Physiological Disturbance in Fatty Liver Energy Metabolism Converges on IGFBP2 Abundance and Regulation in Mice and Men

 , , , and
, , , and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
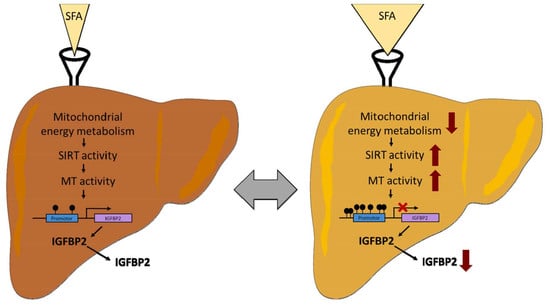
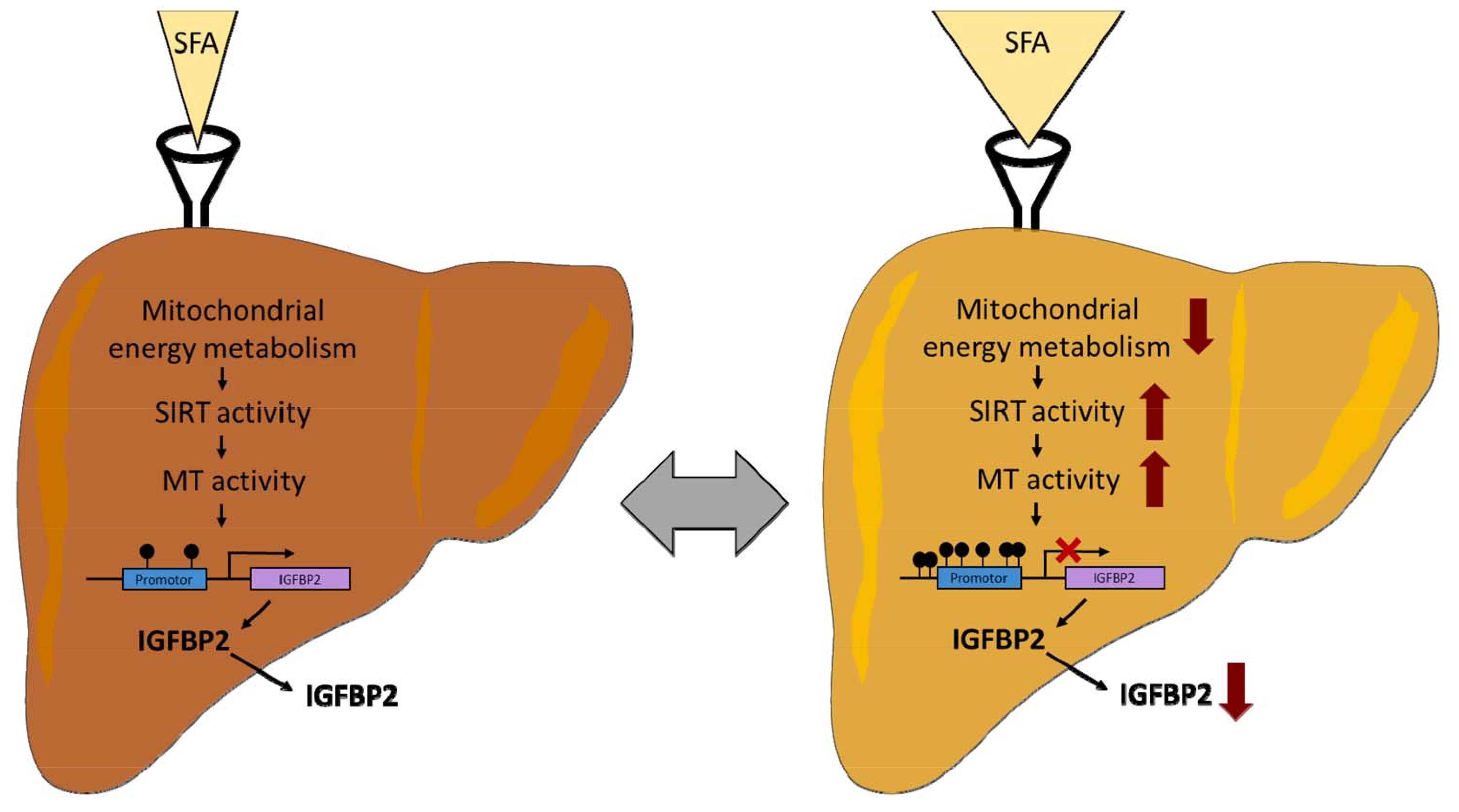
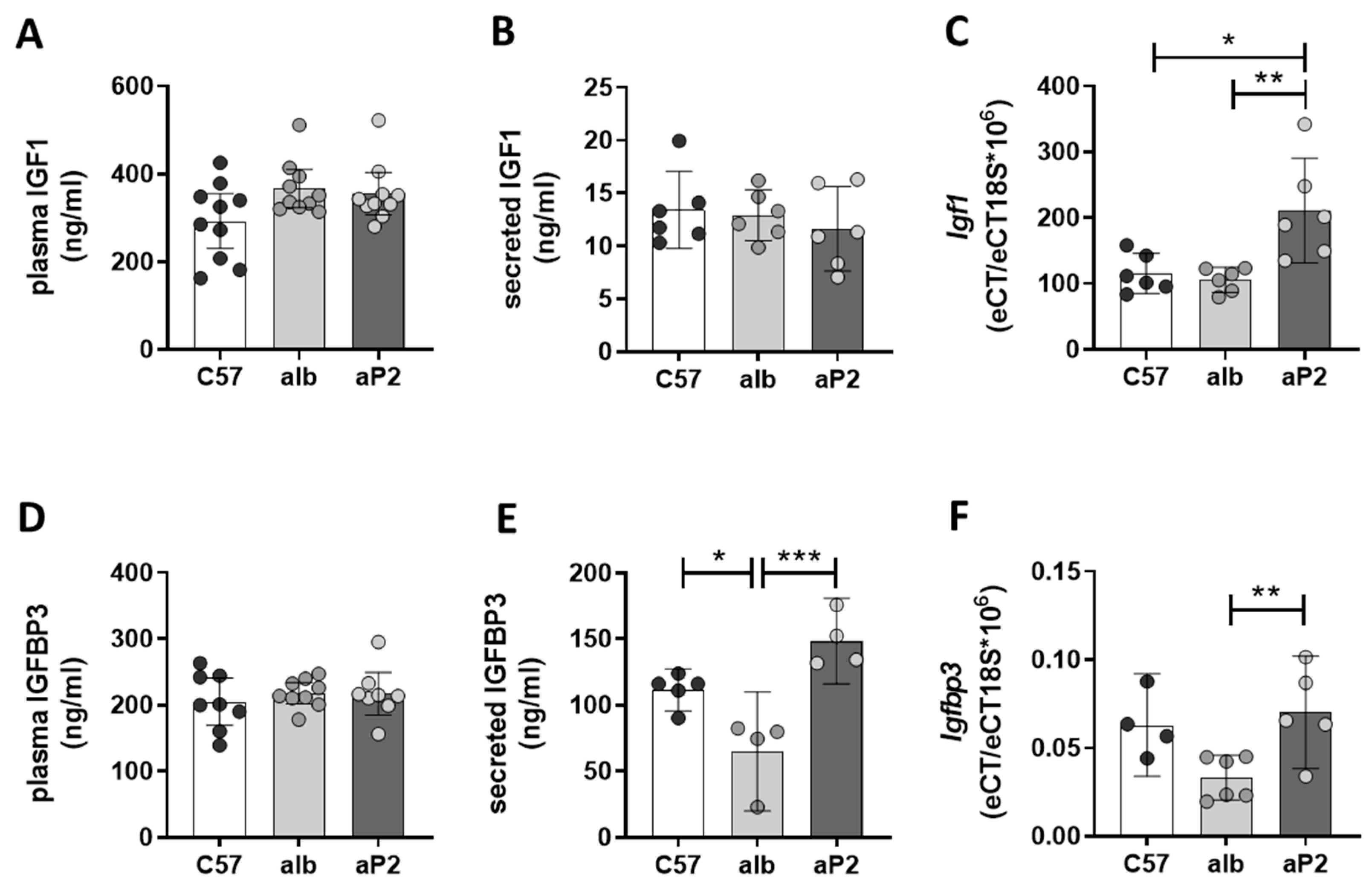
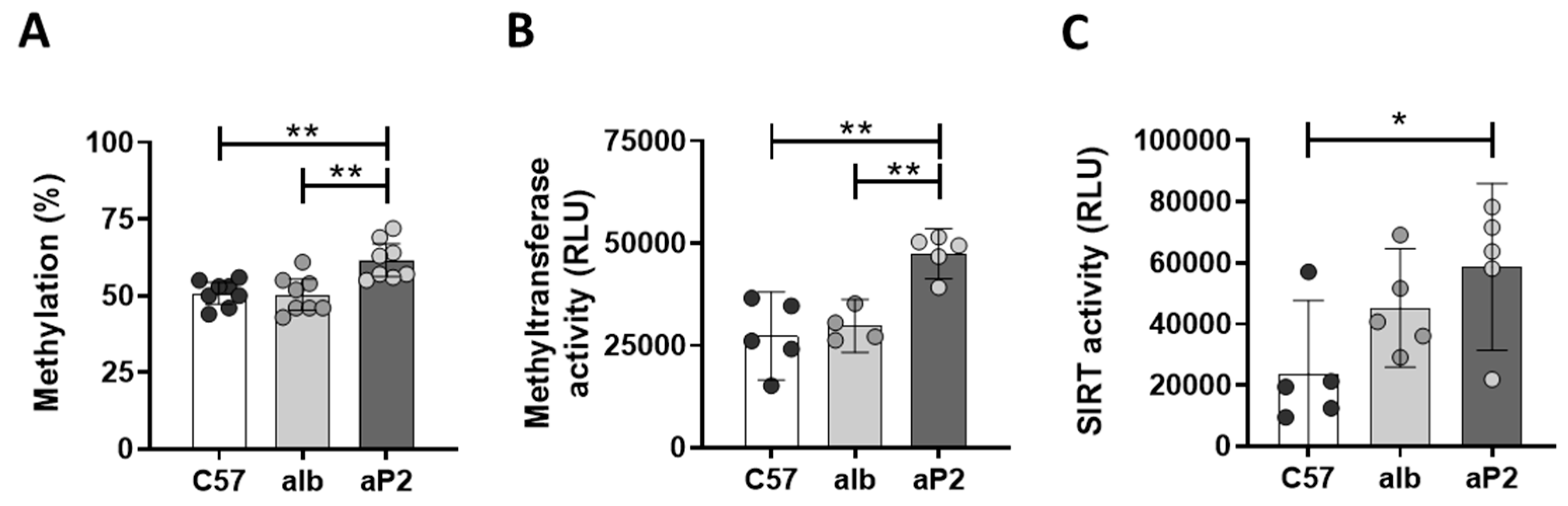
2.1. Analysis of Regulator Networks in Fatty Liver Pathology
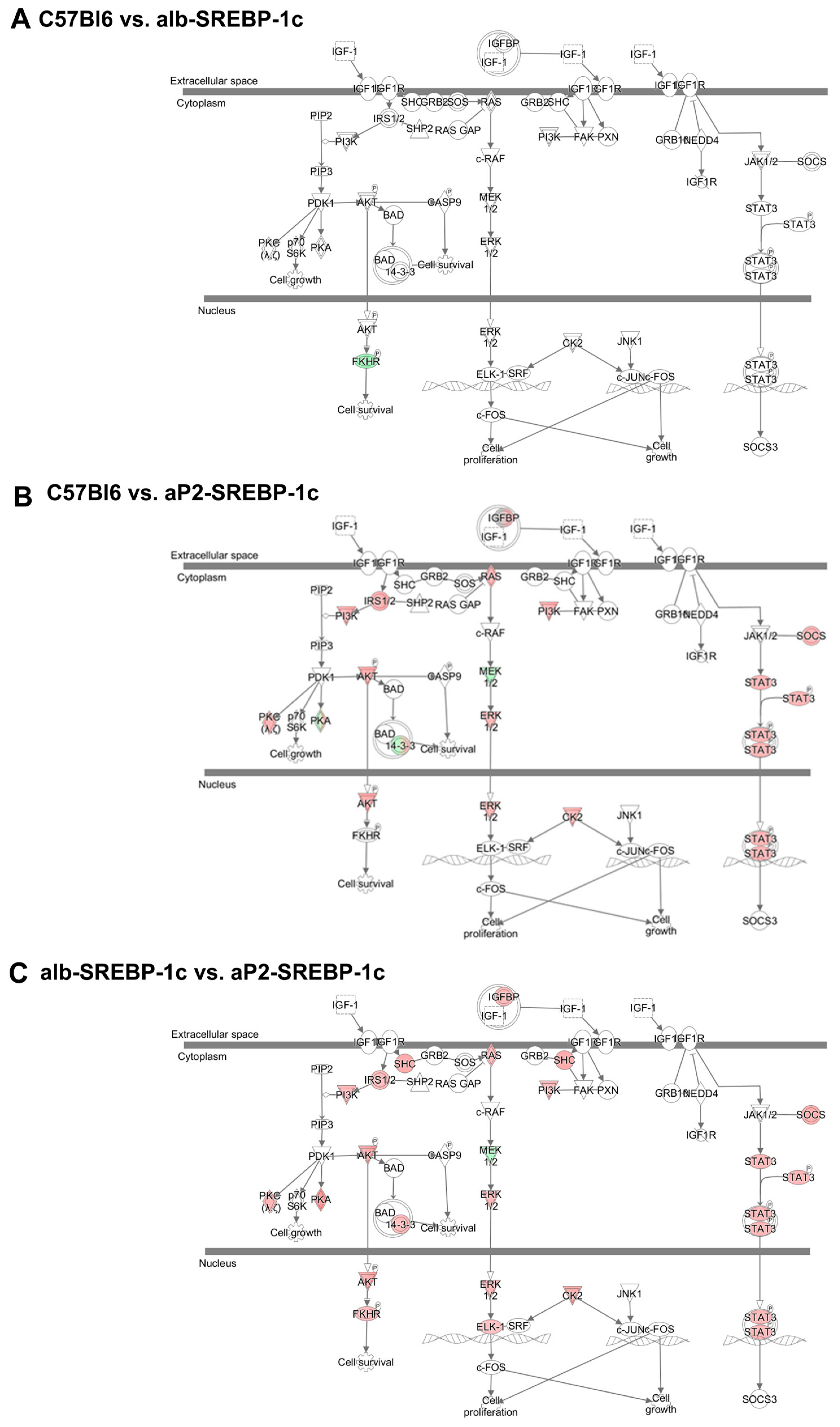
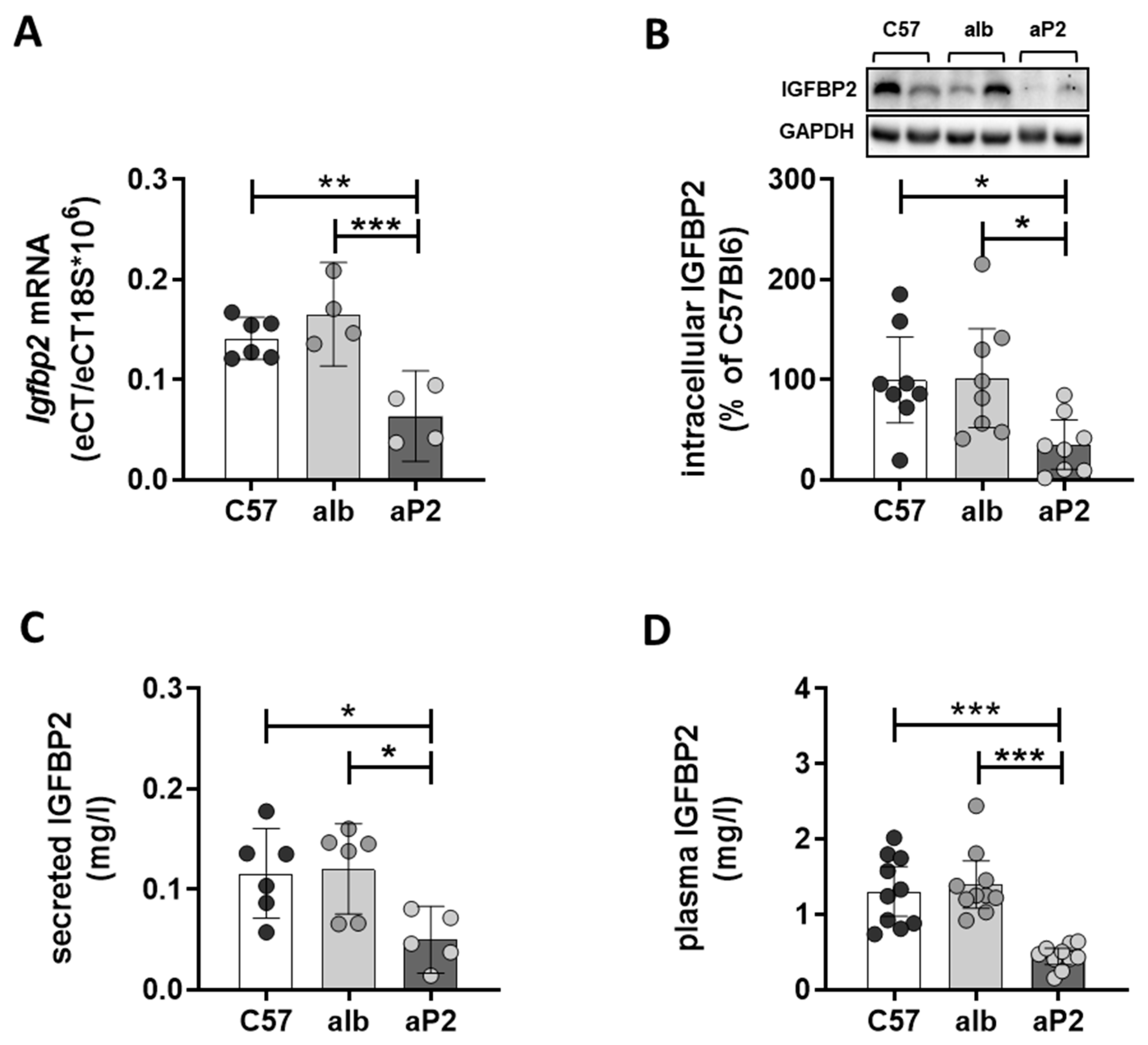
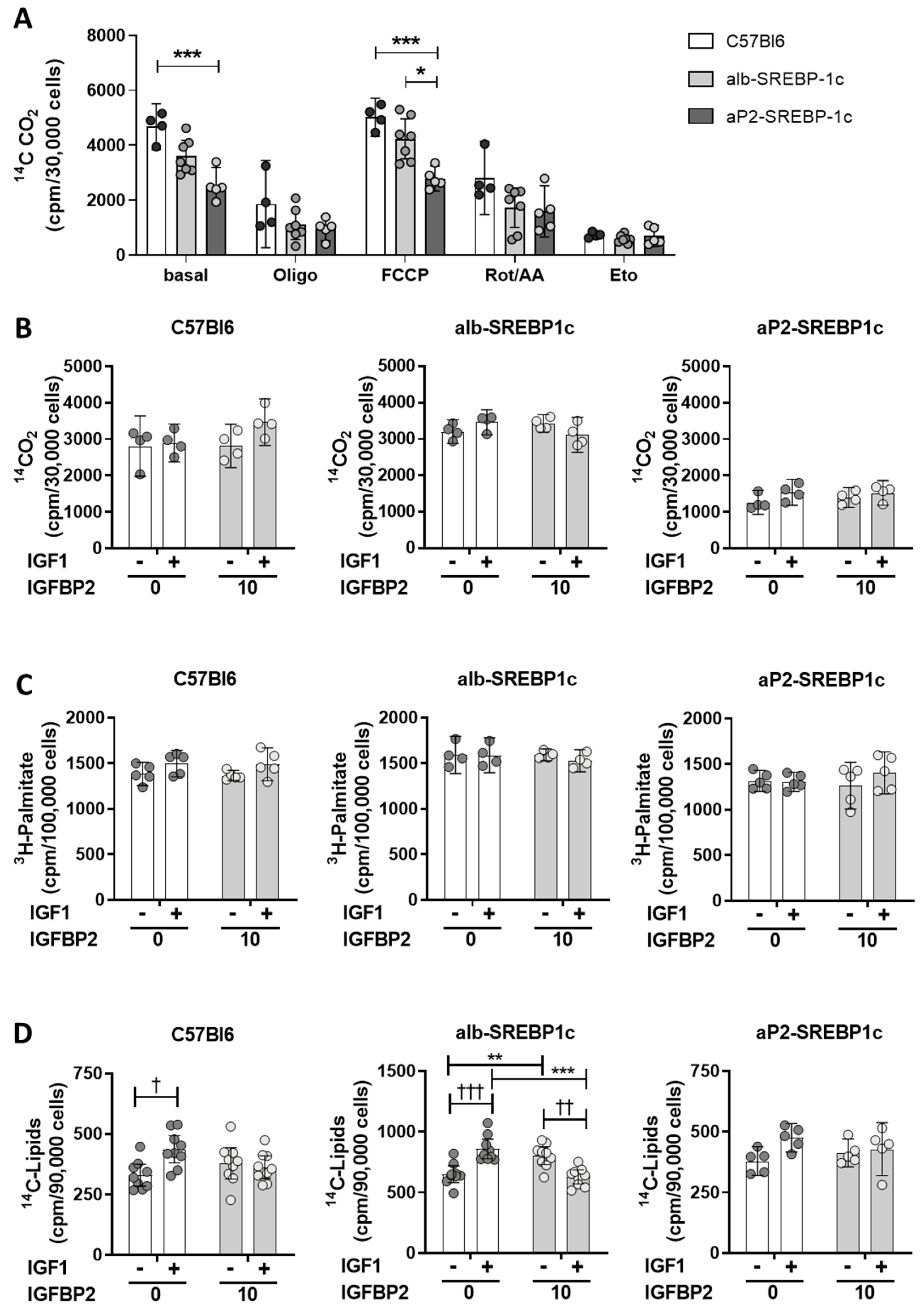
2.2. IGFBP2 Blunts Hepatic IGF1 Action
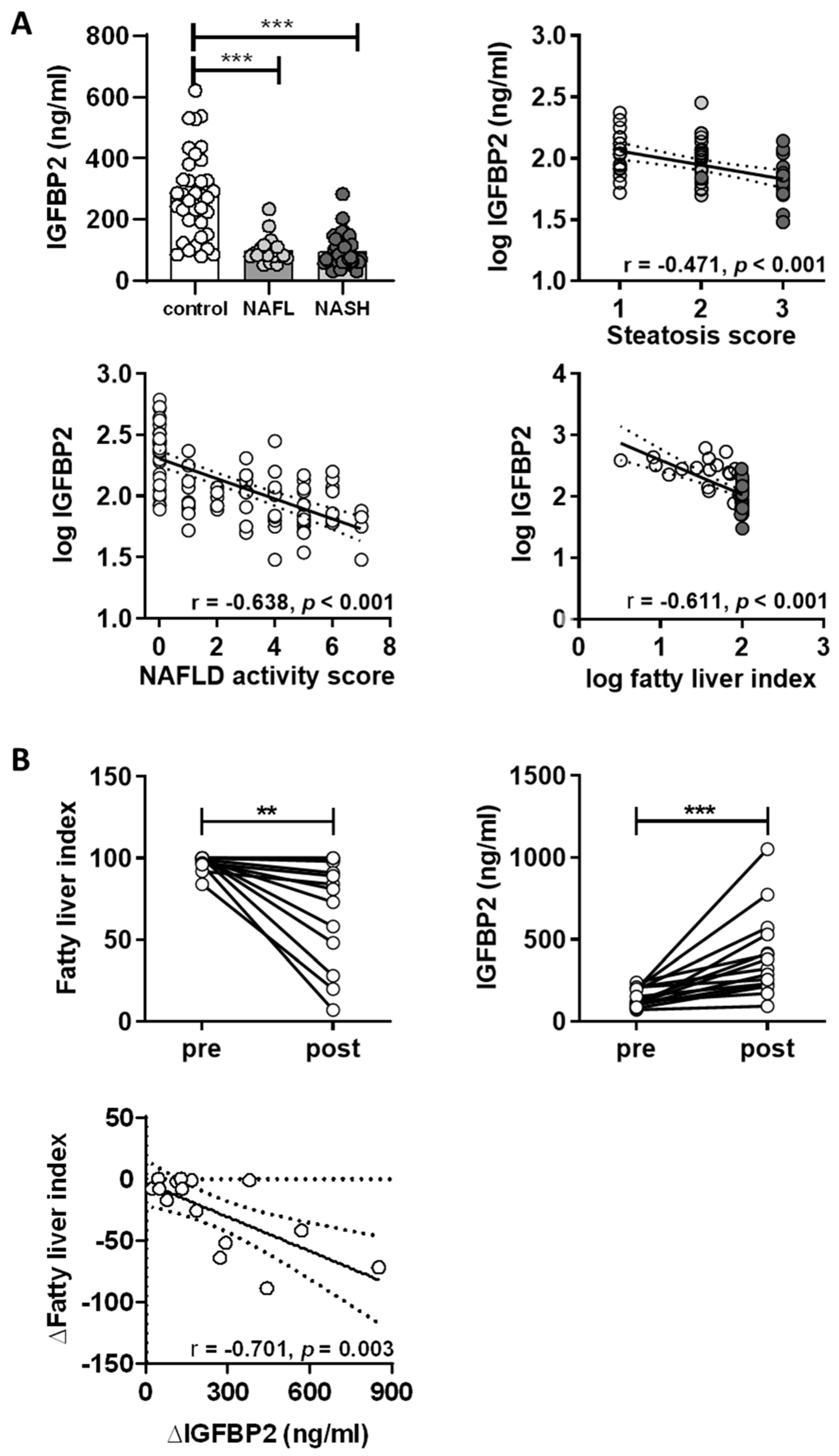
2.3. IGFBP2 Levels in Humans with Non-Alcoholic Fatty Liver Disease
2.4. Impact of Weight Loss Intervention on Serum IGFBP2 Levels
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Transcriptome Analyses of Liver Tissue
4.3. Protein Analysis
4.4. Gene Expression Analysis
4.5. Methylation Analysis
4.6. Lipid Metabolism in Hepatocytes
4.7. Study Cohorts
4.8. Analysis of Clinical Samples
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| IGF | insulin-like growth factor |
| IGFBP | insulin-like growth factor binding protein |
| NAFLD | non-alcoholic fatty liver disease |
| NAFL | non-alcoholic fatty liver |
| NASH | non-alcoholic steatohepatitis |
| SREBP-1c | sterol regulatory element-binding protein 1c |
| ALT | alanine aminotransferase |
| AST | aspartate aminotransferase |
| DNL | de novo lipogenesis |
| NAS | NAFLD activity score |
| BMI | body mass index |
| FLI | fatty liver index |
| HOMA-IR | homeostasis model assessment of insulin resistance |
| γGT | gamma-glutamyl transferase |
| SIRT | sirtuin |
References
- Dietrich, P.; Hellerbrand, C. Non-alcoholic fatty liver disease, obesity and the metabolic syndrome. Best Pr. Res. Clin. Gastroenterol. 2014, 28, 637–653. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef]
