Prediction of Novel Inhibitors of the Main Protease (M-pro) of SARS-CoV-2 through Consensus Docking and Drug Reposition

 , , , , ,
, , , , ,  and
and
Abstract
1. Introduction
2. Results and Discussion
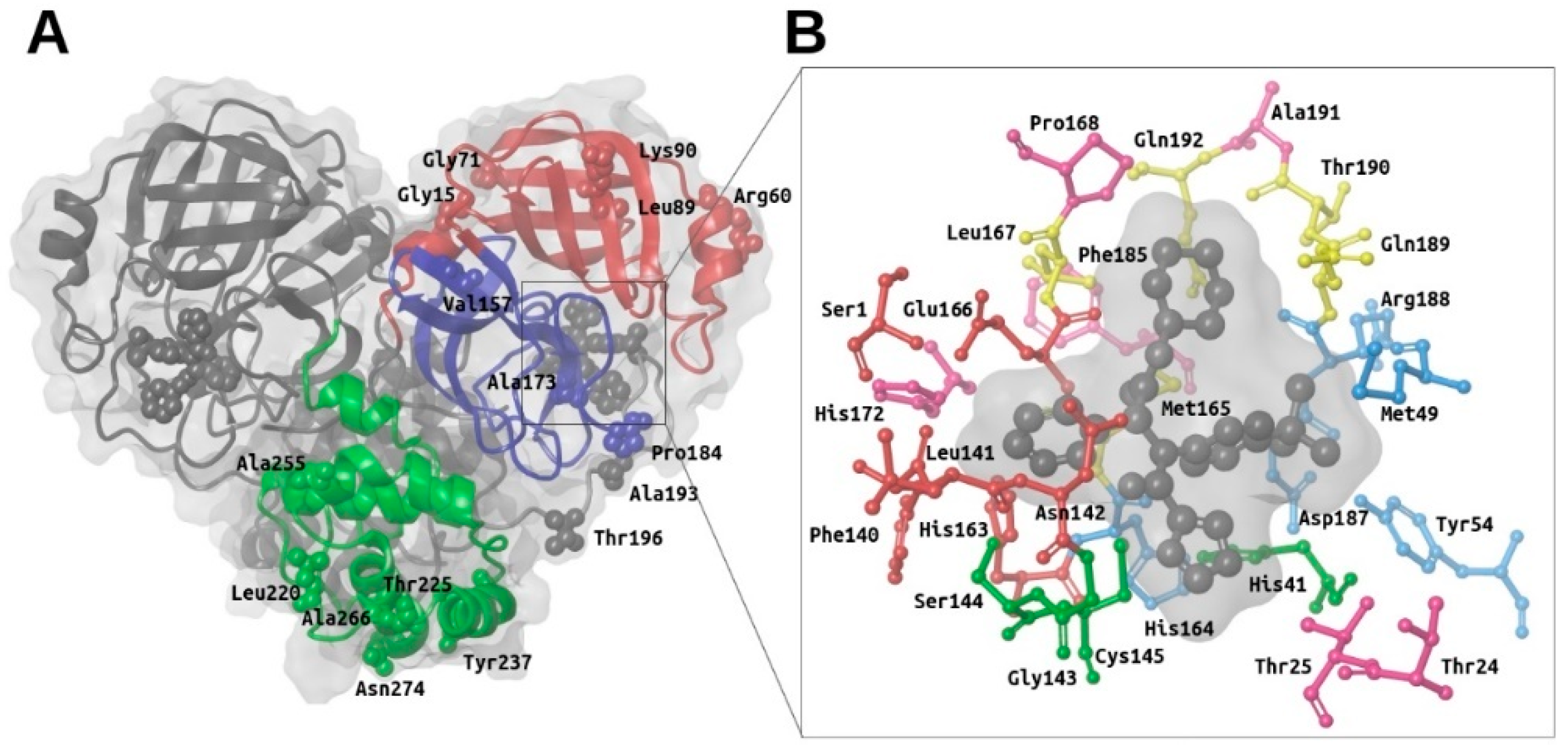
2.1. Structural Description of M-Pro and Report of Known Mutations in Its Structure

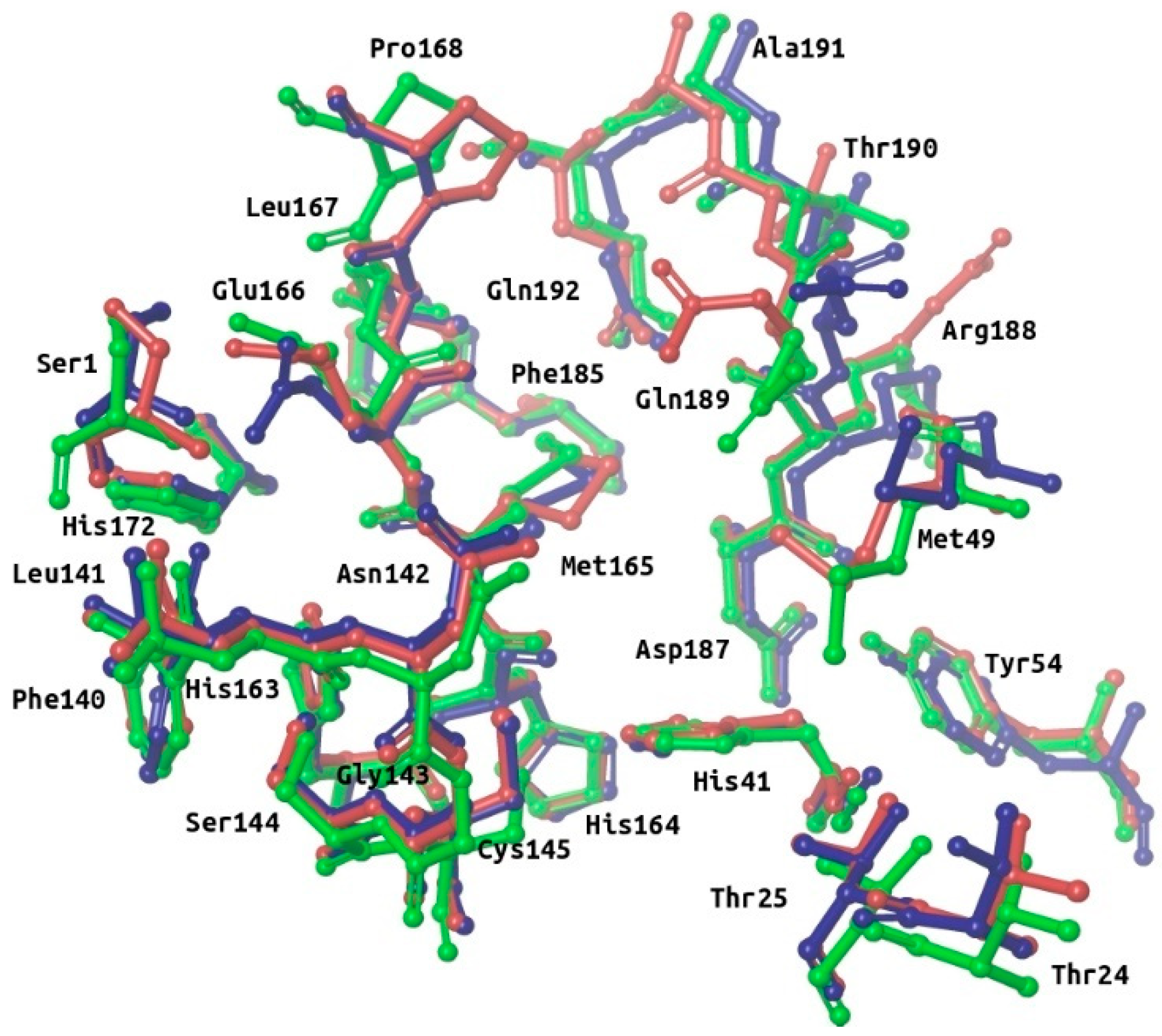
2.2. Description of the Intermolecular Interactions between M-Pro and Cocrystallized or Predicted Inhibitors
- S3 and S2 subsites:
- In the four experimental complexes, Met165 and Gln189 pin the ligand from both sides at the S3 subsite through hydrophobic interactions, with Gln189 interacting with two of its side chain carbons (i.e., CG and CB) and Met165 interacting with two of its side chain atoms (i.e., CB and SD; see Table 2 and Figure 3). Most of the compounds in the reference libraries also interact with Met165 and Gln189 (see Table 3). His41, Met49 and Asp187 also present hydrophobic interactions with most of the ligands around this area (see Table 2 