Global View of Candidate Therapeutic Target Genes in Hormone-Responsive Breast Cancer

 , ,
, ,  , ,
, ,  and
and
Abstract
1. Introduction
2. Estrogen Signaling and Endocrine Resistance in Breast Cancer
3. Mechanisms of Endocrine Resistance in Breast Cancer
4. Dropout Screening Approaches to Dissect Gene Vulnerabilities
5. Gene Essentiality in Estrogen Receptor-Positive Breast Cancers
6. Functional Pathways Involving Estrogen Receptor-Positive Essential Genes
7. Interaction Proteomics as Tool for Estrogen Signaling Protein Network Dissection
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AIs | Aromatase Inhibitors |
| BC | Breast Cancer |
| BFs | Bayesian Factors |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeats |
| DIA | Data-Independent Acquisition |
| DSB | Double-Strand Break |
| ERα+ BC | Estrogen Receptor alpha positive breast cancer |
| ERα− BC | Estrogen Receptor alpha negative breast cancer |
| ERE | Estrogen Response Element |
| FDR | False Discovery Rate |
| ICI | Fulvestrant, ICI 182780 |
| HR | Homologous Recombination |
| RIME | Rapid Immunoprecipitation Mass spectrometry of Endogenous protein |
| RNAi | RNA interference |
| SERDs | Selective Estrogen Receptor Downregulators |
| SERMs | Selective Estrogen Receptor Modulators |
| sgRNA | Single guide RNA |
| TAP | Tandem Affinity Purification |
| tracrRNA | Trans-activating crispr RNA |
References
- Dai, X.; Li, T.; Bai, Z.; Yang, Y.; Liu, X.; Zhan, J.; Shi, B. Breast cancer intrinsic subtype classification, clinical use and future trends. Am. J. Cancer Res. 2015, 5, 2929. [Google Scholar] [PubMed]
- Selli, C.; Dixon, J.M.; Sims, A.H. Accurate prediction of response to endocrine therapy in breast cancer patients: Current and future biomarkers. Breast Cancer Res. 2016, 18, 118. [Google Scholar] [CrossRef] [PubMed]
- Rani, A.; Stebbing, J.; Giamas, G.; Murphy, J. Endocrine resistance in hormone receptor positive breast cancer–from mechanism to therapy. Front. Endocrinol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- AlFakeeh, A.; Brezden-Masley, C. Overcoming endocrine resistance in hormone receptor-positive breast cancer. Curr. Oncol. 2018, 25, S18–S27. [Google Scholar] [CrossRef]
- Unniyampurath, U.; Pilankatta, R.; Krishnan, M.N. RNA Interference in the Age of CRISPR: Will CRISPR Interfere with RNAi? Int. J. Mol. Sci. 2016, 17, 291. [Google Scholar] [CrossRef]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef]
- Behan, F.M.; Iorio, F.; Picco, G.; Goncalves, E.; Beaver, C.M.; Migliardi, G.; Santos, R.; Rao, Y.; Sassi, F.; Pinnelli, M.; et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature 2019, 568, 511–516. [Google Scholar] [CrossRef]
- Dempster, J.M.; Pacini, C.; Pantel, S.; Behan, F.M.; Green, T.; Krill-Burger, J.; Beaver, C.M.; Younger, S.T.; Zhivich, V.; Najgebauer, H.; et al. Agreement between two large pan-cancer CRISPR-Cas9 gene dependency data sets. Nat. Commun. 2019, 10, 5817. [Google Scholar] [CrossRef]
- Aguirre, A.J.; Meyers, R.M.; Weir, B.A.; Vazquez, F.; Zhang, C.Z.; Ben-David, U.; Cook, A.; Ha, G.; Harrington, W.F.; Doshi, M.B.; et al. Genomic Copy Number Dictates a Gene-Independent Cell Response to CRISPR/Cas9 Targeting. Cancer Discov. 2016, 6, 914–929. [Google Scholar] [CrossRef]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576. [Google Scholar] [CrossRef]
- Nagarajan, S.; Rao, S.V.; Sutton, J.; Cheeseman, D.; Dunn, S.; Papachristou, E.K.; Prada, J.G.; Couturier, D.L.; Kumar, S.; Kishore, K.; et al. ARID1A influences HDAC1/BRD4 activity, intrinsic proliferative capacity and breast cancer treatment response. Nat. Genet. 2020, 52, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Li, W.; Wang, X.; Xu, H.; Yang, J.; Wu, Q.; Huang, Y.; Geradts, J.; Jiang, P.; Fei, T.; et al. Estrogen-regulated feedback loop limits the efficacy of estrogen receptor-targeted breast cancer therapy. Proc. Natl. Acad. Sci. USA 2018, 115, 7869–7878. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Chen, A.; Bai, Z. Integrative investigation on breast cancer in ER, PR and HER2-defined subgroups using mRNA and miRNA expression profiling. Sci. Rep. 2014, 4, 6566. [Google Scholar] [CrossRef] [PubMed]
- Kos, M.; Reid, G.; Denger, S.; Gannon, F. Minireview: Genomic organization of the human ERalpha gene promoter region. Mol. Endocrinol. 2001, 15, 2057–2063. [Google Scholar] [CrossRef] [PubMed]
- Ponglikitmongkol, M.; Green, S.; Chambon, P. Genomic organization of the human oestrogen receptor gene. EMBO J. 1988, 7, 3385–3388. [Google Scholar] [CrossRef]
- Pfeffer, U.; Fecarotta, E.; Arena, G.; Forlani, A.; Vidali, G. Alternative splicing of the estrogen receptor primary transcript normally occurs in estrogen receptor positive tissues and cell lines. J. Steroid Biochem. Mol. Biol. 1996, 56, 99–105. [Google Scholar] [CrossRef]
- Flouriot, G.; Brand, H.; Denger, S.; Metivier, R.; Kos, M.; Reid, G.; Sonntag-Buck, V.; Gannon, F. Identification of a new isoform of the human estrogen receptor-alpha (hER-alpha) that is encoded by distinct transcripts and that is able to repress hER-alpha activation function 1. EMBO J. 2000, 19, 4688–4700. [Google Scholar] [CrossRef]