- Samuel, V.T.; Shulman, G.I. The pathogenesis of insulin resistance: Integrating signaling pathways and substrate flux. J. Clin. Investig. 2016, 126, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Saponaro, C.; Gaggini, M.; Carli, F.; Gastaldelli, A. The Subtle Balance between Lipolysis and Lipogenesis: A Critical Point in Metabolic Homeostasis. Nutrients 2015, 7, 9453–9474. [Google Scholar] [CrossRef] [PubMed]
- Boren, J.; Taskinen, M.R.; Olofsson, S.O.; Levin, M. Ectopic lipid storage and insulin resistance: A harmful relationship. J. Intern. Med. 2013, 274, 25–40. [Google Scholar] [CrossRef]
- Cusi, K. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: Pathophysiology and clinical implications. Gastroenterology 2012, 142, 711–725.e6. [Google Scholar] [CrossRef]
- Ahlqvist, E.; Storm, P.; Karajamaki, A.; Martinell, M.; Dorkhan, M.; Carlsson, A.; Vikman, P.; Prasad, R.B.; Aly, D.M.; Almgren, P.; et al. Novel subgroups of adult-onset diabetes and their association with outcomes: A data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol. 2018, 6, 361–369. [Google Scholar] [CrossRef]
- Zaharia, O.P.; Strassburger, K.; Strom, A.; Bonhof, G.J.; Karusheva, Y.; Antoniou, S.; Bodis, K.; Markgraf, D.F.; Burkart, V.; Mussig, K.; et al. Risk of diabetes-associated diseases in subgroups of patients with recent-onset diabetes: A 5-year follow-up study. Lancet Diabetes Endocrinol. 2019, 7, 684–694. [Google Scholar] [CrossRef]
- Hooper, A.J.; Adams, L.A.; Burnett, J.R. Genetic determinants of hepatic steatosis in man. J. Lipid Res. 2011, 52, 593–617. [Google Scholar] [CrossRef]
