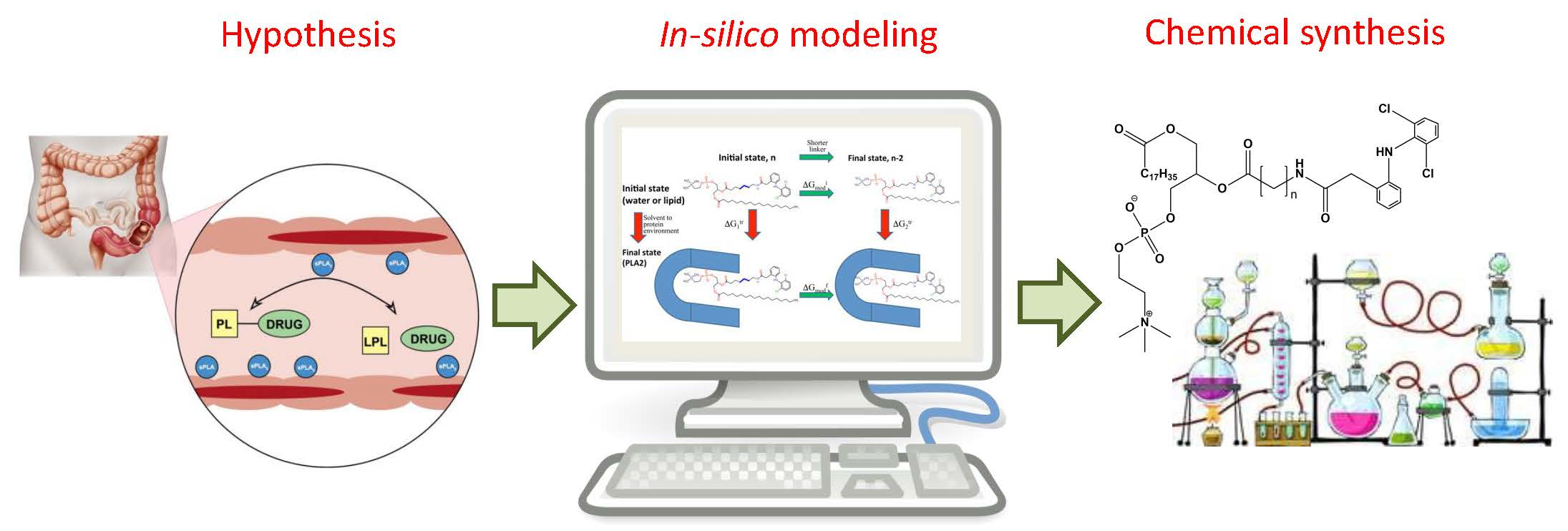
Computational Simulations to Guide Enzyme-Mediated Prodrug Activation




Abstract

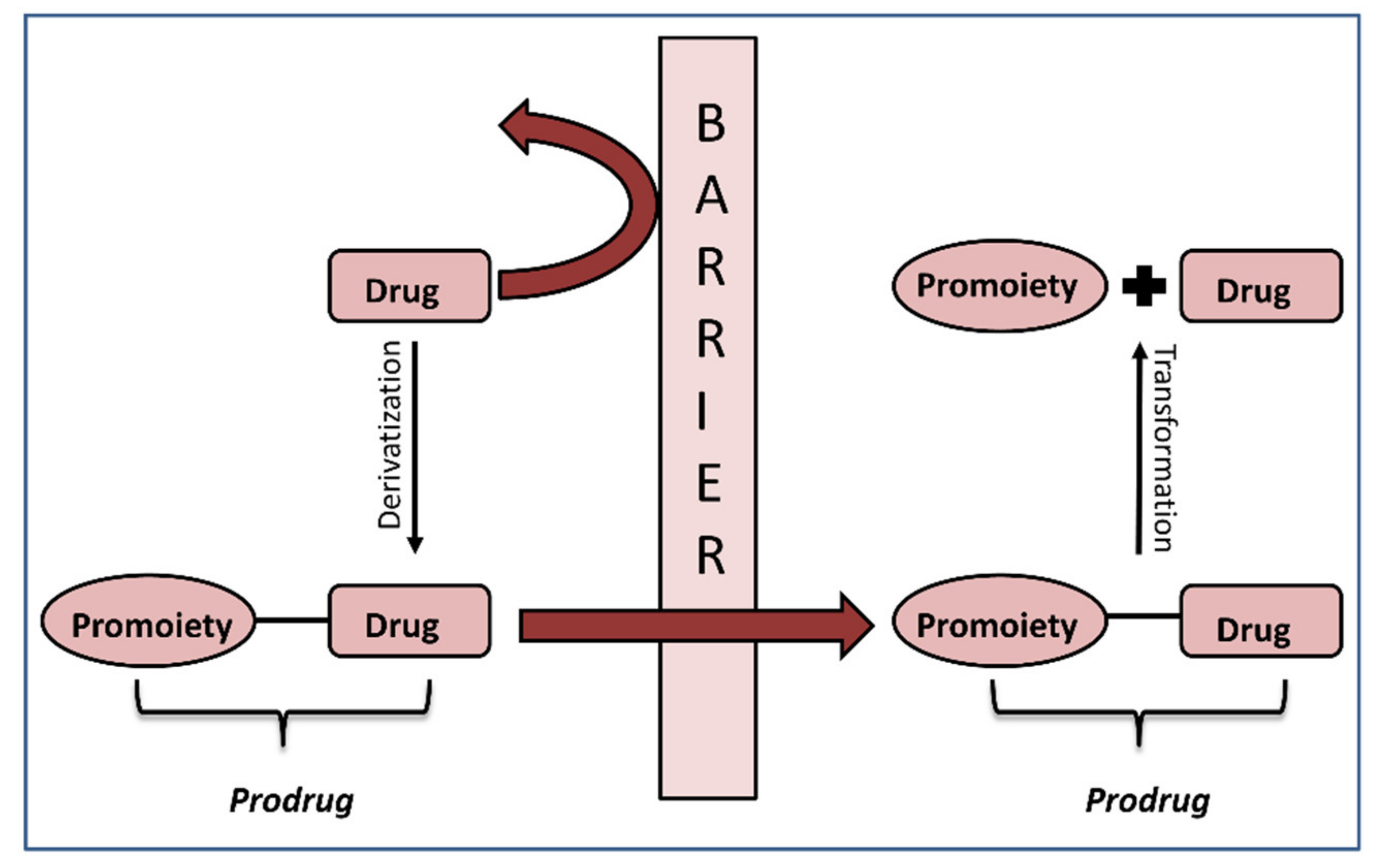
1. Introduction
2. In Silico Methods for Predicting Prodrug Activation
3. Computational Optimization of Enzyme-Mediated Prodrug Activation
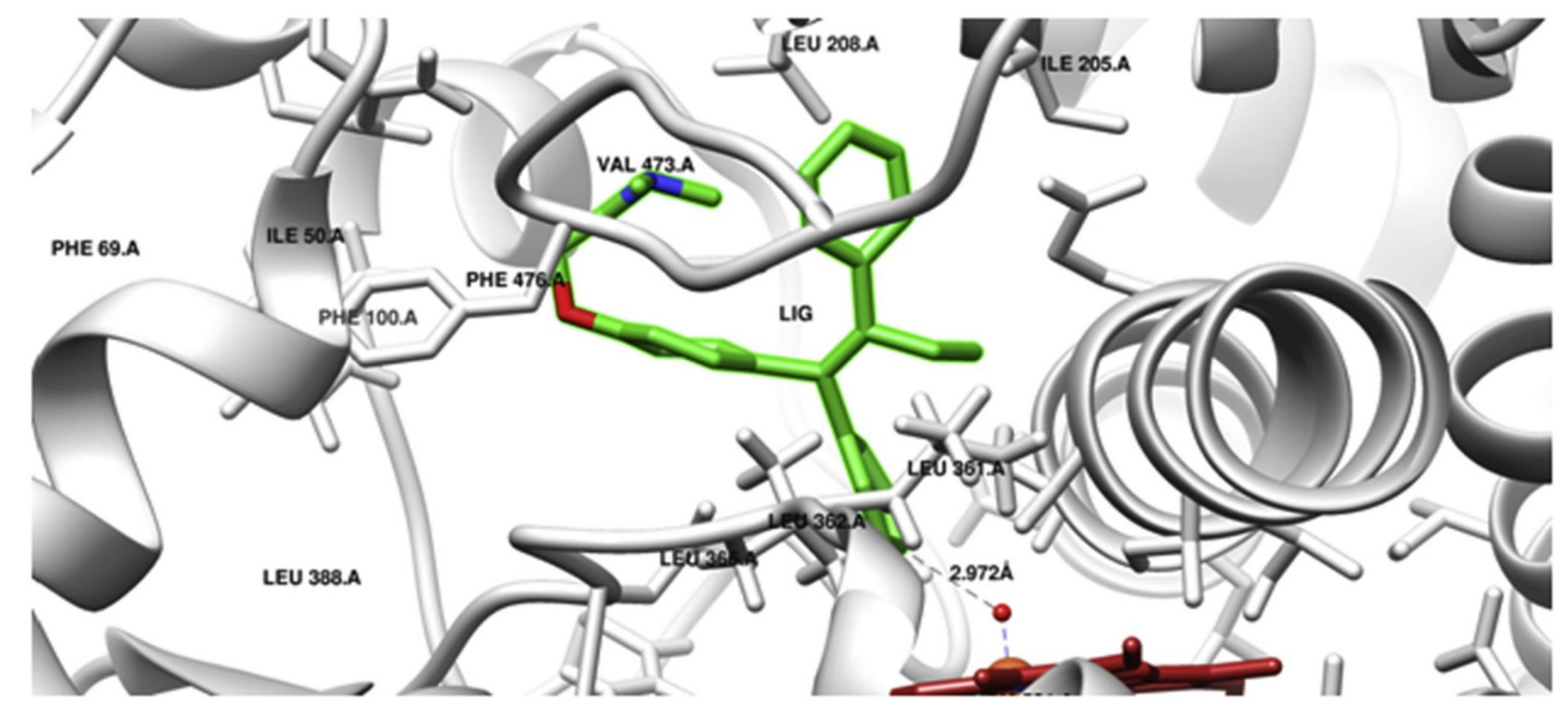
3.1. Cytochrome P450 (CYP450)
3.2. Carboxylesterase
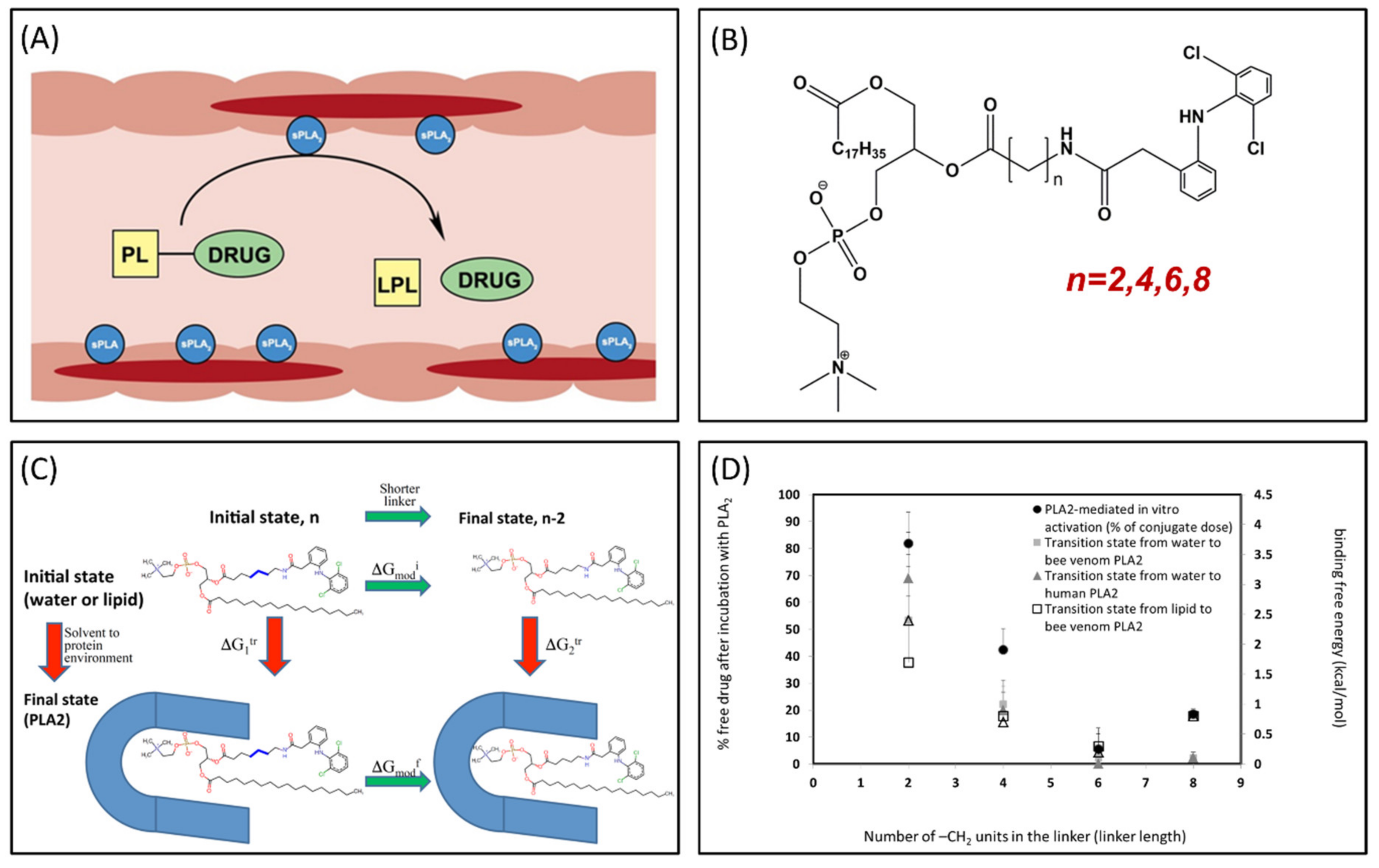
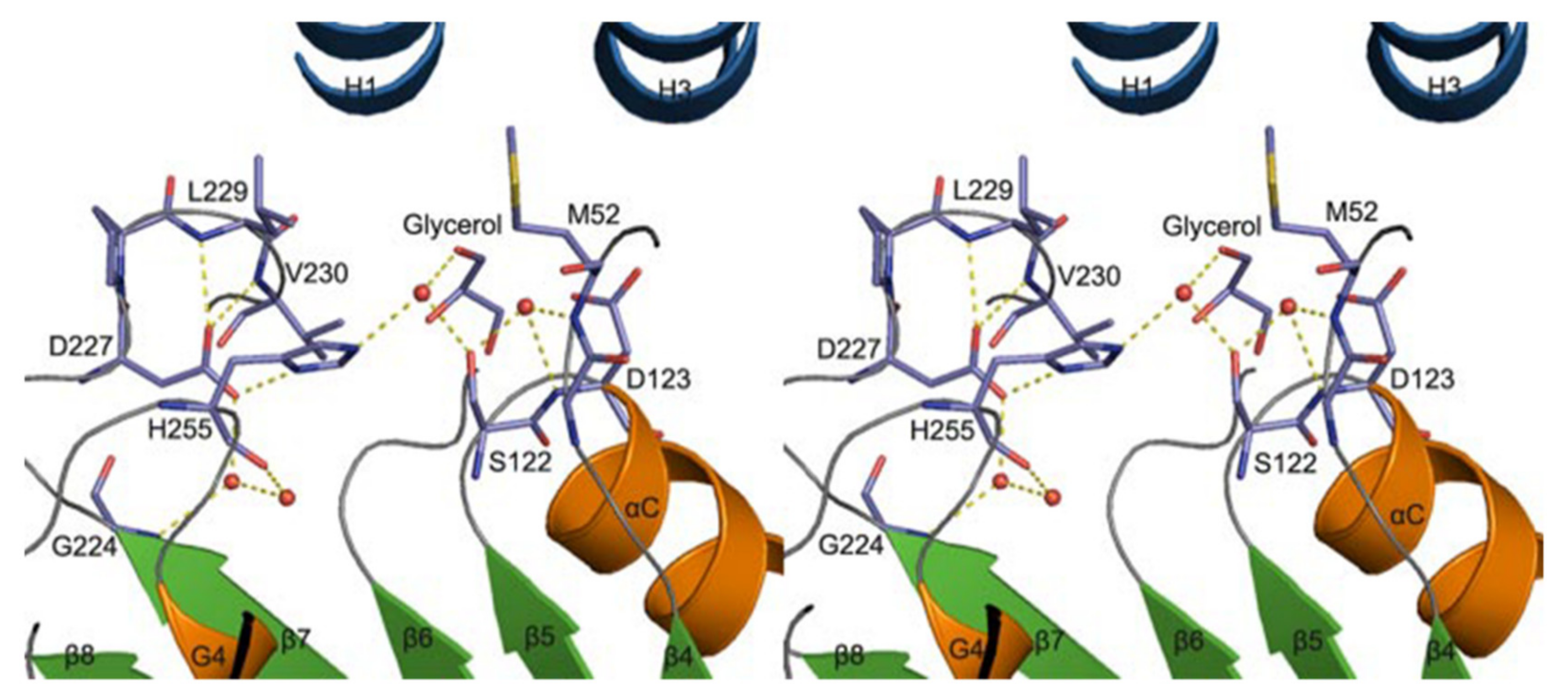
3.3. Phospholipase A2 (PLA2)
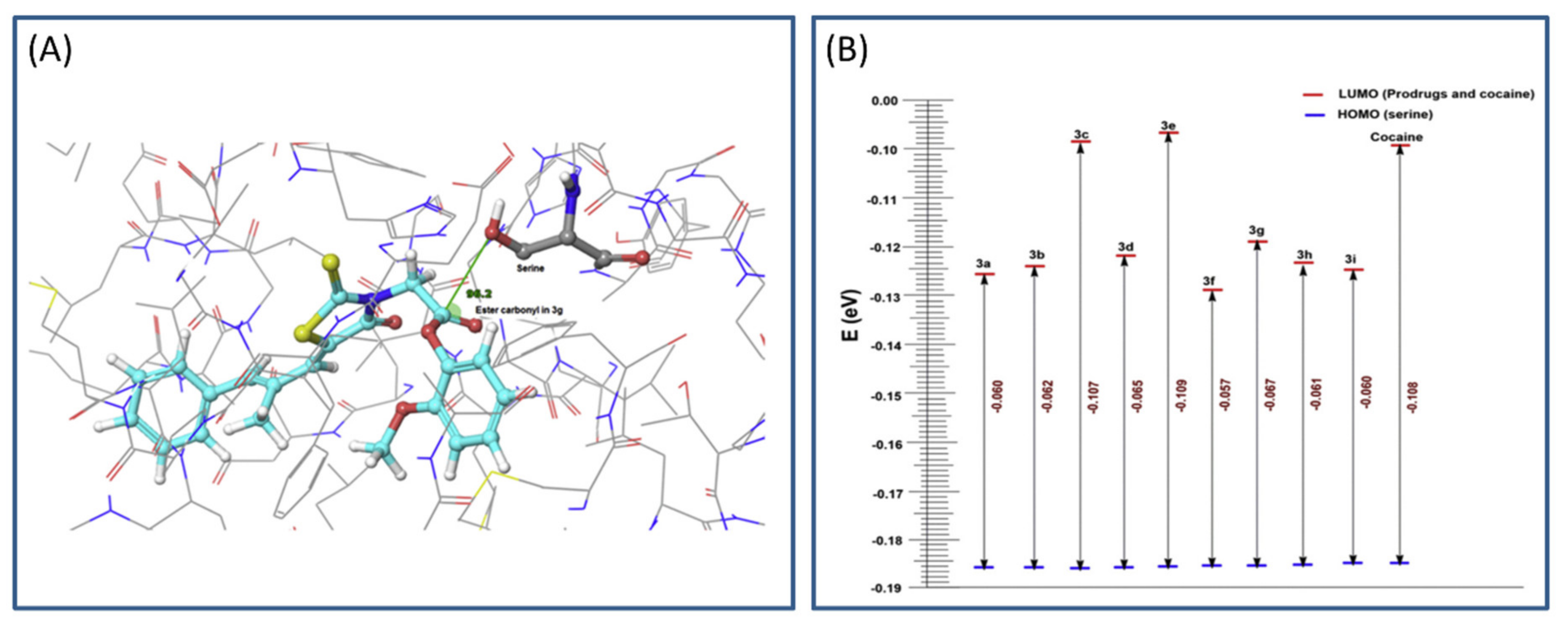
3.4. Cholinesterase
3.5. Alkaline Phosphatase
3.6. Human Valacyclovirase
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rautio, J.; Kumpulainen, H.; Heimbach, T.; Oliyai, R.; Oh, D.; Jarvinen, T.; Savolainen, J. Prodrugs: Design and clinical applications. Nat. Rev. Drug Discov. 2008, 7, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Testa, B. Prodrugs: Bridging pharmacodynamic/pharmacokinetic gaps. Curr. Opin. Chem. Biol. 2009, 13, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Zimmermann, E.M.; Ben-Shabat, S. Modern prodrug design for targeted oral drug delivery. Molecules 2014, 19, 16489–16505. [Google Scholar] [CrossRef] [PubMed]
- Stella, V.J. Prodrugs: Some thoughts and current issues. J. Pharm. Sci. 2010, 99, 4755–4765. [Google Scholar] [CrossRef] [PubMed]
- Stella, V.J.; Nti-Addae, K.W. Prodrug strategies to overcome poor water solubility. Adv. Drug Deliv. Rev. 2007, 59, 677–694. [Google Scholar] [CrossRef] [PubMed]
- Testa, B. Prodrug research: Futile or fertile? Biochem. Pharmacol. 2004, 68, 2097–2106. [Google Scholar] [CrossRef]
- Huttunen, K.M.; Raunio, H.; Rautio, J. Prodrugs—From serendipity to rational design. Pharmacol. Rev. 2011, 63, 750–771. [Google Scholar] [CrossRef]
- Dahan, A.; Markovic, M.; Aponick, A.; Zimmermann, E.M.; Ben-Shabat, S. The prospects of lipidic prodrugs: an old approach with an emerging future. Future Med. Chem. 2019, 11, 2563–2571. [Google Scholar] [CrossRef]
- Yang, Y.-H.; Aloysius, H.; Inoyama, D.; Chen, Y.; Hu, L.-Q. Enzyme-mediated hydrolytic activation of prodrugs. Acta Pharm. Sin. B 2011, 1, 143–159. [Google Scholar] [CrossRef]
- Rautio, J.; Meanwell, N.A.; Di, L.; Hageman, M.J. The expanding role of prodrugs in contemporary drug design and development. Nat. Rev. Drug Discov. 2018, 17, 559–587. [Google Scholar] [CrossRef]
