Investigation of Some Antiviral N-Heterocycles as COVID 19 Drug: Molecular Docking and DFT Calculations




Abstract
1. Introduction
2. Results and Discussion
2.1. DFT Calculations Studies
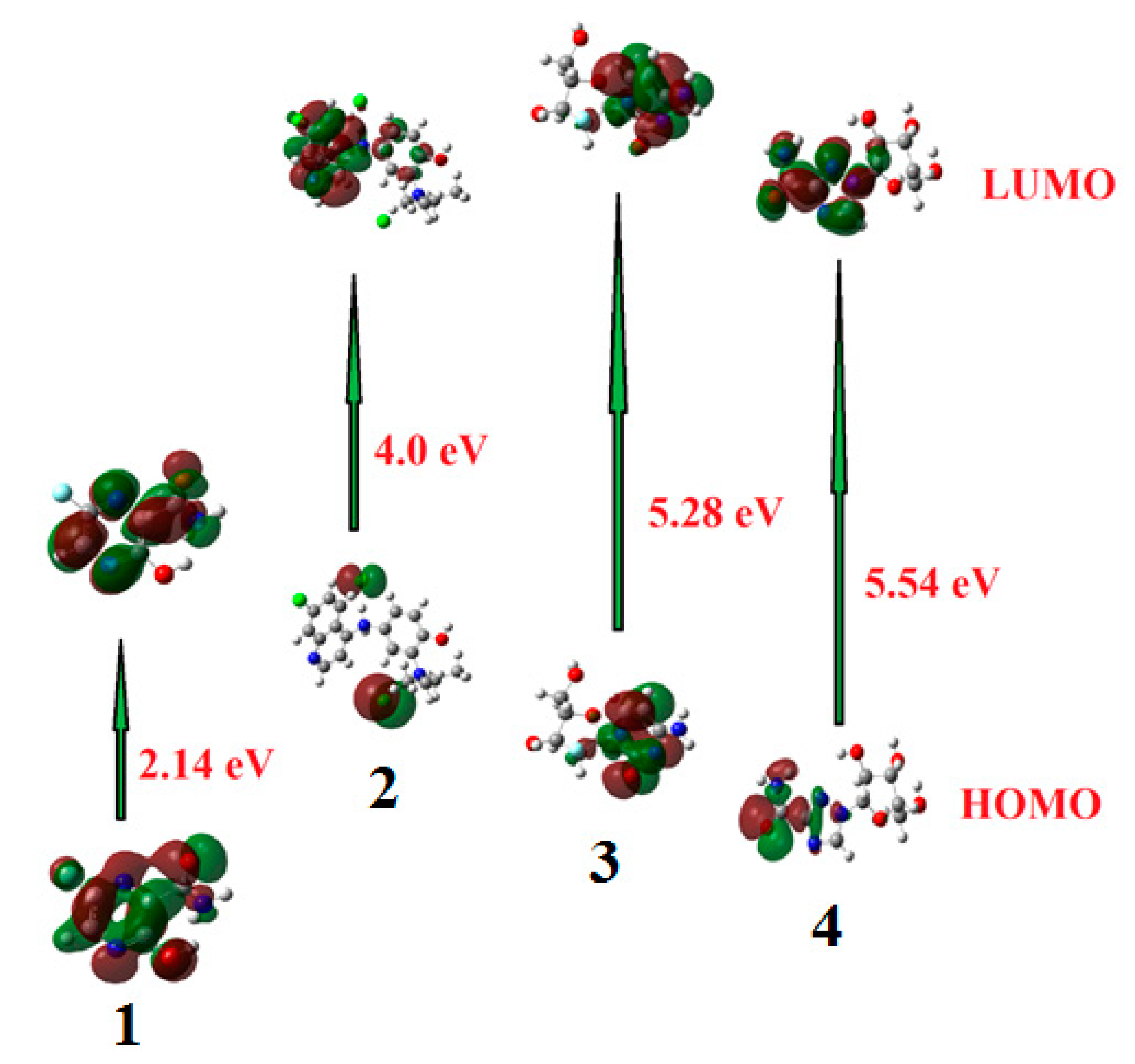
2.1.1. Frontier Molecular Orbitals
2.1.2. Chemical Reactivity Descriptors
2.1.3. Molecular Electrostatic Potential (MEP)
2.1.4. Mulliken Atomic Charges
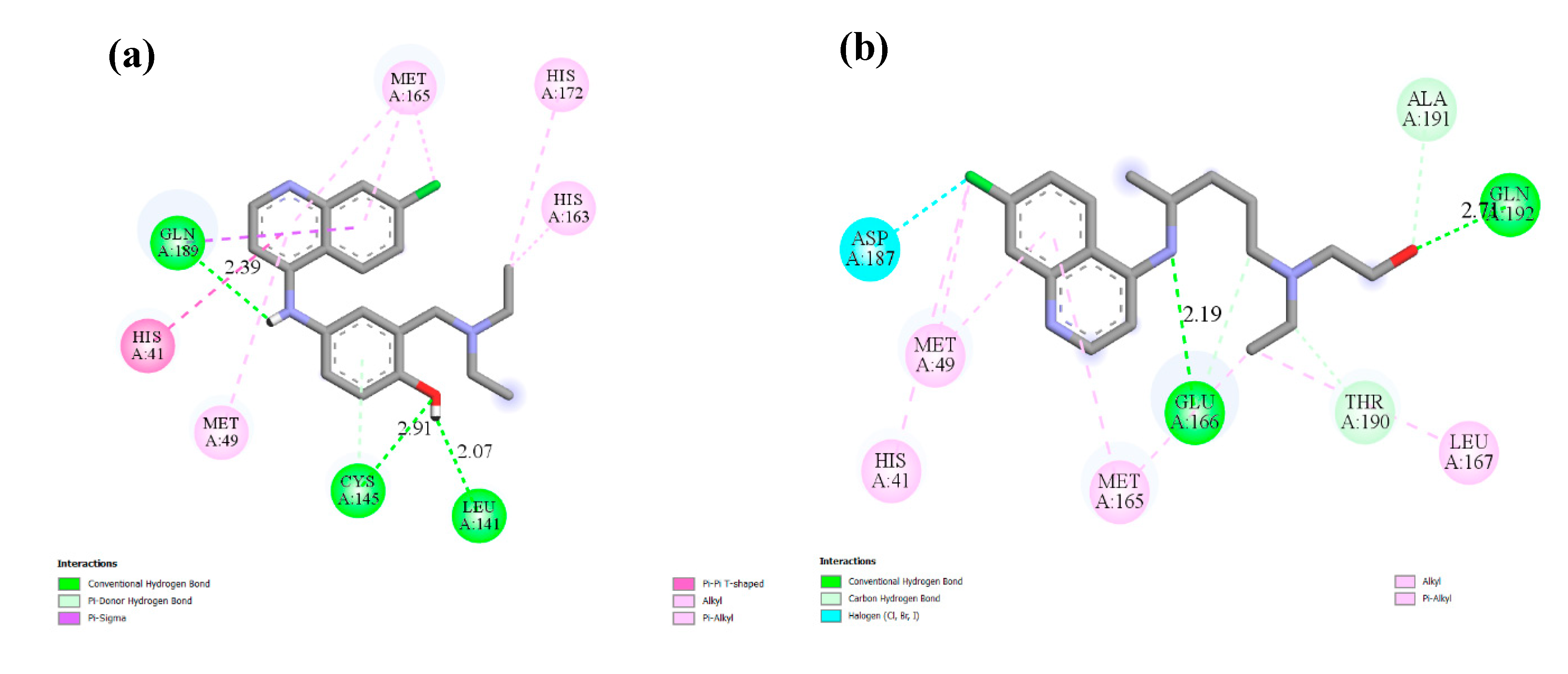
2.2. Molecular Docking
3. Materials and Methods
3.1. DFT Calculations
3.2. Molecular Docking Procedure
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Nishiura, H.; Linton, N.M.; Akhmetzhanov, A.R. Serial interval of novel coronavirus (COVID-19) infections. Int. J. Infect. Dis. 2020, 93, 284–286. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Zhang, P.; Bao, C.; Zhang, Y.; Zhu, N. Emerging understanding of etiology and epidemiology of the novel coronavirus (COVID-19) infection in Wuhan, China. Preprints 2020. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R. China Novel Coronavirus I, Research T. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Wang, X. COVID-19: A new challenge for human beings. Cell. Mol. Immunol. 2020, 17, 555–557. [Google Scholar] [CrossRef]
- Yao, X.; Ye, F.; Zhang, M.; Cui, C.; Huang, B.; Niu, P.; Liu, X.; Zhao, L.; Dong, E.; Song, C. In vitro antiviral activity and projection of optimized dosing design of hydroxychloroquine for the treatment of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Liu, J.; Cao, R.; Xu, M.; Wang, X.; Zhang, H.; Hu, H.; Li, Y.; Hu, Z.; Zhong, W.; Wang, M. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. 2020, 6, 1–4. [Google Scholar] [CrossRef]
- Grimstein, M.; Yang, Y.; Zhang, X.; Grillo, J.; Huang, S.-M.; Zineh, I.; Wang, Y. Physiologically based pharmacokinetic modeling in regulatory science: An update from the US Food and Drug Administration’s Office of Clinical Pharmacology. J. Pharm. Sci. 2019, 108, 21–25. [Google Scholar] [CrossRef]
- Devaux, C.A.; Rolain, J.-M.; Colson, P.; Raoult, D. New insights on the antiviral effects of chloroquine against coronavirus: What to expect for COVID-19? Int. J. Antimicrob. Agents 2020. [Google Scholar] [CrossRef]