- Jelenik, T.; Kaul, K.; Sequaris, G.; Flogel, U.; Phielix, E.; Kotzka, J.; Knebel, B.; Fahlbusch, P.; Horbelt, T.; Lehr, S.; et al. Mechanisms of Insulin Resistance in Primary and Secondary Nonalcoholic Fatty Liver. Diabetes 2017, 66, 2241–2253. [Google Scholar] [CrossRef]
- Knebel, B.; Haas, J.; Hartwig, S.; Jacob, S.; Kollmer, C.; Nitzgen, U.; Muller-Wieland, D.; Kotzka, J. Liver-specific expression of transcriptionally active SREBP-1c is associated with fatty liver and increased visceral fat mass. PLoS ONE 2012, 7, e31812. [Google Scholar] [CrossRef]
- Shimano, H.; Sato, R. SREBP-regulated lipid metabolism: Convergent physiology—divergent pathophysiology. Nat. Rev. Endocrinol. 2017, 13, 710–730. [Google Scholar] [CrossRef]
- Younossi, Z.M. Non-alcoholic fatty liver disease—A global public health perspective. J. Hepatol. 2019, 70, 531–544. [Google Scholar] [CrossRef]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the Complete Program of Cholesterol and Fatty Acid Synthesis in the Liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Bengoechea-Alonso, M.T.; Ericsson, J. SREBP in Signal Transduction: Cholesterol Metabolism and Beyond. Curr. Opin. Cell Biol. 2007, 19, 215–222. [Google Scholar] [CrossRef]
- Knebel, B.; Lehr, S.; Hartwig, S.; Haas, J.; Kaber, G.; Dicken, H.-D.; Susanto, F.; Bohne, L.; Jacob, S.; Nitzgen, U.; et al. Phosphorylation of Sterol Regulatory Element-Binding Protein (SREBP)-1c by p38 Kinases, ERK and JNK Influences Lipid Metabolism and the Secretome of Human Liver Cell Line HepG2. Arch. Physiol. Biochem. 2014, 120, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, I.; Hammer, R.E.; Richardson, J.A.; Ikemoto, S.; Bashmakov, Y.; Goldstein, J.L.; Brown, M.S. Insulin resistance and diabetes mellitus in transgenic mice expressing nuclear SREBP-1c in adipose tissue: Model for congenital generalized lipodystrophy. Genes Dev. 1998, 12, 3182–3194. [Google Scholar] [CrossRef]