- Dahan, A.; Khamis, M.; Agbaria, R.; Karaman, R. Targeted prodrugs in oral drug delivery: The modern molecular biopharmaceutical approach. Expert Opin. Drug Deliv. 2012, 9, 1001–1013. [Google Scholar] [CrossRef]
- Kapetanovic, I.M. Computer-aided drug discovery and development (CADDD): In silico-chemico-biological approach. Chem. Biol. Interact 2008, 171, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Lesyng, B.; McCammon, J.A. Molecular modeling methods. Basic techniques and challenging problems. Pharmacol. Ther. 1993, 60, 149–167. [Google Scholar] [CrossRef]
- Hernandez, B.; Luque, F.J.; Orozco, M. Mixed QM/MM molecular electrostatic potentials. J. Comput. Aided Mol. Des. 2000, 14, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Keinan, S.; Frush, E.H.; Shipman, W.J. Leveraging cloud computing for in-silico drug design using the quantum molecular design (QMD) framework. Comput. Sci. Eng. 2018, 20, 66–73. [Google Scholar] [CrossRef]
- Karaman, R.; Fattash, B.; Qtait, A. The future of prodrugs—Design by quantum mechanics methods. Expert Opin. Drug Deliv. 2013, 10, 713–729. [Google Scholar] [CrossRef]
- Chen, M.; Ko, H.Y.; Remsing, R.C.; Calegari Andrade, M.F.; Santra, B.; Sun, Z.; Selloni, A.; Car, R.; Klein, M.L.; Perdew, J.P.; et al. Ab initio theory and modeling of water. Proc. Natl. Acad. Sci. USA 2017, 114, 10846–10851. [Google Scholar] [CrossRef]
- Kamerlin, S.C.; Haranczyk, M.; Warshel, A. Progress in ab initio QM/MM free-energy simulations of electrostatic energies in proteins: Accelerated QM/MM studies of pKa, redox reactions and solvation free energies. J. Phys. Chem. B 2009, 113, 1253–1272. [Google Scholar] [CrossRef]
- Yeston, J. Whither the density in DFT calculations? Science 2017, 355, 35. [Google Scholar] [CrossRef]
- Hospital, A.; Goñi, J.R.; Orozco, M.; Gelpí, J.L. Molecular dynamics simulations: Advances and applications. Adv. Appl. Bioinform. Chem. 2015, 8, 37–47. [Google Scholar] [CrossRef]
- Field, M.J. Simulating enzyme reactions: Challenges and perspectives. J. Comput. Chem. 2002, 23, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Testa, B.; Kramer, S.D. The biochemistry of drug metabolism—An introduction: Part 2. Redox reactions and their enzymes. Chem. Biodivers 2007, 4, 257–405. [Google Scholar] [CrossRef] [PubMed]