- Omarjee, S.; Jacquemetton, J.; Poulard, C.; Rochel, N.; Dejaegere, A.; Chebaro, Y.; Treilleux, I.; Marangoni, E.; Corbo, L.; Romancer, M.L. The molecular mechanisms underlying the ERalpha-36-mediated signaling in breast cancer. Oncogene 2017, 36, 2503–2514. [Google Scholar] [CrossRef]
- Klinge, C.M. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001, 29, 2905–2919. [Google Scholar] [CrossRef]
- Nilsson, S.; Makela, S.; Treuter, E.; Tujague, M.; Thomsen, J.; Andersson, G.; Enmark, E.; Pettersson, K.; Warner, M.; Gustafsson, J.A. Mechanisms of estrogen action. Physiol. Rev. 2001, 81, 1535–1565. [Google Scholar] [CrossRef]
- Stossi, F.; Madak-Erdogan, Z.; Katzenellenbogen, B.S. Estrogen receptor alpha represses transcription of early target genes via p300 and CtBP1. Mol. Cell. Biol. 2009, 29, 1749–1759. [Google Scholar] [CrossRef][Green Version]
- Casale, F.P.; Giurato, G.; Nassa, G.; Armond, J.W.; Oates, C.J.; Cora, D.; Gamba, A.; Mukherjee, S.; Weisz, A.; Nicodemi, M. Single-cell states in the estrogen response of breast cancer cell lines. PLoS ONE 2014, 9, e88485. [Google Scholar] [CrossRef] [PubMed]
- Weisz, A.; Rosales, R. Identification of an estrogen response element upstream of the human c-fos gene that binds the estrogen receptor and the AP-1 transcription factor. Nucleic Acids Res. 1990, 18, 5097–5106. [Google Scholar] [CrossRef] [PubMed]
- Merenbakh-Lamin, K.; Ben-Baruch, N.; Yeheskel, A.; Dvir, A.; Soussan-Gutman, L.; Jeselsohn, R.; Yelensky, R.; Brown, M.; Miller, V.A.; Sarid, D.; et al. D538G mutation in estrogen receptor-alpha: A novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res. 2013, 73, 6856–6864. [Google Scholar] [CrossRef] [PubMed]
- Shiau, A.K.; Barstad, D.; Loria, P.M.; Cheng, L.; Kushner, P.J.; Agard, D.A.; Greene, G.L. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 1998, 95, 927–937. [Google Scholar] [CrossRef]
- Skafar, D.F. Formation of a powerful capping motif corresponding to start of“helix 12” in agonist-bound estrogen receptor-alpha contributes to increased constitutive activity of the protein. Cell Biochem. Biophys. 2000, 33, 53–62. [Google Scholar] [CrossRef]
- Toy, W.; Weir, H.; Razavi, P.; Lawson, M.; Goeppert, A.U.; Mazzola, A.M.; Smith, A.; Wilson, J.; Morrow, C.; Wong, W.L.; et al. Activating ESR1 Mutations Differentially Affect the Efficacy of ER Antagonists. Cancer Discov. 2017, 7, 277–287. [Google Scholar] [CrossRef]
- Lei, J.T.; Gou, X.; Seker, S.; Ellis, M.J. ESR1 alterations and metastasis in estrogen receptor positive breast cancer. J. Cancer Metastasis Treat 2019, 5. [Google Scholar] [CrossRef]
- Jeselsohn, R.; Buchwalter, G.; De Angelis, C.; Brown, M.; Schiff, R. ESR1 mutations—A mechanism for acquired endocrine resistance in breast cancer. Nat. Rev. Clin. Oncol. 2015, 12, 573–583. [Google Scholar] [CrossRef]
- Jeselsohn, R. Are We Ready to Use ESR1 Mutations in Clinical Practice? Breast Care 2017, 12, 309–313. [Google Scholar] [CrossRef]
- Wardell, S.E.; Ellis, M.J.; Alley, H.M.; Eisele, K.; VanArsdale, T.; Dann, S.G.; Arndt, K.T.; Primeau, T.; Griffin, E.; Shao, J.; et al. Efficacy of SERD/SERM Hybrid-CDK4/6 Inhibitor Combinations in Models of Endocrine Therapy-Resistant Breast Cancer. Clin. Cancer Res. 2015, 21, 5121–5130. [Google Scholar] [CrossRef] [PubMed]
- Bezerra, L.S.; Santos-Veloso, M.A.O.; Bezerra Junior, N.D.S.; Fonseca, L.C.D.; Sales, W.L.A. Impacts of Cytochrome P450 2D6 (CYP2D6) Genetic Polymorphism in Tamoxifen Therapy for Breast Cancer. Rev. Bras. Ginecol. Obstet. 2018, 40, 794–799. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Tyson, J.J.; Dixon, J.M. Endocrine resistance in breast cancer—An overview and update. Mol. Cell. Endocrinol. 2015, 418 Pt 3, 220–234. [Google Scholar] [CrossRef]
- Xu, G.; Chhangawala, S.; Cocco, E.; Razavi, P.; Cai, Y.; Otto, J.E.; Ferrando, L.; Selenica, P.; Ladewig, E.; Chan, C.; et al. ARID1A determines luminal identity and therapeutic response in estrogen-receptor-positive breast cancer. Nat. Genet. 2020, 52, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Mohr, S.E.; Smith, J.A.; Shamu, C.E.; Neumuller, R.A.; Perrimon, N. RNAi screening comes of age: Improved techniques and complementary approaches. Nat. Rev. Mol. Cell. Biol. 2014, 15, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.C.; Doudna, J.A. Molecular mechanisms of RNA interference. Annu. Rev. Biophys. 2013, 42, 217–239. [Google Scholar] [CrossRef]
- Bibikova, M.; Golic, M.; Golic, K.G.; Carroll, D. Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases. Genetics 2002, 161, 1169–1175. [Google Scholar]
- Zhang, M.; Wang, F.; Li, S.; Wang, Y.; Bai, Y.; Xu, X. TALE: A tale of genome editing. Prog. Biophys. Mol. Biol. 2014, 114, 25–32. [Google Scholar] [CrossRef]
- Wiedenheft, B.; Sternberg, S.H.; Doudna, J.A. RNA-guided genetic silencing systems in bacteria and archaea. Nature 2012, 482, 331–338. [Google Scholar] [CrossRef]