- Adamek, A.; Kasprzak, A. Insulin-Like Growth Factor (IGF) System in Liver Diseases. Int. J. Mol. Sci. 2018, 19, 1308. [Google Scholar] [CrossRef] [PubMed]
- Ahrens, M.; Ammerpohl, O.; von Schonfels, W.; Kolarova, J.; Bens, S.; Itzel, T.; Teufel, A.; Herrmann, A.; Brosch, M.; Hinrichsen, H.; et al. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell Metab. 2013, 18, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Kammel, A.; Saussenthaler, S.; Jahnert, M.; Jonas, W.; Stirm, L.; Hoeflich, A.; Staiger, H.; Fritsche, A.; Haring, H.U.; Joost, H.G.; et al. Early hypermethylation of hepatic Igfbp2 results in its reduced expression preceding fatty liver in mice. Hum. Mol. Genet. 2016, 25, 2588–2599. [Google Scholar] [PubMed]
- Li, X. SIRT1 and energy metabolism. Acta Biochim. Biophys. Sin. (Shanghai) 2013, 45, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Bekaert, M.; Ouwens, D.M.; Horbelt, T.; Van de Velde, F.; Fahlbusch, P.; Herzfeld de Wiza, D.; Van Nieuwenhove, Y.; Calders, P.; Praet, M.; Hoorens, A.; et al. Reduced expression of chemerin in visceral adipose tissue associates with hepatic steatosis in patients with obesity. Obesity 2016, 24, 2544–2552. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Bekaert, M.; Van Nieuwenhove, Y.; Calders, P.; Cuvelier, C.A.; Batens, A.H.; Kaufman, J.M.; Ouwens, D.M.; Ruige, J.B. Determinants of testosterone levels in human male obesity. Endocrine 2015, 50, 202–211. [Google Scholar] [CrossRef]
- Wheatcroft, S.B.; Kearney, M.T.; Shah, A.M.; Ezzat, V.A.; Miell, J.R.; Modo, M.; Williams, S.C.; Cawthorn, W.P.; Medina-Gomez, G.; Vidal-Puig, A.; et al. IGF-binding protein-2 protects against the development of obesity and insulin resistance. Diabetes 2007, 56, 285–294. [Google Scholar] [CrossRef]