and Figure 3). Met49 interacts with the ligands via its side chain atoms (i.e., CB, CG, SD and CE), whereas Asp187 and His41 use their CB carbon atom. Table 3 also shows that His41 and Met49 are highly important in the intermolecular interactions with the compounds from the reference libraries (with a more modest role for Asp187). Therefore, all these hydrophobic interactions would act as a hydrophobic grip around the same ligand group and greatly contribute to its binding affinity, which would explain the presence of the highly hydrophobic groups that the cocrystallized ligands present in this position (i.e., cyclohexylmethyl for 13a, cyclopropylmethyl for 13b, isopropylmethyl for N3 and t-butyl for X77; see Figure 3).
- The carbonyl oxygen of His164 (a residue close to the previously described hydrophobic region) provides an anchor point for 13b, N3 and X77 by acting as a hydrogen bond acceptor (see Table 2 and Figure 3). The interaction with His164 also seems important for a high percentage of docked poses in the COVID-Moonshot and DD-top-1000 reference libraries (see Table 3).
- S1 subsite:
- In the S1 subsite, the carboxylic acid group of Glu166 is able to establish either a hydrogen bond interaction with 13b or a salt bridge with N3. Moreover, its main chain oxygen and nitrogen (both oriented towards the S3 subsite) are able to act respectively as a hydrogen bond acceptor with ligands 13a, 13b and N3 or as a hydrogen bond donor with all the ligands (see Table 2 and Figure 3). Therefore, the high number of interactions between this residue and different parts of the ligand suggest that it plays a key role in the binding of compounds. In fact, at least half of the compounds in the four reference libraries interact with this residue (see Table 3).
- S1′ subsite:
- In the S1′ subsite, 13a, 13b and N3 bind covalently to the catalytic residue Cys145, and 13b and N3 effect a hydrogen bond interaction with the NE2 atom of His41 (see Table 2 and Figure 3). As Cys145 and His41 constitute the catalytic dyad of M-pro, interacting with these