- Haft, D.H.; Selengut, J.; Mongodin, E.F.; Nelson, K.E. A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comput. Biol. 2005, 1, e60. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Charpentier, E.; Richter, H.; van der Oost, J.; White, M.F. Biogenesis pathways of RNA guides in archaeal and bacterial CRISPR-Cas adaptive immunity. FEMS Microbiol. Rev. 2015, 39, 428–441. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Cao, Z.; Liu, Z.; He, Y.; Wang, Y.; Yuan, P.; Li, W.; Tian, F.; Bao, Y.; Wei, W. Guide RNAs with embedded barcodes boost CRISPR-pooled screens. Genome Biol. 2019, 20, 20. [Google Scholar] [CrossRef]
- Lino, C.A.; Harper, J.C.; Carney, J.P.; Timlin, J.A. Delivering CRISPR: A review of the challenges and approaches. Drug Deliv. 2018, 25, 1234–1257. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, G.; Amiji, M. Use of CRISPR/Cas9 gene-editing tools for developing models in drug discovery. Drug Discov. Today 2018, 23, 519–533. [Google Scholar] [CrossRef] [PubMed]
- Manghwar, H.; Li, B.; Ding, X.; Hussain, A.; Lindsey, K.; Zhang, X.; Jin, S. CRISPR/Cas Systems in Genome Editing: Methodologies and Tools for sgRNA Design, Off-Target Evaluation, and Strategies to Mitigate Off-Target Effects. Adv. Sci. 2020, 7, 1902312. [Google Scholar] [CrossRef]
- Cameron, P.; Settle, A.; Fuller, C.; Thompson, M.; Cigan, A.; Young, J. SITE-Seq: A genome-wide method to measure Cas9 cleavage. Protoc. Exch. 2017. [Google Scholar] [CrossRef]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef]
- Leach, A.R.; Hann, M.M. The in silico world of virtual libraries. Drug Discov. Today 2000, 5, 326–336. [Google Scholar] [CrossRef]
- Jones, L.H.; Bunnage, M.E. Applications of chemogenomic library screening in drug discovery. Nat. Rev. Drug Discov. 2017, 16, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Hong, A.L.; Tseng, Y.Y.; Cowley, G.S.; Jonas, O.; Cheah, J.H.; Kynnap, B.D.; Doshi, M.B.; Oh, C.; Meyer, S.C.; Church, A.J.; et al. Integrated genetic and pharmacologic interrogation of rare cancers. Nat. Commun. 2016, 7, 11987. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Köster, J.; Xu, H.; Chen, C.H.; Xiao, T.; Liu, J.S.; Brown, M.; Liu, X.S. Quality control, modeling, and visualization of CRISPR screens with MAGeCK-VISPR. Genome Biol. 2015, 16, 281. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Xiao, T.; Fei, T.; Shirley Liu, X.; Li, W. Reducing False Positives in CRISPR/Cas9 Screens from Copy Number Variations. BioRxiv 2018. [Google Scholar] [CrossRef]
- Daley, T.P.; Lin, Z.; Lin, X.; Liu, Y.; Wong, W.H.; Qi, L.S. CRISPhieRmix: A hierarchical mixture model for CRISPR pooled screens. Genome Biol. 2018, 19, 159. [Google Scholar] [CrossRef] [PubMed]
- Iorio, F.; Behan, F.M.; Gonçalves, E.; Bhosle, S.G.; Chen, E.; Shepherd, R.; Beaver, C.; Ansari, R.; Pooley, R.; Wilkinson, P.; et al. Unsupervised correction of gene-independent cell responses to CRISPR-Cas9 targeting. BMC Genom. 2018, 19, 604. [Google Scholar] [CrossRef]
- Gonçalves, E.; Behan, F.M.; Louzada, S.; Arnol, D.; Stronach, E.A.; Yang, F.; Yusa, K.; Stegle, O.; Iorio, F.; Garnett, M.J. Structural rearrangements generate cell-specific, gene-independent CRISPR-Cas9 loss of fitness effects. Genome Biol. 2019, 20, 27. [Google Scholar] [CrossRef]
- Hart, T.; Moffat, J. BAGEL: A computational framework for identifying essential genes from pooled library screens. BMC Bioinform. 2016, 17, 164. [Google Scholar] [CrossRef]
- Diaz, A.A.; Qin, H.; Ramalho-Santos, M.; Song, J.S. HiTSelect: A comprehensive tool for high-complexity-pooled screen analysis. Nucleic Acids Res. 2015, 43, e16. [Google Scholar] [CrossRef]
- Allen, F.; Behan, F.; Khodak, A.; Iorio, F.; Yusa, K.; Garnett, M.; Parts, L. JACKS: Joint analysis of CRISPR/Cas9 knockout screens. Genome Res. 2019, 29, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Xu, H.; Xiao, T.; Cong, L.; Love, M.I.; Zhang, F.; Irizarry, R.A.; Liu, J.S.; Brown, M.; Liu, X.S. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 2014, 15, 554. [Google Scholar] [CrossRef] [PubMed]
- König, R.; Chiang, C.Y.; Tu, B.P.; Yan, S.F.; DeJesus, P.D.; Romero, A.; Bergauer, T.; Orth, A.; Krueger, U.; Zhou, Y.; et al. A probability-based approach for the analysis of large-scale RNAi screens. Nat. Methods 2007, 4, 847–849. [Google Scholar] [CrossRef] [PubMed]
- Bodapati, S.; Daley, T.P.; Lin, X.; Zou, J.; Qi, L.S. A benchmark of algorithms for the analysis of pooled CRISPR screens. Genome Biol. 2020, 21, 62. [Google Scholar] [CrossRef]
- Dempster, J.M.; Rossen, J.; Kazachkova, M.; Pan, J.; Kugener, G.; Root, D.E.; Tsherniak, A. Extracting Biological Insights from the Project Achilles Genome-Scale CRISPR Screens in Cancer Cell Lines. BioRxiv 2019. [Google Scholar] [CrossRef]
- The DepMap Portal. Available online: https://depmap.org/portal (accessed on 14 May 2020).
- The Sanger Project Score. Available online: https://score.depmap.sanger.ac.uk/ (accessed on 14 May 2020).