- Warren, T.K.; Jordan, R.; Lo, M.K.; Ray, A.S.; Mackman, R.L.; Soloveva, V.; Siegel, D.; Perron, M.; Bannister, R.; Hui, H.C. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature 2016, 531, 381–385. [Google Scholar] [CrossRef]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Avorn, J. The $2.6 billion pill–methodologic and policy considerations. N. Engl. J. Med. 2015, 372, 1877–1879. [Google Scholar] [CrossRef]
- Cheng, F. In silico oncology drug repositioning and polypharmacology. In Cancer Bioinformatics; Springer: Cham, Switzerland, 2019; pp. 243–261. [Google Scholar]
- Cheng, F.; Hong, H.; Yang, S.; Wei, Y. Individualized network-based drug repositioning infrastructure for precision oncology in the panomics era. Brief. Bioinform. 2017, 18, 682–697. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Murray, J.L.; Rubin, D.H. Drug repurposing: New treatments for zika virus infection? Trends Mol. Med. 2016, 22, 919–921. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Hou, Y.; Shen, J.; Huang, Y.; Martin, W.; Cheng, F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020, 6, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhou, Q.; Li, Y.; Garner, L.V.; Watkins, S.P.; Carter, L.J.; Smoot, J.; Gregg, A.C.; Daniels, A.D.; Jervey, S. Research and development on Therapeutic Agents and Vaccines for COVID-19 and Related Human Coronavirus Diseases; ACS Publications: Washington, DC, USA, 2020. [Google Scholar]
- Shetty, R.; Ghosh, A.; Honavar, S.G.; Khamar, P.; Sethu, S. Therapeutic opportunities to manage COVID-19/SARS-CoV-2 infection: Present and future. Indian J. Ophthalmol. 2020, 68, 693. [Google Scholar]
- Zhang, L.; Liu, Y. Potential interventions for novel coronavirus in China: A systemic review. J. Med. Virol. 2020. [Google Scholar] [CrossRef]
- Chan, K.W.; Wong, V.T.; Tang, S.C.W. COVID-19: An update on the epidemiological, clinical, preventive and therapeutic evidence and guidelines of integrative Chinese–Western medicine for the management of 2019 novel coronavirus disease. Am. J. Chin. Med. 2020, 1–26. [Google Scholar] [CrossRef]
- Sheahan, T.P.; Sims, A.C.; Leist, S.R.; Schäfer, A.; Won, J.; Brown, A.J.; Montgomery, S.A.; Hogg, A.; Babusis, D.; Clarke, M.O. Comparative therapeutic efficacy of remdesivir and combination lopinavir, ritonavir, and interferon beta against MERS-CoV. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Ortega, J.T.; Serrano, M.L.; Pujol, F.H.; Rangel, H.R. Unrevealing sequence and structural features of novel coronavirus using in silico approaches: The main protease as molecular target. EXCLI J. 2020, 19, 400. [Google Scholar]