- Ortiz de Montellano, P.R. Cytochrome P450-activated prodrugs. Future Med. Chem. 2013, 5, 213–228. [Google Scholar] [CrossRef]
- Huttunen, K.M.; Mahonen, N.; Raunio, H.; Rautio, J. Cytochrome P450-activated prodrugs: Targeted drug delivery. Curr. Med. Chem. 2008, 15, 2346–2365. [Google Scholar] [CrossRef] [PubMed]
- Yin, T.; Maekawa, K.; Kamide, K.; Saito, Y.; Hanada, H.; Miyashita, K.; Kokubo, Y.; Akaiwa, Y.; Otsubo, R.; Nagatsuka, K.; et al. Genetic variations of CYP2C9 in 724 Japanese individuals and their impact on the antihypertensive effects of losartan. Hypertens. Res. 2008, 31, 1549–1557. [Google Scholar] [CrossRef] [PubMed]
- Dodani, S.C.; Kiss, G.; Cahn, J.K.B.; Su, Y.; Pande, V.S.; Arnold, F.H. Discovery of a regioselectivity switch in nitrating P450s guided by molecular dynamics simulations and Markov models. Nat. Chem. 2016, 8, 419–425. [Google Scholar] [CrossRef]
- Louet, M.; Labbé, C.M.; Fagnen, C.; Aono, C.M.; Homem-de-Mello, P.; Villoutreix, B.O.; Miteva, M.A. Insights into molecular mechanisms of drug metabolism dysfunction of human CYP2C9*30. PLoS ONE 2018, 13, e0197249. [Google Scholar] [CrossRef]
- Manish, M.; Lynn, A.M.; Mishra, S. Cytochrome P450 2C9 polymorphism: Effect of amino acid substitutions on protein flexibility in the presence of tamoxifen. Comput. Biol. Chem. 2020, 84, 107166. [Google Scholar] [CrossRef]
- Merali, Z.; Ross, S.; Pare, G. The pharmacogenetics of carboxylesterases: CES1 and CES2 genetic variants and their clinical effect. Drug Metab. Drug Interact. 2014, 29, 143–151. [Google Scholar] [CrossRef]
- Munjal, N.S.; Shukla, R.; Singh, T.R. Chemometric approach to estimate kinetic properties of paclitaxel prodrugs and their substructures for solubility prediction through molecular modelling and simulation studies. J. Chemom. 2019, 33, e3181. [Google Scholar] [CrossRef]
- Vyas, B.; Choudhary, S.; Singh, P.K.; Singh, A.; Singh, M.; Verma, H.; Singh, H.; Bahadur, R.; Singh, B.; Silakari, O. Molecular dynamics/quantum mechanics guided designing of natural products based prodrugs of Epalrestat. J. Mol. Struct. 2018, 1171, 556–563. [Google Scholar] [CrossRef]
- Fukui, K.; Yonezawa, T.; Shingu, H. A molecular orbital theory of reactivity in aromatic hydrocarbons. J. Chem. Phys. 1952, 20, 722–725. [Google Scholar] [CrossRef]