- Ghandi, M.; Huang, F.W.; Jane-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R., 3rd; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef]
- Mahalingaiah, P.K.; Ponnusamy, L.; Singh, K.P. Chronic oxidative stress causes estrogen-independent aggressive phenotype, and epigenetic inactivation of estrogen receptor alpha in MCF-7 breast cancer cells. Breast Cancer Res. Treat 2015, 153, 41–56. [Google Scholar] [CrossRef]
- Okoh, V.O.; Garba, N.A.; Penney, R.B.; Das, J.; Deoraj, A.; Singh, K.P.; Sarkar, S.; Felty, Q.; Yoo, C.; Jackson, R.M.; et al. Redox signalling to nuclear regulatory proteins by reactive oxygen species contributes to oestrogen-induced growth of breast cancer cells. Br. J. Cancer 2015, 112, 1687–1702. [Google Scholar] [CrossRef]
- Falone, S.; Lisanti, M.P.; Domenicotti, C. Oxidative Stress and Reprogramming of Mitochondrial Function and Dynamics as Targets to Modulate Cancer Cell Behavior and Chemoresistance. Oxid. Med. Cell. Longev. 2019, 2019, 4647807. [Google Scholar] [CrossRef]
- Radde, B.N.; Ivanova, M.M.; Mai, H.X.; Alizadeh-Rad, N.; Piell, K.; Van Hoose, P.; Cole, M.P.; Muluhngwi, P.; Kalbfleisch, T.S.; Rouchka, E.C.; et al. Nuclear respiratory factor-1 and bioenergetics in tamoxifen-resistant breast cancer cells. Exp. Cell Res. 2016, 347, 222–231. [Google Scholar] [CrossRef]
- Nassa, G.; Giurato, G.; Salvati, A.; Gigantino, V.; Pecoraro, G.; Lamberti, J.; Rizzo, F.; Nyman, T.A.; Tarallo, R.; Weisz, A. The RNA-mediated estrogen receptor alpha interactome of hormone-dependent human breast cancer cell nuclei. Sci. Data 2019, 6, 173. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, V.; Masjedi, A.; Hazhir-Karzar, B.; Tanomand, A.; Shotorbani, S.S.; Hojjat-Farsangi, M.; Ghalamfarsa, G.; Azizi, G.; Anvari, E.; Baradaran, B.; et al. The role of DEAD-box RNA helicase p68 (DDX5) in the development and treatment of breast cancer. J. Cell. Physiol. 2019, 234, 5478–5487. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Kobayashi, Y.; Wada, O.; Tateishi, Y.; Kitada, L.; Yamamoto, Y.; Takashima, H.; Murayama, A.; Yano, T.; Baba, T.; et al. Full activation of estrogen receptor alpha activation function-1 induces proliferation of breast cancer cells. J. Biol. Chem. 2003, 278, 26704–26714. [Google Scholar] [CrossRef] [PubMed]
- Gyorffy, B.; Bottai, G.; Fleischer, T.; Munkacsy, G.; Budczies, J.; Paladini, L.; Borresen-Dale, A.L.; Kristensen, V.N.; Santarpia, L. Aberrant DNA methylation impacts gene expression and prognosis in breast cancer subtypes. Int. J. Cancer 2016, 138, 87–97. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef]
- Pathiraja, T.N.; Stearns, V.; Oesterreich, S. Epigenetic regulation in estrogen receptor positive breast cancer—Role in treatment response. J. Mammary Gland Biol. Neoplasia 2010, 15, 35–47. [Google Scholar] [CrossRef]
- Cai, Y.; Geutjes, E.J.; de Lint, K.; Roepman, P.; Bruurs, L.; Yu, L.R.; Wang, W.; van Blijswijk, J.; Mohammad, H.; de Rink, I.; et al. The NuRD complex cooperates with DNMTs to maintain silencing of key colorectal tumor suppressor genes. Oncogene 2014, 33, 2157–2168. [Google Scholar] [CrossRef]
- Yan, L.; Nass, S.J.; Smith, D.; Nelson, W.G.; Herman, J.G.; Davidson, N.E. Specific inhibition of DNMT1 by antisense oligonucleotides induces re-expression of estrogen receptor-alpha (ER) in ER-negative human breast cancer cell lines. Cancer Biol. Ther. 2003, 2, 552–556. [Google Scholar] [CrossRef]