residues may be key to establishing a strong binding with this enzyme. Although few of the docked poses of the compounds in the four reference libraries interact with Cys145, most of them interact with His41 (see Table 3).
- In addition, the main chain nitrogen atom of Gly143 effects hydrogen bond interactions with all the cocrystallized ligands, and many compounds in the reference libraries also interact with this residue (see Table 2 and Table 3 and Figure 3). Interacting with Gly143 may be important to orient the compound towards the S1’ subsite and stabilize the binding of the compound in the catalytic site.
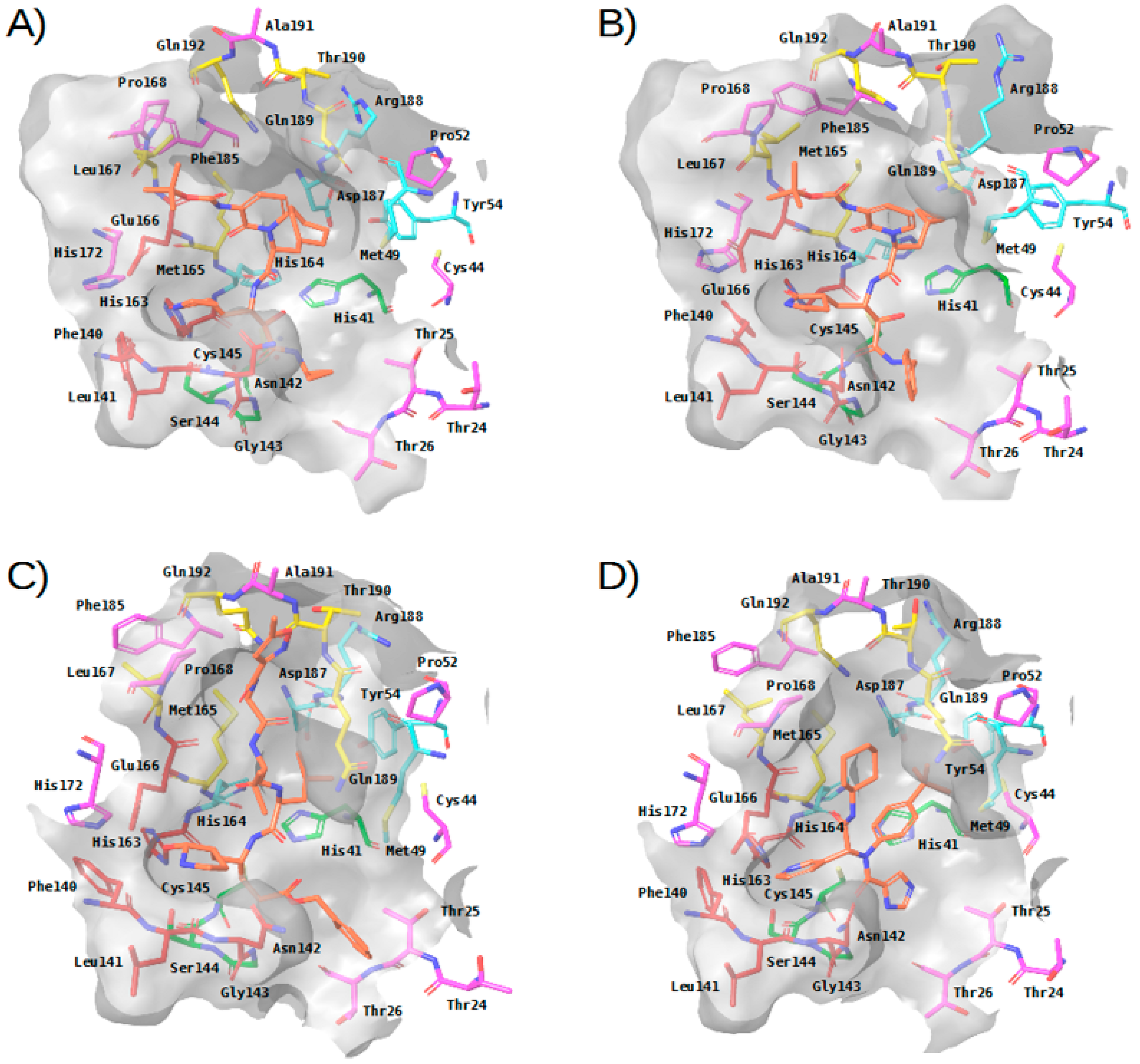
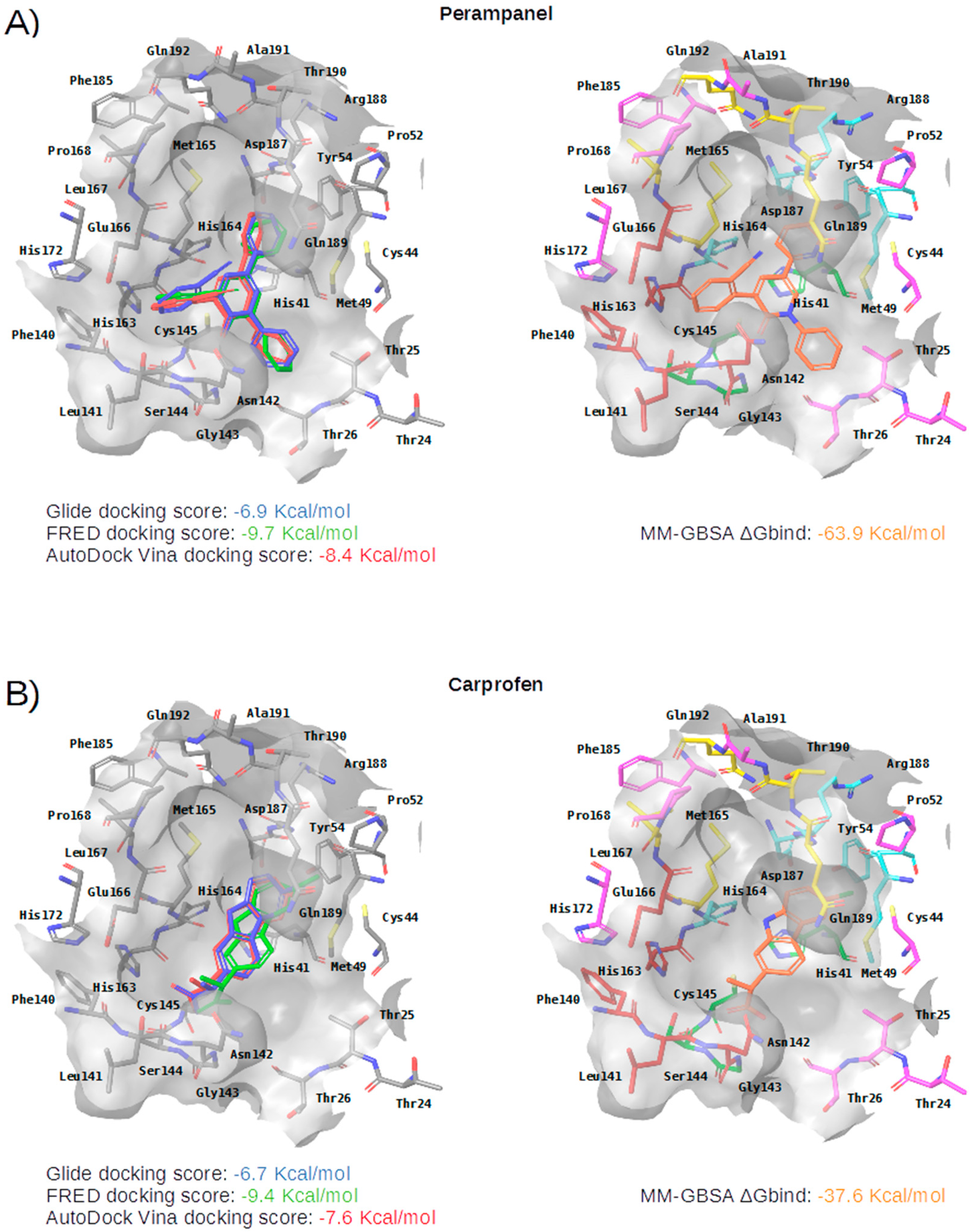
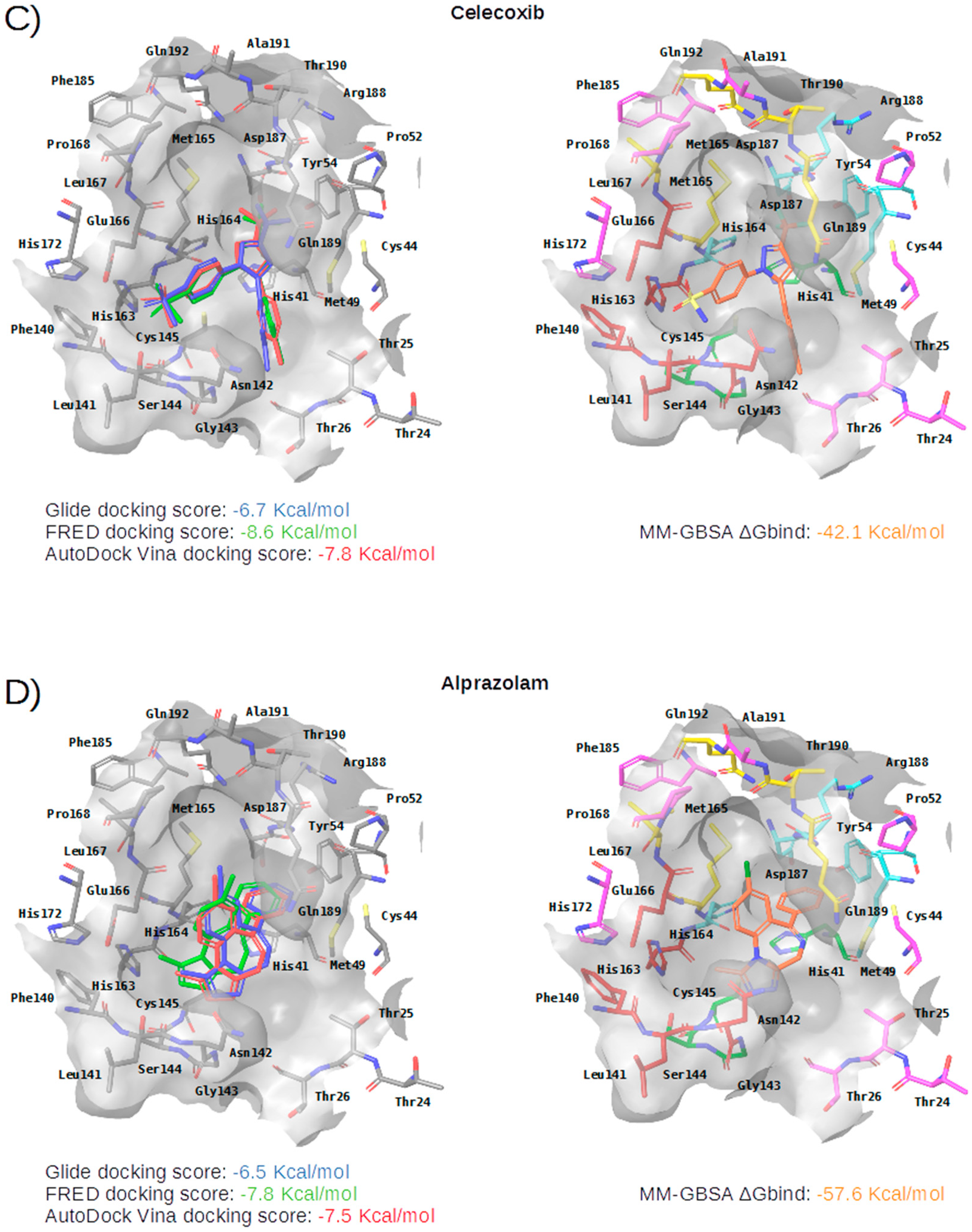
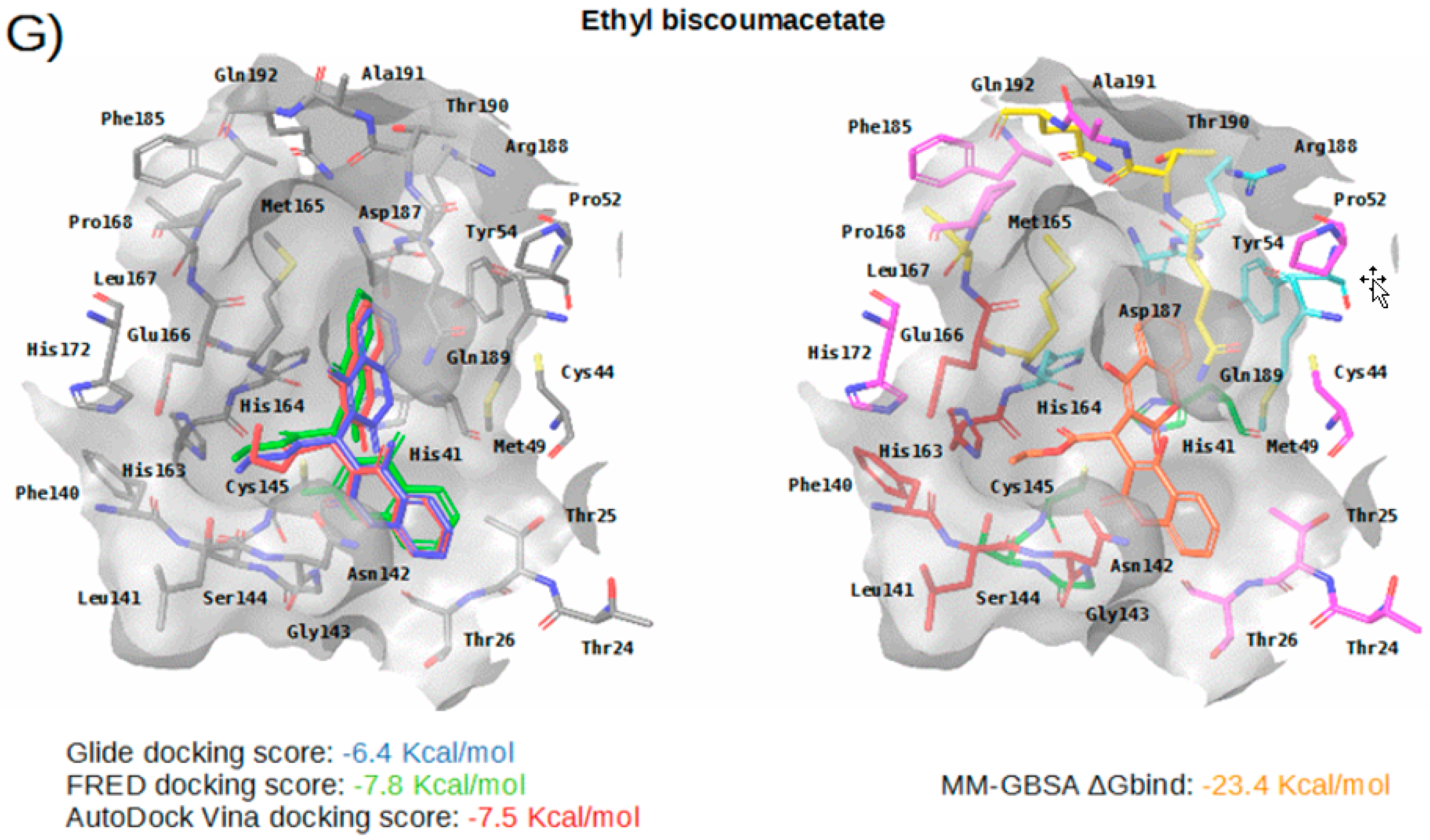
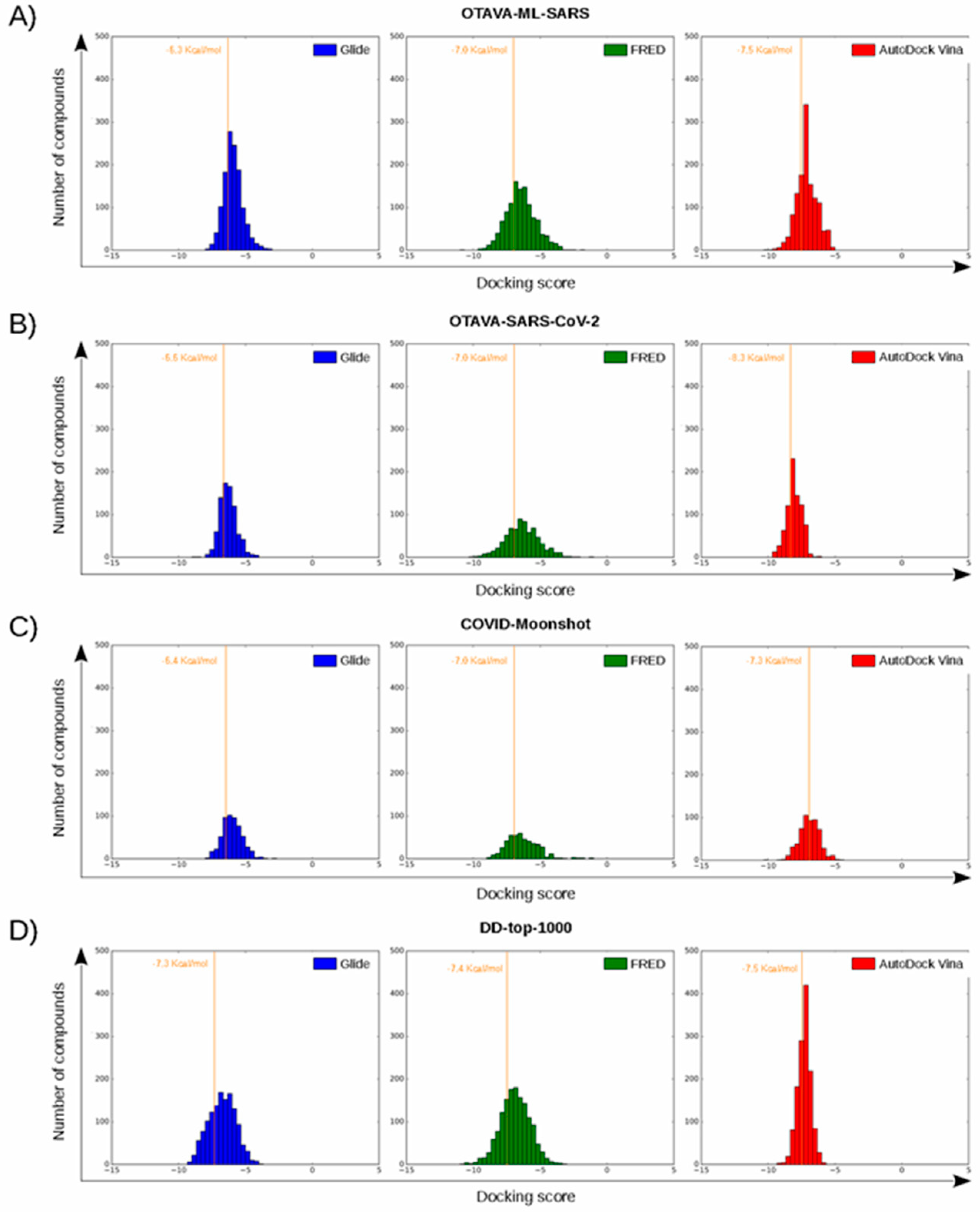
2.3. Virtual Screening of Approved Drugs
2.4. Selectivity of This Virtual Screening Workflow
3. Materials and Methods
3.1. Libraries Description and Preparation
3.2. Visual Inspection of the Fitting of Binding Site Coordinates to the Electron Density Maps