- Minami, T.; Shinomura, Y.; Miyagawa, J.; Tojo, H.; Okamoto, M.; Matsuzawa, Y. Immunohistochemical localization of group II phospholipase A2 in colonic mucosa of patients with inflammatory bowel disease. Am. J. Gastroenterol. 1997, 92, 289–292. [Google Scholar] [PubMed]
- Minami, T.; Tojo, H.; Shinomura, Y.; Matsuzawa, Y.; Okamoto, M. Increased group II phospholipase A2 in colonic mucosa of patients with Crohn’s disease and ulcerative colitis. Gut 1994, 35, 1593–1598. [Google Scholar] [CrossRef]
- Yarla, N.S.; Bishayee, A.; Vadlakonda, L.; Chintala, R.; Duddukuri, G.R.; Reddanna, P.; Dowluru, K.S. Phospholipase A2 isoforms as novel targets for prevention and treatment of inflammatory and oncologic diseases. Curr. Drug Targets 2016, 17, 1940–1962. [Google Scholar] [CrossRef]
- Markovic, M.; Ben-Shabat, S.; Aponick, A.; Zimmermann, E.M.; Dahan, A. Lipids and lipid-processing pathways in drug delivery and therapeutics. Int. J. Mol. Sci. 2020, 21, 3248. [Google Scholar] [CrossRef]
- Markovic, M.; Ben-Shabat, S.; Keinan, S.; Aponick, A.; Zimmermann, E.M.; Dahan, A. Prospects and challenges of phospholipid-based prodrugs. Pharmaceutics 2018, 10, 210. [Google Scholar] [CrossRef]
- Dahan, A.; Duvdevani, R.; Shapiro, I.; Elmann, A.; Finkelstein, E.; Hoffman, A. The oral absorption of phospholipid prodrugs: In Vivo and In Vitro mechanistic investigation of trafficking of a lecithin-valproic acid conjugate following oral administration. J. Control. Release J. Control. Release Soc. 2008, 126, 1–9. [Google Scholar] [CrossRef]
- Dahan, A.; Ben-Shabat, S.; Cohen, N.; Keinan, S.; Kurnikov, I.; Aponick, A.; Zimmermann, E.M. Phospholipid-based prodrugs for drug targeting in inflammatory bowel disease: Computational Optimization and In-Vitro Correlation. Curr. Top. Med. Chem. 2016, 16, 2543–2548. [Google Scholar] [CrossRef]
- Dahan, A.; Markovic, M.; Keinan, S.; Kurnikov, I.; Aponick, A.; Zimmermann, E.M.; Ben-Shabat, S. Computational modeling and in-vitro/in-silico correlation of phospholipid-based prodrugs for targeted drug delivery in inflammatory bowel disease. J. Comput. Aided Mol. Des. 2017, 31, 1021–1028. [Google Scholar] [CrossRef]
- Markovic, M.; Ben-Shabat, S.; Keinan, S.; Aponick, A.; Zimmermann, E.M.; Dahan, A. Molecular modeling-guided design of phospholipid-based prodrugs. Int. J. Mol. Sci. 2019, 20, 2210. [Google Scholar] [CrossRef]