- Majidinia, M.; Yousefi, B. DNA repair and damage pathways in breast cancer development and therapy. DNA Repair 2017, 54, 22–29. [Google Scholar] [CrossRef]
- d’Adda di Fagagna, F. A direct role for small non-coding RNAs in DNA damage response. Trends Cell Biol. 2014, 24, 171–178. [Google Scholar] [CrossRef]
- Moynahan, M.E.; Chiu, J.W.; Koller, B.H.; Jasin, M. Brca1 controls homology-directed DNA repair. Mol. Cell 1999, 4, 511–518. [Google Scholar] [CrossRef]
- Maacke, H.; Opitz, S.; Jost, K.; Hamdorf, W.; Henning, W.; Kruger, S.; Feller, A.C.; Lopens, A.; Diedrich, K.; Schwinger, E.; et al. Over-expression of wild-type Rad51 correlates with histological grading of invasive ductal breast cancer. Int. J. Cancer 2000, 88, 907–913. [Google Scholar] [CrossRef]
- Benedetti, R.; Dell’Aversana, C.; De Marchi, T.; Rotili, D.; Liu, N.Q.; Novakovic, B.; Boccella, S.; Di Maro, S.; Cosconati, S.; Baldi, A.; et al. Inhibition of Histone Demethylases LSD1 and UTX Regulates ERα Signaling in Breast Cancer. Cancers 2019, 11, 2027. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, F.; Nassa, G.; Tarallo, R.; Stellato, C.; De Filippo, M.R.; Ambrosino, C.; Baumann, M.; Nyman, T.A.; Weisz, A. Molecular mechanisms of selective estrogen receptor modulator activity in human breast cancer cells: Identification of novel nuclear cofactors of antiestrogen-ERalpha complexes by interaction proteomics. J. Proteome Res. 2013, 12, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Nassa, G.; Salvati, A.; Tarallo, R.; Gigantino, V.; Alexandrova, E.; Memoli, D.; Sellitto, A.; Rizzo, F.; Malanga, D.; Mirante, T.; et al. Inhibition of histone methyltransferase DOT1L silences ERalpha gene and blocks proliferation of antiestrogen-resistant breast cancer cells. Sci. Adv. 2019, 5, eaav5590. [Google Scholar] [CrossRef] [PubMed]
- Papachristou, E.K.; Kishore, K.; Holding, A.N.; Harvey, K.; Roumeliotis, T.I.; Chilamakuri, C.S.R.; Omarjee, S.; Chia, K.M.; Swarbrick, A.; Lim, E.; et al. A quantitative mass spectrometry-based approach to monitor the dynamics of endogenous chromatin-associated protein complexes. Nat. Commun. 2018, 9, 2311. [Google Scholar] [CrossRef]
- Tarallo, R.; Bamundo, A.; Nassa, G.; Nola, E.; Paris, O.; Ambrosino, C.; Facchiano, A.; Baumann, M.; Nyman, T.A.; Weisz, A. Identification of proteins associated with ligand-activated estrogen receptor alpha in human breast cancer cell nuclei by tandem affinity purification and nano LC-MS/MS. Proteomics 2011, 11, 172–179. [Google Scholar] [CrossRef]
- Morris, J.H.; Knudsen, G.M.; Verschueren, E.; Johnson, J.R.; Cimermancic, P.; Greninger, A.L.; Pico, A.R. Affinity purification-mass spectrometry and network analysis to understand protein-protein interactions. Nat. Protoc. 2014, 9, 2539–2554. [Google Scholar] [CrossRef]
- Mohammed, H.; Taylor, C.; Brown, G.D.; Papachristou, E.K.; Carroll, J.S.; D’Santos, C.S. Rapid immunoprecipitation mass spectrometry of endogenous proteins (RIME) for analysis of chromatin complexes. Nat. Protoc. 2016, 11, 316–326. [Google Scholar] [CrossRef]
- Meyer, J.G.; Schilling, B. Clinical applications of quantitative proteomics using targeted and untargeted data-independent acquisition techniques. Expert Rev. Proteom. 2017, 14, 419–429. [Google Scholar] [CrossRef]