- Liu, C.; Ma, Y.; Zhao, J.; Nussinov, R.; Zhang, Y.-C.; Cheng, F.; Zhang, Z.-K. Computational network biology: Data, model, and applications. Phys. Rep. 2019, 846, 1–66. [Google Scholar] [CrossRef]
- Theerawatanasirikul, S.; Kuo, C.J.; Phetcharat, N.; Lekcharoensuk, P. In silico and in vitro analysis of small molecules and natural compounds targeting the 3CL protease of feline infectious peritonitis virus. Antivir. Res. 2020, 174, 104697. [Google Scholar] [CrossRef] [PubMed]
- Hagar, M.; Chaieb, K.; Parveen, S.; Ahmed, H.; Alnoman, R. N-alkyl 2-pyridone versus O-alkyl 2-pyridol: Ultrasonic synthesis, DFT, docking studies and their antimicrobial evaluation. J. Mol. Struct. 2020, 1199, 126926. [Google Scholar] [CrossRef]
- Alnoman, R.B.; Parveen, S.; Hagar, M.; Ahmed, H.A.; Knight, J.G. A new chiral Boron–dipyrromethene (BODIPY)–based fluorescent probe: Molecular docking, DFT, antibacterial and antioxidant approaches. J. Biomol. Struct. Dyn. 2019, 1–19. [Google Scholar] [CrossRef]
- Alnoman, R.B.; Hagar, M.; Parveen, S.; Ahmed, H.A.; Knight, J.G. Computational and molecular docking approaches of a New axially chiral BODIPY fluorescent dye. J. Photochem. Photobiol. A: Chem. 2020, 395, 112508. [Google Scholar] [CrossRef]
- Oehninger, L.; Rubbiani, R.; Ott, I. N-Heterocyclic carbene metal complexes in medicinal chemistry. Dalton Trans. 2013, 42, 3269–3328. [Google Scholar] [CrossRef]
- Gong, H.-H.; Addla, D.; Lv, J.-S.; Zhou, C.-H. Heterocyclic naphthalimides as new skeleton structure of compounds with increasingly expanding relational medicinal applications. Curr. Top. Med. Chem. 2016, 16, 3303–3364. [Google Scholar] [CrossRef]
- Furuta, Y.; Gowen, B.B.; Takahashi, K.; Shiraki, K.; Smee, D.F.; Barnard, D.L. Favipiravir (T-705), a novel viral RNA polymerase inhibitor. Antivir. Res. 2013, 100, 446–454. [Google Scholar] [CrossRef]
- Olliaro, P.; Nevill, C.; LeBras, J.; Ringwald, P.; Mussano, P.; Garner, P.; Brasseur, P. Systematic review of amodiaquine treatment in uncomplicated malaria. Lancet 1996, 348, 1196–1201. [Google Scholar] [CrossRef]