- Hedbacker, K.; Birsoy, K.; Wysocki, R.W.; Asilmaz, E.; Ahima, R.S.; Farooqi, I.S.; Friedman, J.M. Antidiabetic effects of IGFBP2, a leptin-regulated gene. Cell Metab. 2010, 11, 11–22. [Google Scholar] [CrossRef]
- Frystyk, J.; Skjaerbaek, C.; Vestbo, E.; Fisker, S.; Orskov, H. Circulating levels of free insulin-like growth factors in obese subjects: The impact of type 2 diabetes. Diabetes Metab. Res. Rev. 1999, 15, 314–322. [Google Scholar] [CrossRef]
- Heald, A.H.; Kaushal, K.; Siddals, K.W.; Rudenski, A.S.; Anderson, S.G.; Gibson, J.M. Insulin-like growth factor binding protein-2 (IGFBP-2) is a marker for the metabolic syndrome. Exp. Clin. Endocrinol. Diabetes 2006, 114, 371–376. [Google Scholar] [CrossRef]
- van den Beld, A.W.; Blum, W.F.; Brugts, M.P.; Janssen, J.A.; Grobbee, D.E.; Lamberts, S.W. High IGFBP2 levels are not only associated with a better metabolic risk profile but also with increased mortality in elderly men. Eur. J. Endocrinol. 2012, 167, 111–117. [Google Scholar] [CrossRef]
- van den Beld, A.W.; Carlson, O.D.; Doyle, M.E.; Rizopoulos, D.; Ferrucci, L.; van der Lely, A.J.; Egan, J.M. IGFBP-2 and aging: A 20-year longitudinal study on IGFBP-2, IGF-I, BMI, insulin sensitivity and mortality in an aging population. Eur. J. Endocrinol. 2019, 180, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Fazeli, P.K.; Lawson, E.A.; Prabhakaran, R.; Miller, K.K.; Donoho, D.A.; Clemmons, D.R.; Herzog, D.B.; Misra, M.; Klibanski, A. Effects of recombinant human growth hormone in anorexia nervosa: A randomized, placebo-controlled study. J. Clin. Endocrinol. Metab. 2010, 95, 4889–4897. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.C. Sirtuin biology and relevance to diabetes treatment. Diabetes Manag. (Lond.) 2012, 2, 243–257. [Google Scholar] [CrossRef]