- Markovic, M.; Dahan, A.; Keinan, S.; Kurnikov, I.; Aponick, A.; Zimmermann, E.M.; Ben-Shabat, S. Phospholipid-based prodrugs for colon-targeted drug delivery: Experimental study and in-silico simulations. Pharmaceutics 2019, 11, 186. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Markovic, M.; Epstein, S.; Cohen, N.; Zimmermann, E.M.; Aponick, A.; Ben-Shabat, S. Phospholipid-drug conjugates as a novel oral drug targeting approach for the treatment of inflammatory bowel disease. Eur. J. Pharm. Sci. J. Eur. Fed. Pharm. Sci. 2017, 108, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Duvdevani, R.; Dvir, E.; Elmann, A.; Hoffman, A. A novel mechanism for oral controlled release of drugs by continuous degradation of a phospholipid prodrug along the intestine: In-Vivo and In-Vitro evaluation of an indomethacin-lecithin conjugate. J. Control. Release J. Control. Release Soc. 2007, 119, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Dvir, E.; Elman, A.; Simmons, D.; Shapiro, I.; Duvdevani, R.; Dahan, A.; Hoffman, A.; Friedman, J.E. DP-155, a lecithin derivative of indomethacin, is a novel nonsteroidal antiinflammatory drug for analgesia and Alzheimer’s disease therapy. CNS Drug Rev. 2007, 13, 260–277. [Google Scholar] [CrossRef] [PubMed]
- Dvir, E.; Friedman, J.E.; Lee, J.Y.; Koh, J.Y.; Younis, F.; Raz, S.; Shapiro, I.; Hoffman, A.; Dahan, A.; Rosenberg, G.; et al. A novel phospholipid derivative of indomethacin, DP-155 [mixture of 1-steroyl and 1-palmitoyl-2-{6-[1-(p-chlorobenzoyl)-5-methoxy-2-methyl-3-indolyl acetamido]hexanoyl}-sn-glycero-3-phosophatidyl [corrected] choline], shows superior safety and similar efficacy in reducing brain amyloid beta in an Alzheimer’s disease model. J. Pharmacol. Exp. Ther. 2006, 318, 1248–1256. [Google Scholar] [CrossRef]
- Linderoth, L.; Fristrup, P.; Hansen, M.; Melander, F.; Madsen, R.; Andresen, T.L.; Peters, G.H. Mechanistic study of the sPLA2-mediated hydrolysis of a thio-ester pro anticancer ether lipid. J. Am. Chem. Soc. 2009, 131, 12193–12200. [Google Scholar] [CrossRef]
- Barré, A.; Azzouz, R.; Gembus, V.; Papamicaël, C.; Levacher, V. Design, synthesis, and In Vitro biological activities of a bio-oxidizable prodrug to deliver both ChEs and DYRK1A inhibitors for AD therapy. Molecules 2019, 24, 1264. [Google Scholar] [CrossRef]
- Chen, K.; Aowad, A.F.; Adelstein, S.J.; Kassis, A.I. Molecular-docking-guided design, synthesis, and biologic evaluation of radioiodinated quinazolinone prodrugs. J. Med. Chem. 2007, 50, 663–673. [Google Scholar] [CrossRef]