- Wohlrab, F.; Jamieson, A.; Hay, J.; Mengel, R.; Guschlbauer, W. The effect of 2′-fluoro-2′-deoxycytidine on herpes virus growth. Biochim. Biophys. Acta (BBA)-Gene Struct. Expr. 1985, 824, 233–242. [Google Scholar] [CrossRef]
- Glue, P. The clinical pharmacology of ribavirin. Semin. Liver Dis. 1999, 19, 17–24. [Google Scholar] [PubMed]
- Sato, R.; Vohra, S.; Yamamoto, S.; Suzuki, K.; Pavel, K.; Shulga, S.; Blume, Y.; Kurita, N. Specific interactions between tau protein and curcumin derivatives: Molecular docking and ab initio molecular orbital simulations. J. Mol. Graph. Model. 2020, 98, 107611. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, D.; Singh, D.K.; Pandey, D.K.; Parai, D.; Bankura, K.; Mishra, D. DFT investigations of linear Zn3-type complex with compartmental N/O-donor Schiff base: Synthesis, characterizations, crystal structure, fluorescence and molecular docking. J. Mol. Struct. 2020, 1209, 127936. [Google Scholar] [CrossRef]
- Almutairi, M.S.; Leenaraj, D.; Ghabbour, H.A.; Joe, I.H.; Attia, M.I. Spectroscopic identification, structural features, Hirshfeld surface analysis and molecular docking studies on stiripentol: An orphan antiepileptic drug. J. Mol. Struct. 2019, 1180, 110–118. [Google Scholar] [CrossRef]
- Xavier, S.; Periandy, S.; Ramalingam, S. NBO, conformational, NLO, HOMO–LUMO, NMR and electronic spectral study on 1-phenyl-1-propanol by quantum computational methods. Spectrochim. Acta Part A: Mol. Biomol. Spectrosc. 2015, 137, 306–320. [Google Scholar] [CrossRef]
- Nasief, N.N.; Said, A.M.; Hangauer, D. Modulating hydrogen-bond basicity within the context of protein-ligand binding: A case study with thrombin inhibitors that reveals a dominating role for desolvation. Eur. J. Med. Chem. 2017, 125, 975–991. [Google Scholar] [CrossRef]
- Kouza, M.; Banerji, A.; Kolinski, A.; Buhimschi, I.; Kloczkowski, A. Role of resultant dipole moment in mechanical dissociation of biological complexes. Molecules 2018, 23, 1995. [Google Scholar] [CrossRef]
- Shawon, J.; Khan, A.M.; Rahman, A.; Hoque, M.M.; Khan, M.A.K.; Sarwar, M.G.; Halim, M.A. Molecular recognition of azelaic acid and related molecules with DNA polymerase I investigated by molecular modeling calculations. Interdiscip. Sci.: Comput. Life Sci. 2018, 10, 525–537. [Google Scholar] [CrossRef]