- Lomb, D.J.; Laurent, G.; Haigis, M.C. Sirtuins regulate key aspects of lipid metabolism. Biochim. Biophys. Acta 2010, 1804, 1652–1657. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Canto, C.; Wanders, R.J.; Auwerx, J. The secret life of NAD+: An old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 2010, 31, 194–223. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Sinclair, D.A. Mammalian sirtuins: Biological insights and disease relevance. Annu. Rev. Pathol. 2010, 5, 253–295. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Auwerx, J. Exploring the therapeutic space around NAD+. J. Cell Biol. 2012, 199, 205–209. [Google Scholar] [CrossRef]
- Peng, L.; Yuan, Z.; Ling, H.; Fukasawa, K.; Robertson, K.; Olashaw, N.; Koomen, J.; Chen, J.; Lane, W.S.; Seto, E. SIRT1 deacetylates the DNA methyltransferase 1 (DNMT1) protein and alters its activities. Mol. Cell Biol. 2011, 31, 4720–4734. [Google Scholar] [CrossRef]
- Drew, J.E.; Farquharson, A.J.; Horgan, G.W.; Williams, L.M. Tissue-specific regulation of sirtuin and nicotinamide adenine dinucleotide biosynthetic pathways identified in C57Bl/6 mice in response to high-fat feeding. J. Nutr. Biochem. 2016, 37, 20–29. [Google Scholar] [CrossRef]
- Wittenbecher, C.; Ouni, M.; Kuxhaus, O.; Jahnert, M.; Gottmann, P.; Teichmann, A.; Meidtner, K.; Kriebel, J.; Grallert, H.; Pischon, T.; et al. Insulin-Like Growth Factor Binding Protein 2 (IGFBP-2) and the Risk of Developing Type 2 Diabetes. Diabetes 2019, 68, 188–197. [Google Scholar] [CrossRef]
