33 in Sjögren’s Syndrome: Promising Inhibitory Effect of IL-37. International Journal of Molecular Sciences, 21(12), 4297. https://doi.org/10.3390/ijms21124297