- Uzzaman, M.; Hoque, M.J. Physiochemical, molecular docking, and pharmacokinetic studies of Naproxen and its modified derivatives based on DFT. Int. J. Sci. Res. Manag. 2018, 6, C. [Google Scholar] [CrossRef]
- Gaussian 09, Revision a. 02; gaussian. Inc.: Wallingford, CT, USA, 2009.
- Dennington, R.; Keith, T.; Millam, J. GaussView, version 5; Semichem Inc.: Shawnee Mission, KS, USA, 2009.
- Morris, G.; Huey, R.; Lindstrom, W.; Sanner, M.; Belew, R.; Goodsell, D.; Olson, A. AutoDock4 and AutoDockTools4: Automated docking with selective receptor 828 flexibility. J. Comput. Chem 2009, 30, 2785–2791. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Ecorr | 0.10 | 0.38 | 0.23 | 0.23 |
| ZPVE | −607.34 | −2357.00 | −914.90 | −906.88 |
| Etot | −607.34 | −2356.97 | −914.89 | −906.86 |
| H | −607.33 | −2356.97 | −914.89 | −906.86 |
| G | −607.38 | −2357.05 | −914.94 | −906.92 |
| Total Dipole Moment | 4.32 | 14.44 | 2.26 | 5.94 |
| Polarizability α | 76.39 | 268.38 | 127.41 | 123.52 |
| Drugs | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| EHOMO | −7.61 | −5.65 | −6.34 | −7.22 |
| ELUMO | −5.47 | −1.65 | −1.05 | −1.68 |
| ΔE | 2.14 | 4.00 | 5.28 | 5.54 |
| χ | 2.06 | 3.70 | 3.65 | 3.04 |
| η | 1.07 | 2.00 | 2.64 | 2.77 |
| δ | 0.94 | 0.50 | 0.38 | 0.36 |
| ω | 2.27 | 13.69 | 17.59 | 12.80 |
| I | 7.61 | 5.65 | 6.34 | 7.22 |
| A | 5.47 | 1.65 | 1.05 | 1.68 |
| 1 | 2 | 3 | 4 |
|---|---|---|---|
| 1 C 0.395842 | 1 C −0.062356 | 1 C 0.495609 | 1 N −0.335624 |
| 2 C −0.045572 | 2 C −0.295910 | 2 C −0.095415 | 2 C 0.510567 |
| 3 N −0.238512 | 3 C 0.034382 | 3 C 0.456261 | 3 N−0.512630 |
| 4 C 0.446897 | 4 C 0.046151 | 4 N−0.653167 | 4 N −0.206644 |
| 5 C −0.112549 | 5 C −0.014008 | 5 C 0.557768 | 5 C 0.104039 |
| 6 N −0.220205 | 6 C −0.063072 | 6 N −0.407585 | 6 C 0.500391 |
| 7 C 0.507551 | 7 N −0.272785 | 7 C 0.428075 | 7 O −0.489846 |
| 8 N −0.831622 | 8 C 0.010866 | 8 C 0.347162 | 8 C 0.203009 |
| 9 O −0.278205 | 9 C −0.079472 | 9 C 0.173018 | 9 C 0.204580 |
| 10 O−0.571957 | 10 C 0.071445 | 10 C 0.176333 | 10 C 0.218767 |
| 11 F −0.327241 | 11 N −0.729443 | 11 O −0.486554 | 11 C 0.285414 |
| 12 Cl 0.053556 | 12 C 0.345234 | 12 O −0.197979 | |