- Han, J.; Kaufman, R.J. The role of ER stress in lipid metabolism and lipotoxicity. J. Lipid Res. 2016, 57, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Knebel, B.; Hartwig, S.; Jacob, S.; Kettel, U.; Schiller, M.; Passlack, W.; Koellmer, C.; Lehr, S.; Muller-Wieland, D.; Kotzka, J. Inactivation of SREBP-1a Phosphorylation Prevents Fatty Liver Disease in Mice: Identification of Related Signaling Pathways by Gene Expression Profiles in Liver and Proteomes of Peroxisomes. Int. J. Mol. Sci. 2018, 19, 980. [Google Scholar] [CrossRef] [PubMed]
- Knebel, B.; Fahlbusch, P.; Dille, M.; Wahlers, N.; Hartwig, S.; Jacob, S.; Kettel, U.; Schiller, M.; Herebian, D.; Koellmer, C.; et al. Fatty Liver Due to Increased de novo Lipogenesis: Alterations in the Hepatic Peroxisomal Proteome. Front. Cell Dev. Biol. 2019, 7, 248. [Google Scholar] [CrossRef] [PubMed]
- Horbelt, T.; Knebel, B.; Fahlbusch, P.; Barbosa, D.; de Wiza, D.H.; Van de Velde, F.; Van Nieuwenhove, Y.; Lapauw, B.; Thoresen, G.H.; Al-Hasani, H.; et al. The adipokine sFRP4 induces insulin resistance and lipogenesis in the liver. Biochim. Biophys. Acta 2019, 1865, 2671–2684. [Google Scholar] [CrossRef]
- ACE/ADA Task Force on Inpatient Diabetes. American College of Endocrinology and American Diabetes Association Consensus statement on inpatient diabetes and glycemic control. Diabetes Care 2006, 29, 1955–1962. [Google Scholar] [CrossRef]
- Bedogni, G.; Bellentani, S.; Miglioli, L.; Masutti, F.; Passalacqua, M.; Castiglione, A.; Tiribelli, C. The Fatty Liver Index: A simple and accurate predictor of hepatic steatosis in the general population. BMC Gastroenterol. 2006, 6, 33. [Google Scholar] [CrossRef]
- Matthews, D.R.; Hosker, J.P.; Rudenski, A.S.; Naylor, B.A.; Treacher, D.F.; Turner, R.C. Homeostasis model assessment: Insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985, 28, 412–419. [Google Scholar] [CrossRef]