- Happonen, L.; Hauri, S.; Svensson Birkedal, G.; Karlsson, C.; de Neergaard, T.; Khakzad, H.; Nordenfelt, P.; Wikström, M.; Wisniewska, M.; Björck, L.; et al. A quantitative Streptococcus pyogenes-human protein-protein interaction map reveals localization of opsonizing antibodies. Nat. Commun. 2019, 10, 2727. [Google Scholar] [CrossRef] [PubMed]
- Fuller-Pace, F.V.; Ali, S. The DEAD box RNA helicases p68 (Ddx5) and p72 (Ddx17): Novel transcriptional co-regulators. Biochem. Soc. Trans. 2008, 36, 609–612. [Google Scholar] [CrossRef] [PubMed]
- Simabuco, F.M.; Morello, L.G.; Aragao, A.Z.; Paes Leme, A.F.; Zanchin, N.I. Proteomic characterization of the human FTSJ3 preribosomal complexes. J. Proteome Res. 2012, 11, 3112–3126. [Google Scholar] [CrossRef] [PubMed]
- Manning, M.; Jiang, Y.; Wang, R.; Liu, L.; Rode, S.; Bonahoom, M.; Kim, S.; Yang, Z.Q. Pan-cancer analysis of RNA methyltransferases identifies FTSJ3 as a potential regulator of breast cancer progression. RNA Biol. 2020, 17, 474–486. [Google Scholar] [CrossRef]
- Mohammed, H.; D’Santos, C.; Serandour, A.A.; Ali, H.R.; Brown, G.D.; Atkins, A.; Rueda, O.M.; Holmes, K.A.; Theodorou, V.; Robinson, J.L.; et al. Endogenous purification reveals GREB1 as a key estrogen receptor regulatory factor. Cell Rep. 2013, 3, 342–349. [Google Scholar] [CrossRef]
- Ghosh, M.G.; Thompson, D.A.; Weigel, R.J. PDZK1 and GREB1 are estrogen-regulated genes expressed in hormone-responsive breast cancer. Cancer Res. 2000, 60, 6367–6375. [Google Scholar]
- Rae, J.M.; Johnson, M.D.; Scheys, J.O.; Cordero, K.E.; Larios, J.M.; Lippman, M.E. GREB 1 is a critical regulator of hormone dependent breast cancer growth. Breast Cancer Res. Treat 2005, 92, 141–149. [Google Scholar] [CrossRef]
- Carroll, J.S.; Liu, X.S.; Brodsky, A.S.; Li, W.; Meyer, C.A.; Szary, A.J.; Eeckhoute, J.; Shao, W.; Hestermann, E.V.; Geistlinger, T.R.; et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell 2005, 122, 33–43. [Google Scholar] [CrossRef]
- Theodorou, V.; Stark, R.; Menon, S.; Carroll, J.S. GATA3 acts upstream of FOXA1 in mediating ESR1 binding by shaping enhancer accessibility. Genome Res. 2013, 23, 12–22. [Google Scholar] [CrossRef]
- Stossi, F.; Likhite, V.S.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Estrogen-occupied estrogen receptor represses cyclin G2 gene expression and recruits a repressor complex at the cyclin G2 promoter. J. Biol. Chem. 2006, 281, 16272–16278. [Google Scholar] [CrossRef]
- Mashtalir, N.; D’Avino, A.R.; Michel, B.C.; Luo, J.; Pan, J.; Otto, J.E.; Zullow, H.J.; McKenzie, Z.M.; Kubiak, R.L.; St Pierre, R.; et al. Modular Organization and Assembly of SWI/SNF Family Chromatin Remodeling Complexes. Cell 2018, 175, 1272–1288. [Google Scholar] [CrossRef] [PubMed]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Basavapathruni, A.; Jin, L.; Boriack-Sjodin, P.A.; Allain, C.J.; Klaus, C.R.; Raimondi, A.; Scott, M.P.; et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 2013, 122, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Lin, C.; Jin, C.; Yang, J.C.; Tanasa, B.; Li, W.; Merkurjev, D.; Ohgi, K.A.; Meng, D.; Zhang, J.; et al. lncRNA-dependent mechanisms of androgen-receptor-regulated gene activation programs. Nature 2013, 500, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Salvati, A.; Gigantino, V.; Nassa, G.; Giurato, G.; Alexandrova, E.; Rizzo, F.; Tarallo, R.; Weisz, A. The Histone Methyltransferase DOT1L Is a Functional Component of Estrogen Receptor Alpha Signaling in Ovarian Cancer Cells. Cancers 2019, 11, 1720. [Google Scholar] [CrossRef] [PubMed]