- Sun, J.; Dahan, A.; Walls, Z.F.; Lai, L.; Lee, K.D.; Amidon, G.L. Specificity of a prodrug-activating enzyme hVACVase: The leaving group effect. Mol. Pharm. 2010, 7, 2362–2368. [Google Scholar] [CrossRef]
- Kim, I.; Chu, X.Y.; Kim, S.; Provoda, C.J.; Lee, K.D.; Amidon, G.L. Identification of a human valacyclovirase: Biphenyl hydrolase-like protein as valacyclovir hydrolase. J. Biol. Chem. 2003, 278, 25348–25356. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Crippen, G.M.; Amidon, G.L. Structure and specificity of a human valacyclovir activating enzyme: A homology model of BPHL. Mol. Pharm. 2004, 1, 434–446. [Google Scholar] [CrossRef] [PubMed]
- Lai, L.; Xu, Z.; Zhou, J.; Lee, K.-D.; Amidon, G.L. Molecular basis of prodrug activation by human valacyclovirase, an alpha-amino acid ester hydrolase. J. Biol. Chem. 2008, 283, 9318–9327. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.V.; Gupta, D.; Sun, J.; Dahan, A.; Tsume, Y.; Hilfinger, J.; Lee, K.D.; Amidon, G.L. Enhancing the intestinal membrane permeability of zanamivir: A carrier mediated prodrug approach. Mol. Pharm. 2011, 8, 2358–2367. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Dahan, A.; Amidon, G.L. Enhancing the intestinal absorption of molecules containing the polar guanidino functionality: a double-targeted prodrug approach. J. Med. Chem. 2010, 53, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Karaman, R.; Dajani, K.K.; Qtait, A.; Khamis, M. Prodrugs of acyclovir--a computational approach. Chem. Biol. Drug Des. 2012, 79, 819–834. [Google Scholar] [CrossRef]
- Sousa, S.F.; Ribeiro, A.J.; Coimbra, J.T.; Neves, R.P.; Martins, S.A.; Moorthy, N.S.; Fernandes, P.A.; Ramos, M.J. Protein-ligand docking in the new millennium—A retrospective of 10 years in the field. Curr. Med. Chem. 2013, 20, 2296–2314. [Google Scholar] [CrossRef]
- Kohen, A.; Cannio, R.; Bartolucci, S.; Klinman, J.P. Enzyme dynamics and hydrogen tunnelling in a thermophilic alcohol dehydrogenase. Nature 1999, 399, 496–499. [Google Scholar] [CrossRef]
- Mulholland, A.J. Modelling enzyme reaction mechanisms, specificity and catalysis. Drug Discov. Today 2005, 10, 1393–1402. [Google Scholar] [CrossRef]