3.3. M-pro Structure Preparation, Grid Generation and Protein-ligand Docking Setup
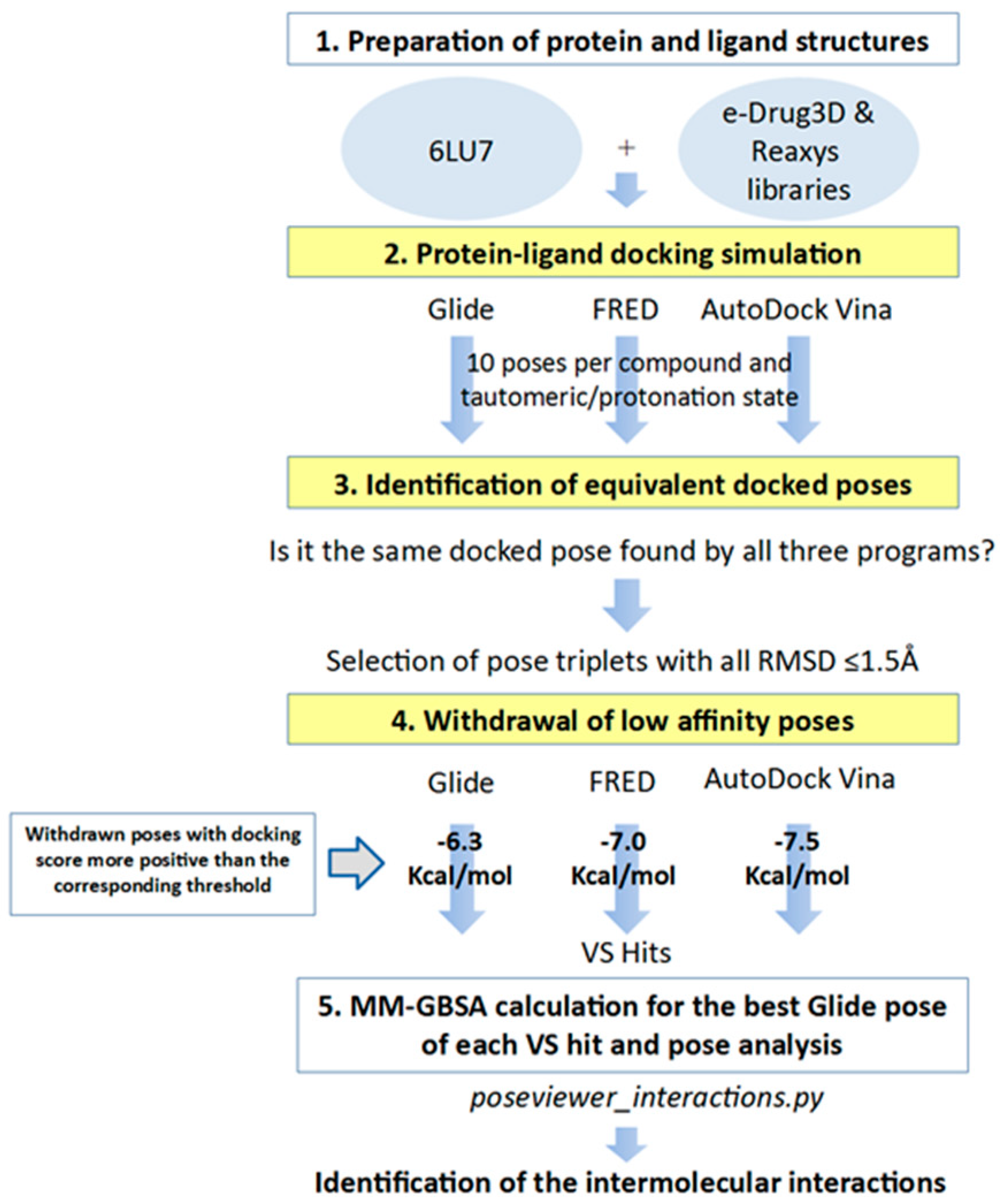
3.4. Identification of Equivalent Docked Poses among the Three Protein-Ligand Docking Programs
3.5. Apply Docking Score Thresholds to Keep Only the Equivalent Docked Poses with the Highest Affinity for M-pro
3.6. Virtual Screening Workflow Validation
3.7. Analysis of the Intermolecular Interactions between M-pro and Its Inhibitors
3.8. Analysis of Known Mutations of the M-pro Gene
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M.; et al. A Trial of Lopinavir-Ritonavir in Adults Hospitalized with Severe Covid-19. N. Engl. J. Med. 2020, 382, 1787–1799. [Google Scholar] [CrossRef]
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]
- Statista. Coronavirus (COVID-19) Death Rate in Italy as of April 11, 2020, by Age Group. Available online: https://www.statista.com/statistics/1106372/coronavirus-death-rate-by-age-group-italy/ (accessed on 12 April 2020).