- Hajmirza, A.; Emadali, A.; Gauthier, A.; Casasnovas, O.; Gressin, R.; Callanan, M.B. BET Family Protein BRD4: An Emerging Actor in NFkappaB Signaling in Inflammation and Cancer. Biomedicines 2018, 6, 16. [Google Scholar] [CrossRef]
- Lu, L.; Chen, Z.; Lin, X.; Tian, L.; Su, Q.; An, P.; Li, W.; Wu, Y.; Du, J.; Shan, H.; et al. Inhibition of BRD4 suppresses the malignancy of breast cancer cells via regulation of Snail. Cell Death Differ. 2020, 27, 255–268. [Google Scholar] [CrossRef]
- Feng, Q.; Zhang, Z.; Shea, M.J.; Creighton, C.J.; Coarfa, C.; Hilsenbeck, S.G.; Lanz, R.; He, B.; Wang, L.; Fu, X.; et al. An epigenomic approach to therapy for tamoxifen-resistant breast cancer. Cell Res. 2014, 24, 809–819. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
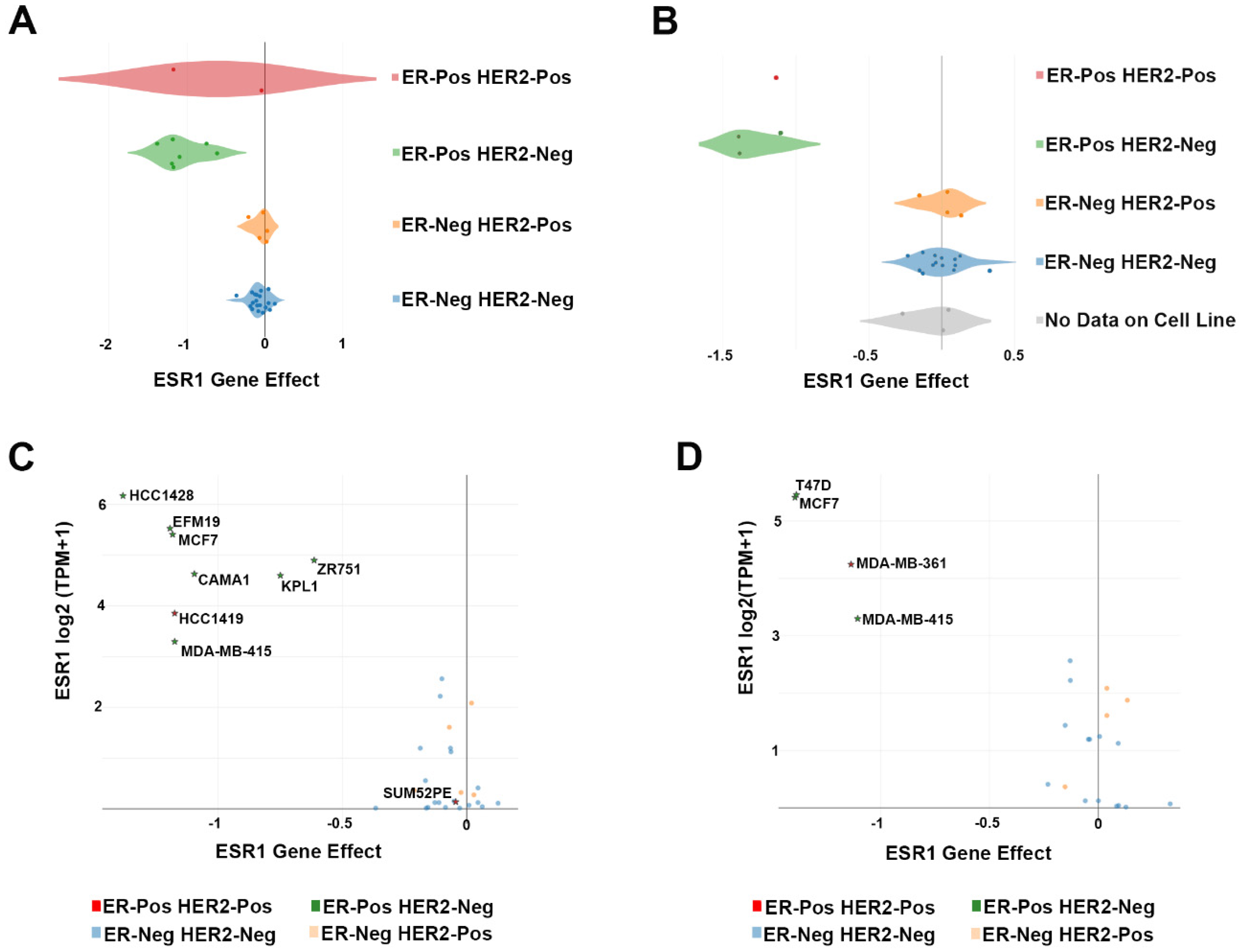
| Cell Line | Lineage | Lineage Subtype | Lineage Sub-Subtype | Tumor Type | CRISPR Screening |
|---|---|---|---|---|---|
| CAMA1 | Breast | Breast Carcinoma | ER-Pos HER2-Neg | Metastasis | [6] |
| EFM19 | Breast | Breast Ductal Carcinoma | ER-Pos HER2-Neg | Primary | [6] |
| HCC1419 | Breast | Breast Ductal Carcinoma | ER-Pos HER2-Pos | Metastasis | [6] |
| HCC1428 | Breast | Breast Carcinoma | ER-Pos HER2-Neg | Metastasis | [6] |
| KPL1 | Breast | Breast Carcinoma | ER-Pos HER2-Neg | Metastasis | [6] |
| MCF7 | Breast | Breast Carcinoma | ER-Pos HER2-Neg | Metastasis | [6,7] |
| MDA-MB-361 | Breast | Breast Carcinoma | ER-Pos HER2-Pos | Metastasis | [7] |
| MDA-MB-415 | Breast | Breast Carcinoma | ER-Pos HER2-Neg | Metastasis | [6,7] |
| SUM52PE | Breast | Breast Carcinoma | ER-Pos HER2-Pos | Metastasis | [6] |
| T47D | Breast | Breast Ductal Carcinoma | ER-Pos HER2-Neg | Metastasis | [7] |
| ZR751 | Breast | Breast Ductal Carcinoma | ER-Pos HER2-Neg | Metastasis | [6] |
| Project | ERα+ BC Cell Line | Number of Essential Genes |
|---|---|---|
| [6] | CAMA1 | 2292 |
| EFM19 | 2278 | |
| HCC1419 | 2279 | |
| HCC1428 | 2042 | |
| MCF7 | 2463 | |
| MDA-MB-415 | 2162 | |
| KPL1 | 2305 | |
| SUM52PE | 3089 | |
| ZR75.1 | 2149 | |
| [7] | MDA-MB-361 | 1494 |
| MDA-MB-415 | 1156 | |
| MCF7 | 761 | |
| T47D | 1191 | |
| [9] | T47D | 1915 |
| [10] | BT474 | 433 |
| EFM19 | 515 | |
| HCC1428 | 804 | |
| HCC1500 | 817 | |
| KPL1 | 794 | |
| MCF7 | 527 | |
| MDA-MB-175VII | 771 | |
| MDA-MB-361 | 697 | |
| MDA-MB-415 | 415 | |
| T47D | 803 | |
| UACC812 | 744 | |
| ZR75.1 | 799 | |
| ZR75.30 | 510 |
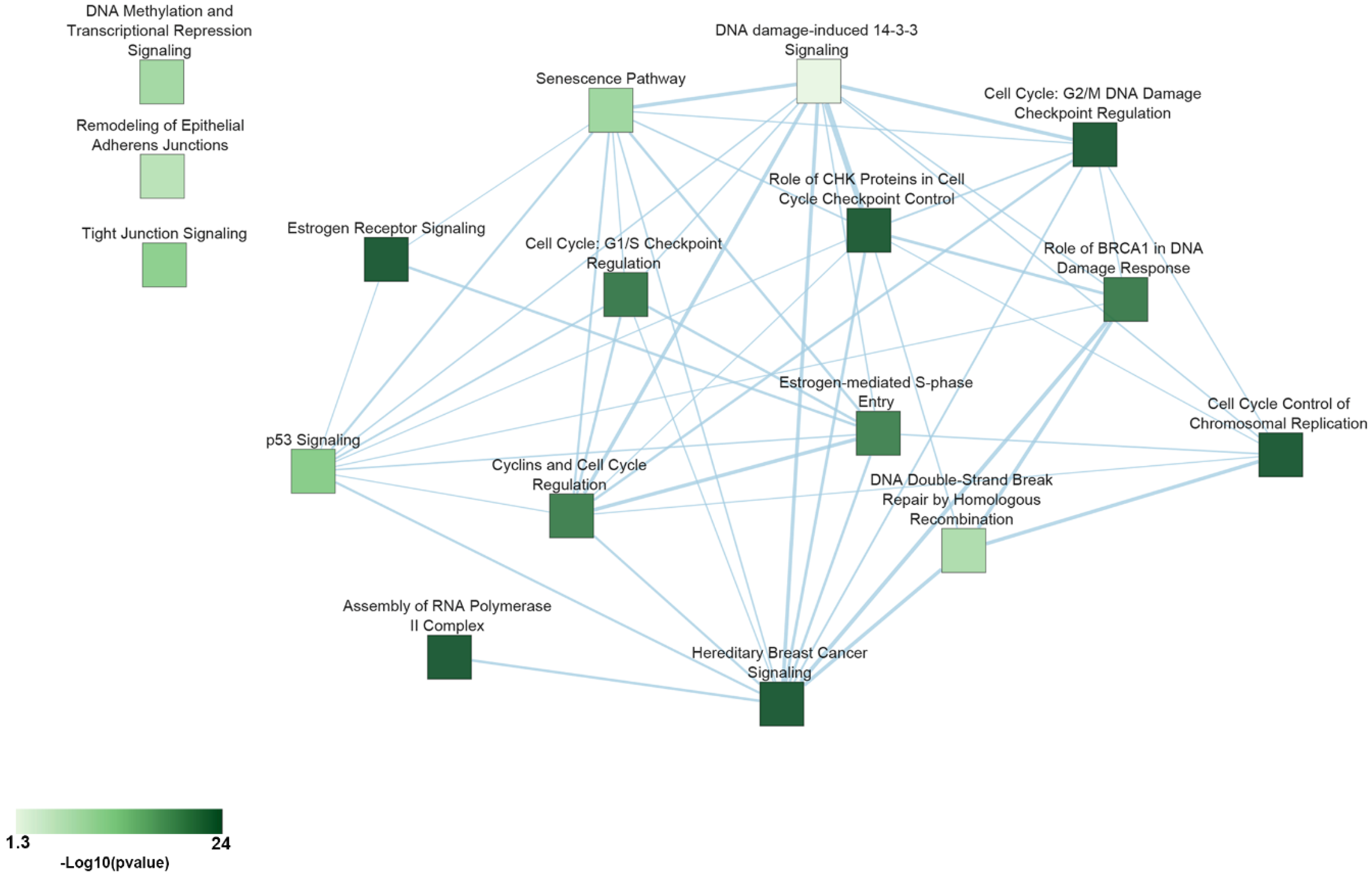
| Pathway | p-Value | Essential Genes |
|---|---|---|
| Cell Cycle Control of Chromosomal Replication | 5.01 × 10−24 | CDC45, CDC6, CDC7, CDK1, CDK11A, CDK4, CDK7, CDK9, CDT1, DBF4, MCM2, MCM3, MCM4, MCM5, MCM6, MCM7, ORC1, ORC6, PCNA, POLA1, POLA2, POLD1, POLE, PRIM1, RPA1, RPA2, RPA3, TOP2A |
| Assembly of RNA Polymerase II Complex | 3.16 × 10−15 | CCNH, CDK7, DR1, ERCC3, GTF2A1, GTF2A2, GTF2B, GTF2E1, GTF2E2, POLR2B, POLR2C, POLR2D, POLR2E, POLR2F, POLR2G, POLR2H, POLR2I, POLR2K, POLR2L, TAF1 |
| Hereditary Breast Cancer Signaling | 3.16 × 10−11 | ATR, CCND, CDK1, CDK4, CHEK1, KRAS, PIK3CA, POLR2B, POLR2C, POLR2D, POLR2E, POLR2F, POLR2G, POLR2H, POLR2I, POLR2K, POLR2L, RAD51, RFC3, RFC5, RPA1, RPS27A, SMARCB1, SMARCE1, TUBG1, UBA52, WEE1 |