- Ridder, L.; Harvey, J.N.; Rietjens, I.M.C.M.; Vervoort, J.; Mulholland, A.J. Ab Initio QM/MM modeling of the hydroxylation step in p-Hydroxybenzoate hydroxylase. J. Phys. Chem. B 2003, 107, 2118–2126. [Google Scholar] [CrossRef]
- Ridder, L.; Rietjens, I.M.C.M.; Vervoort, J.; Mulholland, A.J. Quantum mechanical/molecular mechanical free energy simulations of the glutathione S-Transferase (M1-1) reaction with phenanthrene 9,10-oxide. J. Am. Chem. Soc. 2002, 124, 9926–9936. [Google Scholar] [CrossRef] [PubMed]
- Pinzi, L.; Rastelli, G. Molecular docking: Shifting paradigms in drug discovery. Int. J. Mol. Sci. 2019, 20, 4331. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Truhlar, D.G. Quantum mechanical methods for enzyme kinetics. Annu. Rev. Phys. Chem. 2002, 53, 467–505. [Google Scholar] [CrossRef] [PubMed]
- Kollman, P. Free energy calculations: Applications to chemical and biochemical phenomena. Chem. Rev. 1993, 93, 2395–2417. [Google Scholar] [CrossRef]
- Najjar, A.; Karaman, R. The prodrug approach in the era of drug design. Expert Opin. Drug Deliv. 2019, 16, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Markovic, M.; Ben-Shabat, S.; Keinan, S.; Aponick, A.; Zimmermann, E.M.; Dahan, A. Lipidic prodrug approach for improved oral drug delivery and therapy. Med. Res. Rev. 2019, 39, 579–607. [Google Scholar] [CrossRef]
- Han, H.K.; Amidon, G.L. Targeted prodrug design to optimize drug delivery. AAPS Pharmsci 2000, 2, E6. [Google Scholar] [CrossRef]
- Pedersen, P.J.; Adolph, S.K.; Subramanian, A.K.; Arouri, A.; Andresen, T.L.; Mouritsen, O.G.; Madsen, R.; Madsen, M.W.; Peters, G.H.; Clausen, M.H. Liposomal formulation of retinoids designed for enzyme triggered release. J. Med. Chem. 2010, 53, 3782–3792. [Google Scholar] [CrossRef]
- Linderoth, L.; Andresen, T.L.; Jørgensen, K.; Madsen, R.; Peters, G.H. Molecular basis of phospholipase A2 activity toward phospholipids with sn-1 substitutions. Biophys. J. 2008, 94, 14–26. [Google Scholar] [CrossRef]
- Karaman, R.; Hallak, H. Computer-assisted design of pro-drugs for antimalarial atovaquone. Chem. Biol. Drug Des. 2010, 76, 350–360. [Google Scholar] [CrossRef]
- Chen, K.; Wang, K.; Kirichian, A.M.; Al Aowad, A.F.; Iyer, L.K.; Adelstein, S.J.; Kassis, A.I. In silico design, synthesis, and biological evaluation of radioiodinated quinazolinone derivatives for alkaline phosphatase-mediated cancer diagnosis and therapy. Mol. Cancer Ther. 2006, 5, 3001–3013. [Google Scholar] [CrossRef] [PubMed]