- Mirza, M.U.; Froeyen, M. Structural elucidation of SARS-CoV-2 vital proteins: Computational methods reveal potential drug candidates against Main protease, Nsp12 RNA-dependent RNA polymerase and Nsp13 helicase. Preprints 2020, 2020030085. [Google Scholar] [CrossRef]
- Ton, A.-T.; Gentile, F.; Hsing, M.; Ban, F.; Cherkasov, A. Rapid Identification of Potential Inhibitors of SARS-CoV-2 Main Protease by Deep Docking of 1.3 Billion Compounds. Mol. Inform. 2020, 39, 202000028. [Google Scholar] [CrossRef]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) In Vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Han, M.; Li, T.; Sun, W.; Wang, D.; Fu, B.; Zheng, X.; Yang, Y.; Li, X.; Zhang, X.; et al. Effective Treatment of Severe COVID-19 Patients with Tocilizumab. Proc. Natl. Acad. Sci. USA 2020, 117, 10970–10975. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature 2020. [Google Scholar] [CrossRef]
- Kong, R.; Yang, G.; Xue, R.; Liu, M.; Wang, F.; Hu, J.; Guo, X.; Chang, S. COVID-19 Docking Server: An interactive server for docking small molecules, peptides and antibodies against potential targets of COVID-19. arXiv 2020, arXiv:2003.00163v1. [Google Scholar]
- Dang, M.; Song, J. 2019-nCoV 3C-Like Protease carries an activity-enhancing T285/A variation which may contribute to its high infectivity. Preprints 2020. [Google Scholar] [CrossRef]
- Tang, B.; He, F.; Liu, D.; Fang, M.; Wu, Z.; Xu, D. AI-aided design of novel targeted covalent inhibitors against SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Gimeno, A.; Ojeda-Montes, M.; Tomás-Hernández, S.; Cereto-Massagué, A.; Beltrán-Debón, R.; Mulero, M.; Pujadas, G.; Garcia-Vallvé, S. The Light and Dark Sides of Virtual Screening: What Is There to Know? Int. J. Mol. Sci. 2019, 20, 1375. [Google Scholar] [CrossRef]
- Chen, Y.W.; Yiu, C.-P.B.; Wong, K.-Y. Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CL pro) structure: Virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. F1000Research 2020, 9, 129. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, X.-J. Potential inhibitors against 2019-nCoV coronavirus M protease from clinically approved medicines. J. Genet. Genom. 2020, 47, 119–121. [Google Scholar] [CrossRef] [PubMed]
- Adem, S.; Eyupoglu, V.; Sarfraz, I.; Rasul, A.; Ali, M. Identification of potent COVID-19 main protease (Mpro) inhibitors from natural polyphenols: An in silico strategy unveils a hope against CORONA. Preprints 2020, 2020030333. [Google Scholar] [CrossRef]
- Yoshino, R.; Yasuo, N.; Sekijima, M. Identification of key interactions between SARS-CoV-2 Main Protease and inhibitor drug candidates. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Hosseini, F.S.; Amanlou, M. Simeprevir, potential candidate to repurpose for coronavirus infection: Virtual screening and molecular docking study. Preprints 2020, 2020020438. [Google Scholar] [CrossRef]
- Bzowka, M.; Mitusinska, K.; Raczynska, A.; Samol, A.; Tuszynski, J.A.; Gora, A. Molecular Dynamics Simulations Indicate the SARS-CoV-2 Mpro Is Not a Viable Target for Small-Molecule Inhibitors Design. bioRxiv 2020. [Google Scholar] [CrossRef]
- Xu, Z.; Peng, C.; Shi, Y.; Zhu, Z.; Mu, K.; Wang, X.; Zhu, W. Nelfinavir was predicted to be a potential inhibitor of 2019-nCov main protease by an integrative approach combining homology modelling, molecular docking and binding free energy calculation. bioRxiv 2020. [Google Scholar] [CrossRef]
- Khaerunnisa, S.; Kurniawan, H.; Awaluddin, R.; Suhartati, S. Potential Inhibitor of COVID-19 Main Protease (M pro) from Several Medicinal Plant Compounds by Molecular Docking Study. Preprints 2020, 2020030226. [Google Scholar] [CrossRef]
- Liu, Z.; Fang, H.; Reagan, K.; Xu, X.; Mendrick, D.L.; Slikker, W.; Tong, W. In silico drug repositioning: What we need to know. Drug Discov. Today 2013, 18, 110–115. [Google Scholar] [CrossRef] [PubMed]
- GISAID. Available online: https://www.gisaid.org/ (accessed on 31 March 2020).
- Schrödinger Release 2019-3: Maestro; Schrödinger, LLC.: New York, NY, USA, 2019.
- Macchiagodena, M.; Pagliai, M.; Procacci, P. Inhibition of the Main Protease 3CL-pro of the Coronavirus Disease 19 via Structure-Based Ligand Design and Molecular Modeling. arXiv 2020, arXiv:2002.09937. [Google Scholar]
- Bouchentouf, S.; Missoum, N. Identification of Compounds from Nigella Sativa as New PotentialInhibitors of 2019 Novel Coronasvirus (Covid-19): Molecular DockingStudy. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Structure of COVID-19 Main Protease Bound to Potent Broad-Spectrum Non-Covalent Inhibitor X77. Available online: http://www.rcsb.org/structure/6W63 (accessed on 26 March 2020).
- Novič, M.; Tibaut, T.; Anderluh, M.; Borišek, J.; Tomašič, T. The Comparison of Docking Search Algorithms and Scoring Functions. In Methods and Algorithms for Molecular Docking-Based Drug Design and Discovery; Medical Information Science Reference: Hershey, PA, USA, 2016; pp. 99–127. [Google Scholar]
- Ul-Haq, Z.; Madura, J.D. Frontiers in Computational Chemistry; Ul-Haq, Z., Madura, J.D., Eds.; Bentham Science Publishers: Sharjah, UAE, 2017; Volume 3, ISBN 9781681081670. [Google Scholar]
- McGann, M. FRED pose prediction and virtual screening accuracy. J. Chem. Inf. Model. 2011, 51, 578–596. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Torres, P.H.M.; Sodero, A.C.R.; Jofily, P.; Silva, F.P., Jr. Key Topics in Molecular Docking for Drug Design. Int. J. Mol. Sci. 2019, 20, 4574. [Google Scholar] [CrossRef]
- Chaput, L.; Mouawad, L. Efficient conformational sampling and weak scoring in docking programs? Strategy of the wisdom of crowds. J. Cheminform. 2017, 9, 37. [Google Scholar] [CrossRef]
- Li, J.; Fu, A.; Zhang, L. An Overview of Scoring Functions Used for Protein-Ligand Interactions in Molecular Docking. Interdiscip. Sci. 2019, 11, 320–328. [Google Scholar] [CrossRef]
- SchrSchrdinger Release 2020-2: Prime; Schrödinger, LLC.: New York, NY, USA, 2020.