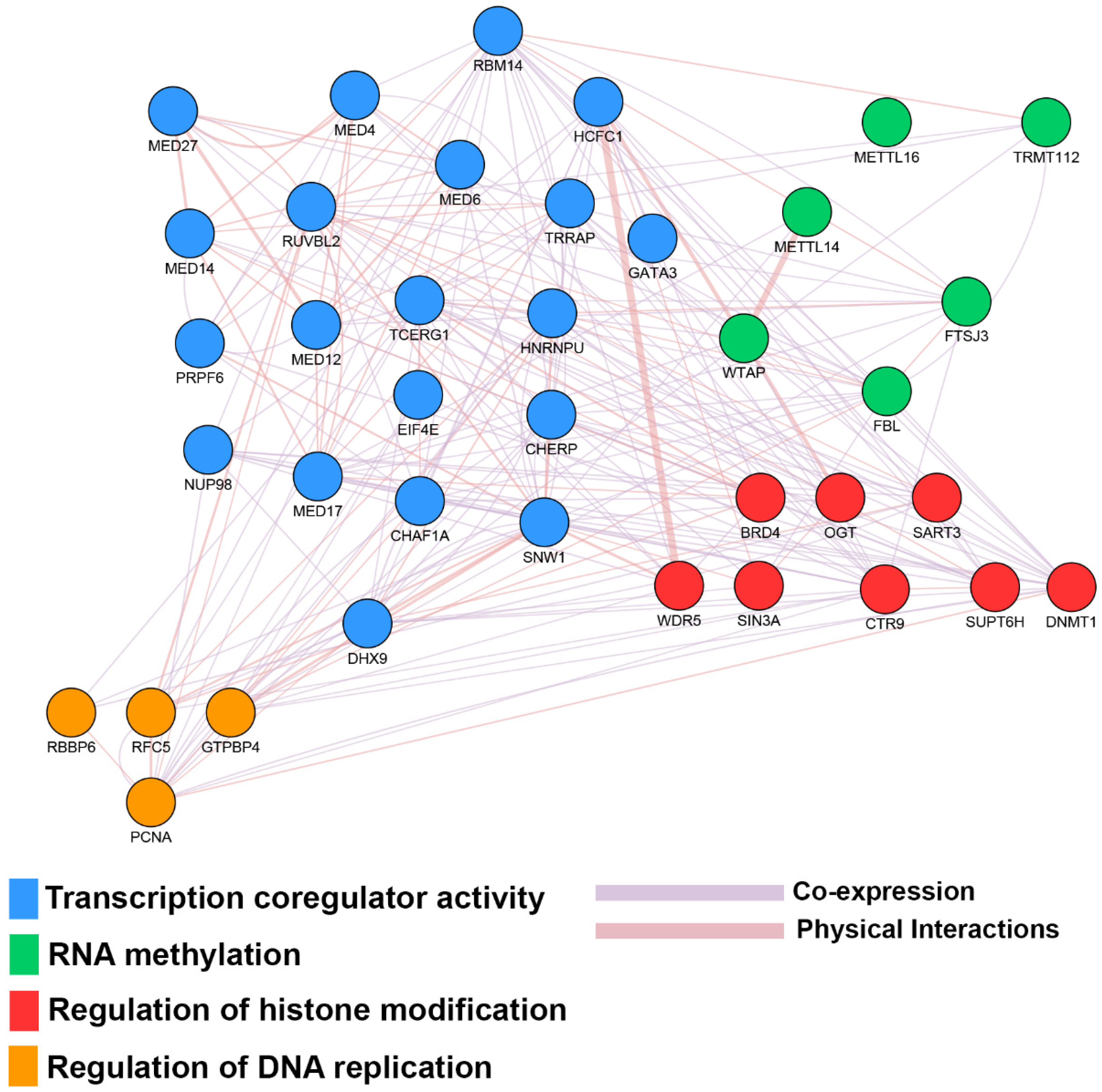
| Estrogen Receptor Signaling | 1.32 × 10−6 | CCND1, DDX5, EIF2B1, EIF2B2, EIF2B3, EIF2B4, EIF2B5, EIF4E, ESR1, FOXA1, KRAS, MED12, MED14, MED17, MED18, MED20, MED21, MED27, MED30, MED31, MED4, MED6, MTOR, MYC, NRF1, PCNA, PIK3CA, POLR2B, PPP1CB, PPP1R12A, SDHC, TFAM, TRRAP, UQCRFS1 |
| Role of CHK Proteins in Cell Cycle Checkpoint Control | 2.13 × 10−5 | ATR, CDK1, CHEK1, CLSPN, PCNA, PLK1, PPP2CA, RAD17, RFC3, RFC5, RPA1 |
| Cell Cycle: G2/M DNA Damage Checkpoint Regulation | 3.01 × 10−5 | ATR, AURKA, CDK1, CDK7, CHEK1, PKMYT1, PLK1, SKP1, TOP2A, WEE1 |
| Cell Cycle: G1/S Checkpoint Regulation | 4 × 10−4 | ATR, CCND1, CDK4, GNL3, MYC, PAK1IP1, RPL11, RPL5, SIN3A, SKP1 |
| Role of BRCA1 in DNA Damage Response | 5 × 10−4 | ATR, CHEK1, PLK1, RAD51, RBBP8, RFC3, RFC5, RPA1, SMARCB1, SMARCE1, TOPBP1 |
| Cyclins and Cell Cycle Regulation | 6 × 10−4 | ATR, CCNA2, CCND1, CCNH, CDK1, CDK4, CDK7, PPP2CA, SIN3A, SKP1, WEE1 |
| Estrogen-mediated S-phase Entry | 6 × 10−4 | CCNA2, CCND1, CDK1, CDK4, ESR1, MYC |
| p53 Signaling | 2 × 10−3 | ATR, BCL2L1, BIRC5, CCND1, CCNK, CDK4, CHEK1, GNL3, PCNA, PIK3CA, TOPBP1 |
| Tight Junction Signaling | 5 × 10−3 | CDC42, CDK4, CPSF2, CPSF3, CPSF6, CSTF3, GOSR2, NAPA, NSF, NUDT21, PPP2CA, RAC1, STX4, SYMPK, YKT6 |
| Senescence Pathway | 0.01 | ANAPC1, ANAPC10, ANAPC11, ANAPC2, ANAPC4, ANAPC5, ATR, CCND1, CDC16, CDC23, CDC26, CDC27, CDK1, CDK4, CHEK1, EIF4E, KRAS, MTOR, PIK3CA, PPP2CA |
| DNA Methylation and Transcriptional Repression Signaling | 0.01 | CHD4, DNMT1, RBBP4, SAP18, SIN3A |
| DNA Double-Strand Break Repair by Homologous Recombination | 0.02 | POLA1, RAD51, RPA1 |
| Remodeling of Epithelial Adherens Junctions | 0.02 | ACTR2, DNM1L, DNM2, TUBA1B, TUBA1C, TUBB, TUBG1 |
| DNA damage-induced 14-3-3 Signaling | 0.04 | ATR, CDK1, RAD17 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salvati, A.; Gigantino, V.; Nassa, G.; Mirici Cappa, V.; Ventola, G.M.; Cracas, D.G.C.; Mastrocinque, R.; Rizzo, F.; Tarallo, R.; Weisz, A.; et al. Global View of Candidate Therapeutic Target Genes in Hormone-Responsive Breast Cancer. Int. J. Mol. Sci. 2020, 21, 4068. https://doi.org/10.3390/ijms21114068
Salvati A, Gigantino V, Nassa G, Mirici Cappa V, Ventola GM, Cracas DGC, Mastrocinque R, Rizzo F, Tarallo R, Weisz A, et al. Global View of Candidate Therapeutic Target Genes in Hormone-Responsive Breast Cancer. International Journal of Molecular Sciences. 2020; 21(11):4068. https://doi.org/10.3390/ijms21114068
Chicago/Turabian StyleSalvati, Annamaria, Valerio Gigantino, Giovanni Nassa, Valeria Mirici Cappa, Giovanna Maria Ventola, Daniela Georgia Cristina Cracas, Raffaella Mastrocinque, Francesca Rizzo, Roberta Tarallo, Alessandro Weisz, and et al. 2020. "Global View of Candidate Therapeutic Target Genes in Hormone-Responsive Breast Cancer" International Journal of Molecular Sciences 21, no. 11: 4068. https://doi.org/10.3390/ijms21114068
APA StyleSalvati, A., Gigantino, V., Nassa, G., Mirici Cappa, V., Ventola, G. M., Cracas, D. G. C., Mastrocinque, R., Rizzo, F., Tarallo, R., Weisz, A., & Giurato, G. (2020). Global View of Candidate Therapeutic Target Genes in Hormone-Responsive Breast Cancer. International Journal of Molecular Sciences, 21(11), 4068. https://doi.org/10.3390/ijms21114068