| 20 C 0.021858 | 13 O −0.221785 | 13 O −0.172743 | |
| 21 C −0.034281 | 14 O −0.210208 | 14 O −0.219698 | |
| 22 C 0.117931 | 15 F −0.340011 | 15 C 0.585341 | |
| 23 C −0.148508 | 16 N −0.126724 | 16 N −0.102687 | |
| 25 C 0.030298 | 17 O −0.438011 | 17 O −0.374256 | |
| 27 C 0.290148 | |||
| 29 O −0.612097 | |||
| 31 N −0.742655 | |||
| 32 C −0.283151 | |||
| 35 C −0.516801 | |||
| 39 C −0.296192 | |||
| 42 C −0.522817 | |||
| 46 Cl −0.904072 | |||
| 48 Cl −0.788808 |
| Drug | PubChem CID | Binding Affinity (Kcal/mol) | Amino Acids Residue of SARS-CoV-2 Mpro |
|---|---|---|---|
| 1 | 492405, favipiravir  | −4.06 | His64, Phe66, Leu67, Gln74, Arg76, Val77 |
| 2 | 6246, amodiaquine  | −7.77 | His41, Met49, Leu141, Cys145, His163, Met165,His172 |
| 3 | 101507, 2′-fluoro-2′-deoxycytidine  | −4.47 | Met165, Glu166, Pro168, Arg188, Gln189,Thr190 |
| 4 | 37542, ribavirin  | −4.69 | Thr26, His41, Phe140, Leu141, Asn142, Gly143, Ser144, Cys145, His163 |
Hydroxychloroquine  | −6.06 | His41, Met49, Met165, Glu166, Lue167, Asp187, Ala191, Gln192 | |
Remdesivir  | −4.96 | Cys145, His163, Met165, Glu166, His172, Gln189, Ala191 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hagar, M.; Ahmed, H.A.; Aljohani, G.; Alhaddad, O.A. Investigation of Some Antiviral N-Heterocycles as COVID 19 Drug: Molecular Docking and DFT Calculations. Int. J. Mol. Sci. 2020, 21, 3922. https://doi.org/10.3390/ijms21113922
Hagar M, Ahmed HA, Aljohani G, Alhaddad OA. Investigation of Some Antiviral N-Heterocycles as COVID 19 Drug: Molecular Docking and DFT Calculations. International Journal of Molecular Sciences. 2020; 21(11):3922. https://doi.org/10.3390/ijms21113922
Chicago/Turabian StyleHagar, Mohamed, Hoda A. Ahmed, Ghadah Aljohani, and Omaima A. Alhaddad. 2020. "Investigation of Some Antiviral N-Heterocycles as COVID 19 Drug: Molecular Docking and DFT Calculations" International Journal of Molecular Sciences 21, no. 11: 3922. https://doi.org/10.3390/ijms21113922
APA StyleHagar, M., Ahmed, H. A., Aljohani, G., & Alhaddad, O. A. (2020). Investigation of Some Antiviral N-Heterocycles as COVID 19 Drug: Molecular Docking and DFT Calculations. International Journal of Molecular Sciences, 21(11), 3922. https://doi.org/10.3390/ijms21113922