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Singh, B.K.; Haque, S.E.; Pillai, K.K. Assessment of nonsteroidal anti-inflammatory drug-induced cardiotoxicity. Expert Opin. Drug Metab. Toxicol. 2014, 10, 143–156. [Google Scholar] [CrossRef]
- Basille, D.; Plouvier, N.; Trouve, C.; Duhaut, P.; Andrejak, C.; Jounieaux, V. Non-steroidal Anti-inflammatory Drugs may Worsen the Course of Community-Acquired Pneumonia: A Cohort Study. Lung 2017, 195, 201–208. [Google Scholar] [CrossRef]
- Hada, M. Chemotherapeutic Strategy with Synbiotics, Thalidomide and Celecoxib for severe COVID-19 Pneumonia. Association between microbiota, chronic inflammation and pneumonia. Preprint 2020. [Google Scholar] [CrossRef]
- Decision Memo—Analysis and Recommendations for Agency Action-COX-2 Selective and Non-Selective NSAIDs. Available online: https://www.fda.gov/media/74279/download (accessed on 6 April 2020).
- CDER Statement: FDA Approves Labeling Supplement for Celebrex (Celecoxib). Available online: https://www.fda.gov/drugs/drug-safety-and-availability/cder-statement-fda-approves-labeling-supplement-celebrex-celecoxib (accessed on 6 April 2020).
- Douguet, D. Data Sets Representative of the Structures and Experimental Properties of FDA-Approved Drugs. ACS Med. Chem. Lett. 2018, 9, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Elsevier. Reaxys Database. 2020. Available online: https://www.reaxys.com (accessed on 16 March 2020).
- OTAVA Chemicals. Available online: https://otavachemicals.com/targets/sars-cov-2-targeted-libraries (accessed on 16 March 2020).
- COVID Moonshot. Available online: https://covid.postera.ai/covid (accessed on 16 March 2020).
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2019-3: Glide; Schrödinger, LLC.: New York, NY, USA, 2019.
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- OMEGA 3.1.1.2: OpenEye Scientific Software; OpenEye Scientific Software: Santa Fe, NM. USA, 2019.
- Schrödinger Release 2019-3: LigPrep; Schrödinger, LLC.: New York, NY, USA, 2019.
- OEDOCKING 3.4.0.2: OpenEye Scientific Software; OpenEye Scientific Software: Santa Fe, NM, USA, 2019.
- QUACPAC 2.0.2.2: OpenEye Scientific Software; OpenEye Scientific Software: Santa Fe, NM, USA, 2019.
- Herráez, A. Biomolecules in the computer: Jmol to the rescue. Biochem. Mol. Biol. Educ. 2006, 34, 255–261. [Google Scholar] [CrossRef]
- Protein Data Bank in Europe. Available online: https://www.ebi.ac.uk/pdbe (accessed on 31 March 2020).
- Schrödinger Release 2019-3: Protein Preparation Wizard; Schrödinger, LLC.: New York, NY, USA, 2019.
- Schrödinger Release 2019-3: Impact; Schrödinger, LLC.: New York, NY, USA, 2019.
- Schrödinger Release 2019-3: Epik; Schrödinger, LLC.: New York, NY, USA, 2019.
- Schrödinger Release 2019-3: Prime; Schrödinger, LLC.: New York, NY, USA, 2019.
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Hevener, K.E.; Zhao, W.; Ball, D.M.; Babaoglu, K.; Qi, J.; White, S.W.; Lee, R.E. Validation of molecular docking programs for virtual screening against dihydropteroate synthase. J. Chem. Inf. Model. 2009, 49, 444–460. [Google Scholar] [CrossRef]
- Schrödinger Release 2019-3; Schrödinger, LLC.: New York, NY, USA, 2019.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | Type Mutation | AA Change |
|---|---|---|
| G10097A | missense | Gly15Ser |
| C10138T | synonymous | Asn28Asn |
| C10228T | synonymous | Leu58Leu |
| C10232T | missense | Arg60Cys |
| G10265A | missense | Gly71Ser |
| C10319T | missense | Leu89Phe |
| A10323G | missense | Lys90Arg |
| C10369T | synonymous | Arg105Arg |
| C10450T | synonymous | Pro132Pro |
| T10480C | synonymous | Asn142Asn |
| C10507T | synonymous | Asn151Asn |
| G10523A | missense | Val157Ile |
| C10572T | missense | Ala173Val |
| C10582T | synonymous | Asp176Asp |
| C10604T | missense | Pro184Ser |
| C10632T | missense | Ala193Val |
| C10641T | missense | Thr196Met |
| C10712T | missense | Leu220Phe |
| C10728T | missense | Thr225Ile |
| C10741T | synonymous | Asp229Asp |
| T10763C | missense | Tyr237His |
| T10771C | synonymous | Tyr239Tyr |
| C10789T | synonymous | Asp245Asp |
| C10818T | missense | Ala255Val |
| T10825A | synonymous | Thr257Thr |
| C10834T | synonymous | Ala260Ala |
| C10851T | missense | Ala266Val |
| G10870T | synonymous | Leu272Leu |
| A10874G | missense | Asn274Asp |
| A10912G | synonymous | Leu286Leu |
| Subsite | Residue | 13a 6Y7M | 13b 6Y2F, 6Y2G | N3 6LU7 | X77 6W63 |
|---|---|---|---|---|---|
| S3 | Met165 | CBh, SDh | CBh, 1SDh | CBh,SDh | CBh |
| Leu167 | |||||
| Gln189 | CGh | CBh, CGh | CGh | CBh, CGh | |
| Thr190 | Od | ||||
| Gln192 | |||||
| S2 | Met49 | CEh, SDh | CEh, 2SDh | CBh, CGh, SDh | |
| Tyr54 | |||||
| His164 | Od | Od | OAr | ||
| Asp187 | CBh | CBh | |||
| Arg188 | |||||
| S1 | Phe140 | Od | Od | Od | |
| Leu141 | |||||
| Asn142 | CBh | OD1Ar | |||
| His163 | NE2a | NE2a | |||
| Glu166 | Na, Od | Na, Od, OE2d, CGh | Na, Od, OE2s | Na, CBh | |
| S1’ | His41 | CBh | 1NE2d, 1CBh | CBh, 3NE2a | CBh |
| Gly143 | Na | Na | Na | Na | |
| Ser144 | |||||
| Cys145 | †SG, Na | †SG, Na | †SG, CBh | ||
| Thr25 | CG2h | ||||
| Thr26 | OAr | OAr | OAr | ||
| Pro168 | 1CBh | CGh | |||
| His172 | 3CD2a | 3CD2a |
| Sub-Site | Residue | OTAVA-ML-SARS | OTAVA-SARS-CoV-2 | COVID-Moonshot | DD-top-1000 |
|---|---|---|---|---|---|
| S3 | Met165 | 88.5 | 91.7 | 67.1 | 66.4 |