- Bedossa, P.; Poitou, C.; Veyrie, N.; Bouillot, J.L.; Basdevant, A.; Paradis, V.; Tordjman, J.; Clement, K. Histopathological algorithm and scoring system for evaluation of liver lesions in morbidly obese patients. Hepatology 2012, 56, 1751–1759. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fahlbusch, P.; Knebel, B.; Hörbelt, T.; Barbosa, D.M.; Nikolic, A.; Jacob, S.; Al-Hasani, H.; Van de Velde, F.; Van Nieuwenhove, Y.; Müller-Wieland, D.; et al. Physiological Disturbance in Fatty Liver Energy Metabolism Converges on IGFBP2 Abundance and Regulation in Mice and Men. Int. J. Mol. Sci. 2020, 21, 4144. https://doi.org/10.3390/ijms21114144
Fahlbusch P, Knebel B, Hörbelt T, Barbosa DM, Nikolic A, Jacob S, Al-Hasani H, Van de Velde F, Van Nieuwenhove Y, Müller-Wieland D, et al. Physiological Disturbance in Fatty Liver Energy Metabolism Converges on IGFBP2 Abundance and Regulation in Mice and Men. International Journal of Molecular Sciences. 2020; 21(11):4144. https://doi.org/10.3390/ijms21114144
Chicago/Turabian StyleFahlbusch, Pia, Birgit Knebel, Tina Hörbelt, David Monteiro Barbosa, Aleksandra Nikolic, Sylvia Jacob, Hadi Al-Hasani, Frederique Van de Velde, Yves Van Nieuwenhove, Dirk Müller-Wieland, and et al. 2020. "Physiological Disturbance in Fatty Liver Energy Metabolism Converges on IGFBP2 Abundance and Regulation in Mice and Men" International Journal of Molecular Sciences 21, no. 11: 4144. https://doi.org/10.3390/ijms21114144
APA StyleFahlbusch, P., Knebel, B., Hörbelt, T., Barbosa, D. M., Nikolic, A., Jacob, S., Al-Hasani, H., Van de Velde, F., Van Nieuwenhove, Y., Müller-Wieland, D., Lapauw, B., Ouwens, D. M., & Kotzka, J. (2020). Physiological Disturbance in Fatty Liver Energy Metabolism Converges on IGFBP2 Abundance and Regulation in Mice and Men. International Journal of Molecular Sciences, 21(11), 4144. https://doi.org/10.3390/ijms21114144