- Hermann, J.C.; Hensen, C.; Ridder, L.; Mulholland, A.J.; Holtje, H.D. Mechanisms of antibiotic resistance: QM/MM modeling of the acylation reaction of a class A beta-lactamase with benzylpenicillin. J. Am. Chem. Soc. 2005, 127, 4454–4465. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecular Docking | Quantum Mechanics | Molecular Mechanics | Free Energy Perturbation | |
|---|---|---|---|---|
| Purpose | Predicting ligand-target interactions at a molecular level [62] | Study reactions in molecular systems [63] | Protein structure and dynamics (not applicable to chemical reactions) [59] | Computing free energy differences between an enzyme and a substrate [64] |
| Calculation | Algorithms | Schrödinger equation | Empirical methods | Laws of classical dynamics |
| Applications | Various systems | Small system size (e.g., isolated active sites) | Large systems (proteins, large crystal structures and relatively large solvated systems) | Proteins and other biological macromolecules |
| Limitations | Low reliability, cannot be used when protein dynamics should not be ignored [62] | Difficulty in understanding large biological system (used for up to a few hundred atoms); high computational time and resource [63] | Large number of unique torsion angles present in different molecules [65] | High computational costs; absence of optimized tools and representation standards [20] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Markovic, M.; Ben-Shabat, S.; Dahan, A. Computational Simulations to Guide Enzyme-Mediated Prodrug Activation. Int. J. Mol. Sci. 2020, 21, 3621. https://doi.org/10.3390/ijms21103621
Markovic M, Ben-Shabat S, Dahan A. Computational Simulations to Guide Enzyme-Mediated Prodrug Activation. International Journal of Molecular Sciences. 2020; 21(10):3621. https://doi.org/10.3390/ijms21103621
Chicago/Turabian StyleMarkovic, Milica, Shimon Ben-Shabat, and Arik Dahan. 2020. "Computational Simulations to Guide Enzyme-Mediated Prodrug Activation" International Journal of Molecular Sciences 21, no. 10: 3621. https://doi.org/10.3390/ijms21103621
APA StyleMarkovic, M., Ben-Shabat, S., & Dahan, A. (2020). Computational Simulations to Guide Enzyme-Mediated Prodrug Activation. International Journal of Molecular Sciences, 21(10), 3621. https://doi.org/10.3390/ijms21103621