| Leu167 | 4.2 | 5.8 | 11.4 | 5.9 | |
| Gln189 | 95.5 | 92.1 | 90.4 | 96.4 | |
| Thr190 | 9.7 | 14.5 | 15.0 | 11.2 | |
| Gln192 | 2.9 | 3.7 | 7.8 | 1.3 | |
| S2 | Met49 | 74.1 | 78.5 | 67.1 | 68.4 |
| Tyr54 | 0.3 | 0.0 | 0.6 | 4.3 | |
| His164 | 17.3 | 18.2 | 49.1 | 76.3 | |
| Asp187 | 25.9 | 22.3 | 38.3 | 47.3 | |
| Arg188 | 14.1 | 14.0 | 10.2 | 30.5 | |
| S1 | Phe140 | 12.3 | 7.4 | 14.4 | 18.8 |
| Leu141 | 14.4 | 22.7 | 14.4 | 42.7 | |
| Asn142 | 18.3 | 19.8 | 22.8 | 9.2 | |
| His163 | 4.5 | 3.3 | 4.8 | 4.8 | |
| Glu166 | 50.0 | 62.0 | 70.7 | 59.8 | |
| S1′ | His41 | 77.7 | 81.0 | 82.6 | 79.9 |
| Gly143 | 42.7 | 49.2 | 24.6 | 78.1 | |
| Ser144 | 0.5 | 1.7 | 6.0 | 0.3 | |
| Cys145 | 5.2 | 3.7 | 7.8 | 2.3 | |
| Thr25 | 24.1 | 30.6 | 15.0 | 7.4 | |
| Thr26 | 27.2 | 42.1 | 22.2 | 16.8 | |
| Leu27 | 10.7 | 9.9 | 7.8 | 3.8 | |
| Pro168 | 11.3 | 18.2 | 15.0 | 2.0 |
| Compound | Drugbank and COVID MoonShot IDs (with % of Inhibition at 50 µM When Available) | Status | Mechanism | Indication | Adverse Effects |
|---|---|---|---|---|---|
 Perampanel | DB08883 GER-UNI-cfb | Approved | AMPA glutamate receptor antagonist. | Anticonvulsant: treatment of partial-onset seizures that may or may not occur with generalized seizures | Serious or life-threatening behavioral and psychiatric reactions |
 Carprofen | DB00821 GER-UNI-ec7-1 (3.97 ± 0.60%) | Approved; Withdrawn 1 | selective cyclooxygenase-2 (COX-2) inhibitor | Pain reliever in the treatment of joint pain and postsurgical pain | Mild, such as gastro-intestinal pain and nausea, similar to those recorded with aspirin and other nonsteroidal anti-inflammatory drugs (NSAIDS) |
 Celecoxib | DB00482 GER-UNI-05c (11.90 ± 0.59%) | Approved | selective COX-2 inhibitor | Arthritis pain and in familial adenomatous polyposis (FAP) to reduce precancerous polyps in the colon | Like other NSAIDS it is not advisable to administer it to patients with previous cardiovascular events |
 Alprazolam | DB00404 GER-UNI-cad | Approved | acts on benzodiazepine receptors BNZ-1 and BNZ-2 | Treatment of anxiety and panic disorders | Generally related to its sedative effects. Mixed with alcohol it may lead to coma and death |
 Trovafloxacin | DB00685 GER-UNI-c28 | Approved; Withdrawn | inhibition of DNA gyrase and topoisomerase IV. | Broad spectrum antibiotic | It was withdrawn in 1999 due to its hepatotoxic potential. |
 Sarafloxacin | DB11491 GER-UNI-cae | Vet approved; Withdrawn 2 | Antibiotic | ||
 Ethyl biscoumacetate | DB08794 GER-UNI-9e0 | Withdrawn | Vitamin K anatgonist | Anticoagulant | It is contraindicated in conditions like myocardial infarction, liver diseases, postpartum, hypersensitivity, pregnancy, bleeding, kidney disease, breast feeding and duodenal ulcer. It can produce increased blood clotting time, prolonged bleeding and severe hemorrhage. |
| Subsite | Residue | Perampanel | Carprofen | Celecoxib | Alprazolam | Trovafloxacin | Sarafloxacin | Ethyl Biscoumacetate |
|---|---|---|---|---|---|---|---|---|
| S3 | Met165 | CBh | CBh | CBh | CBh | CBh, SDh | ||
| Leu167 | ||||||||
| Gln189 | CGh | CGh | NE2a, CGh | CGh | CGh | |||
| Thr190 | Od | |||||||
| Gln192 | ||||||||
| S2 | Met49 | CEh, SDh | CBh, CGh, SDh | SDh | CBh, CEh, CGh, SDh | CGh, SDh | CBh, CGh, SDh | SDh |
| Tyr54 | ||||||||
| His164 | OAr | Od | OAr | OAr | ||||
| Asp187 | CBh | CBh | CBh | CBh | ||||
| Arg188 | ||||||||
| S1 | Phe140 | |||||||
| Leu141 | OAr | OAr | ||||||
| Asn142 | OD1d, CBh | CBh | ||||||
| His163 | ||||||||
| Glu166 | CBh | CBh | OAr | CBh | ||||
| S1’ | His41 | CGp, CBh | CGp, CGp, CBh | CGp, CBh | NE2a, CBh, CGp | NE2a, CD2Ar, CBh | ||
| Gly143 | Na | Na | Na | Na | ||||
| Ser144 | Na | Na | ||||||
| Cys145 | Na | Na | ||||||
| Thr26 | OAr | |||||||
| Cys44 | SGx, CBh | |||||||
| Pro52 | CGh |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gimeno, A.; Mestres-Truyol, J.; Ojeda-Montes, M.J.; Macip, G.; Saldivar-Espinoza, B.; Cereto-Massagué, A.; Pujadas, G.; Garcia-Vallvé, S. Prediction of Novel Inhibitors of the Main Protease (M-pro) of SARS-CoV-2 through Consensus Docking and Drug Reposition. Int. J. Mol. Sci. 2020, 21, 3793. https://doi.org/10.3390/ijms21113793
Gimeno A, Mestres-Truyol J, Ojeda-Montes MJ, Macip G, Saldivar-Espinoza B, Cereto-Massagué A, Pujadas G, Garcia-Vallvé S. Prediction of Novel Inhibitors of the Main Protease (M-pro) of SARS-CoV-2 through Consensus Docking and Drug Reposition. International Journal of Molecular Sciences. 2020; 21(11):3793. https://doi.org/10.3390/ijms21113793
Chicago/Turabian StyleGimeno, Aleix, Júlia Mestres-Truyol, María José Ojeda-Montes, Guillem Macip, Bryan Saldivar-Espinoza, Adrià Cereto-Massagué, Gerard Pujadas, and Santiago Garcia-Vallvé. 2020. "Prediction of Novel Inhibitors of the Main Protease (M-pro) of SARS-CoV-2 through Consensus Docking and Drug Reposition" International Journal of Molecular Sciences 21, no. 11: 3793. https://doi.org/10.3390/ijms21113793
APA StyleGimeno, A., Mestres-Truyol, J., Ojeda-Montes, M. J., Macip, G., Saldivar-Espinoza, B., Cereto-Massagué, A., Pujadas, G., & Garcia-Vallvé, S. (2020). Prediction of Novel Inhibitors of the Main Protease (M-pro) of SARS-CoV-2 through Consensus Docking and Drug Reposition. International Journal of Molecular Sciences, 21(11), 3793. https://doi.org/10.3390/ijms21113793