Proprotein Convertase Subtilisin/Kexin Type 9, Angiopoietin-Like Protein 8, Sortilin, and Cholesteryl Ester Transfer Protein—Friends of Foes for Psoriatic Patients at the Risk of Developing Cardiometabolic Syndrome?




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
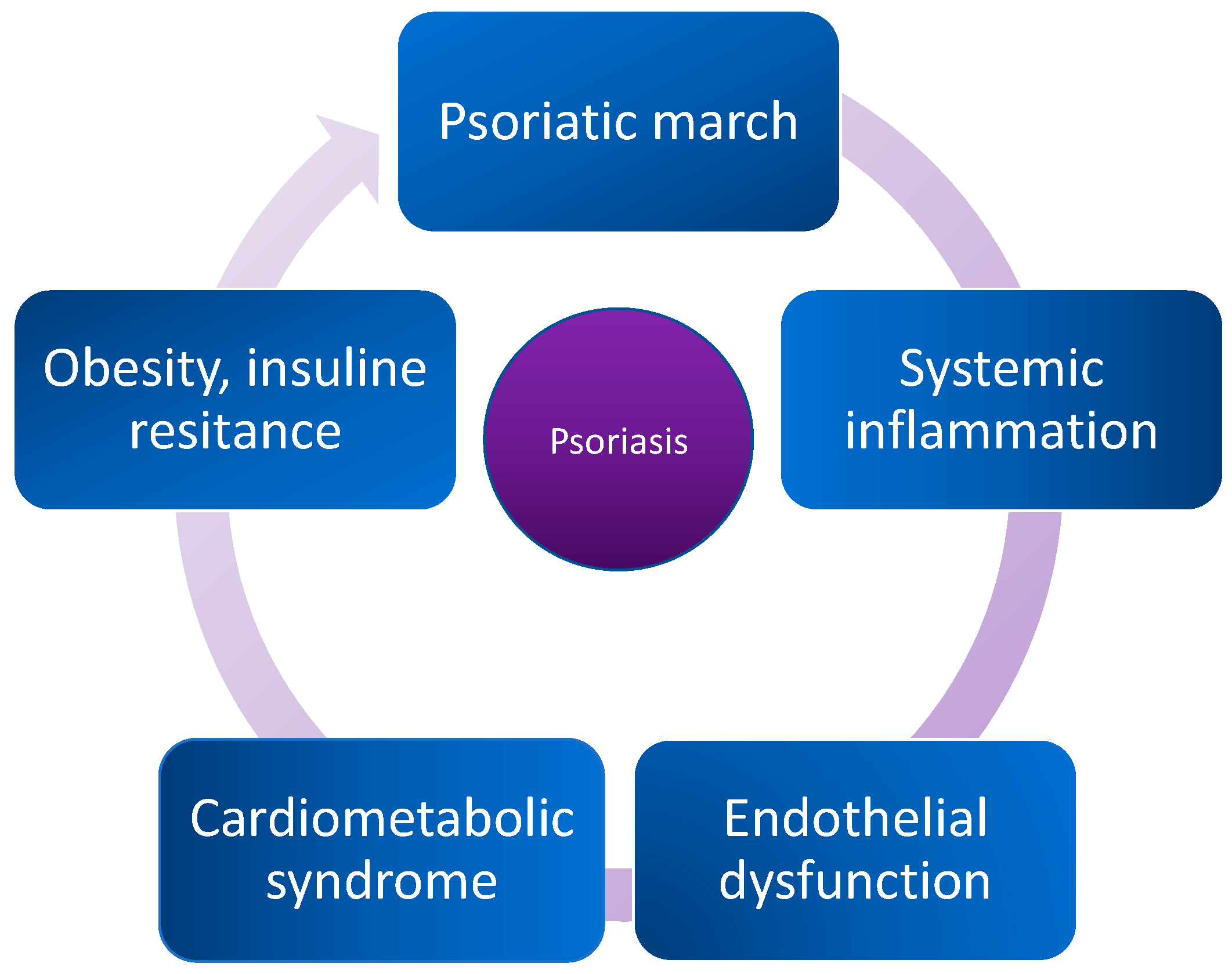
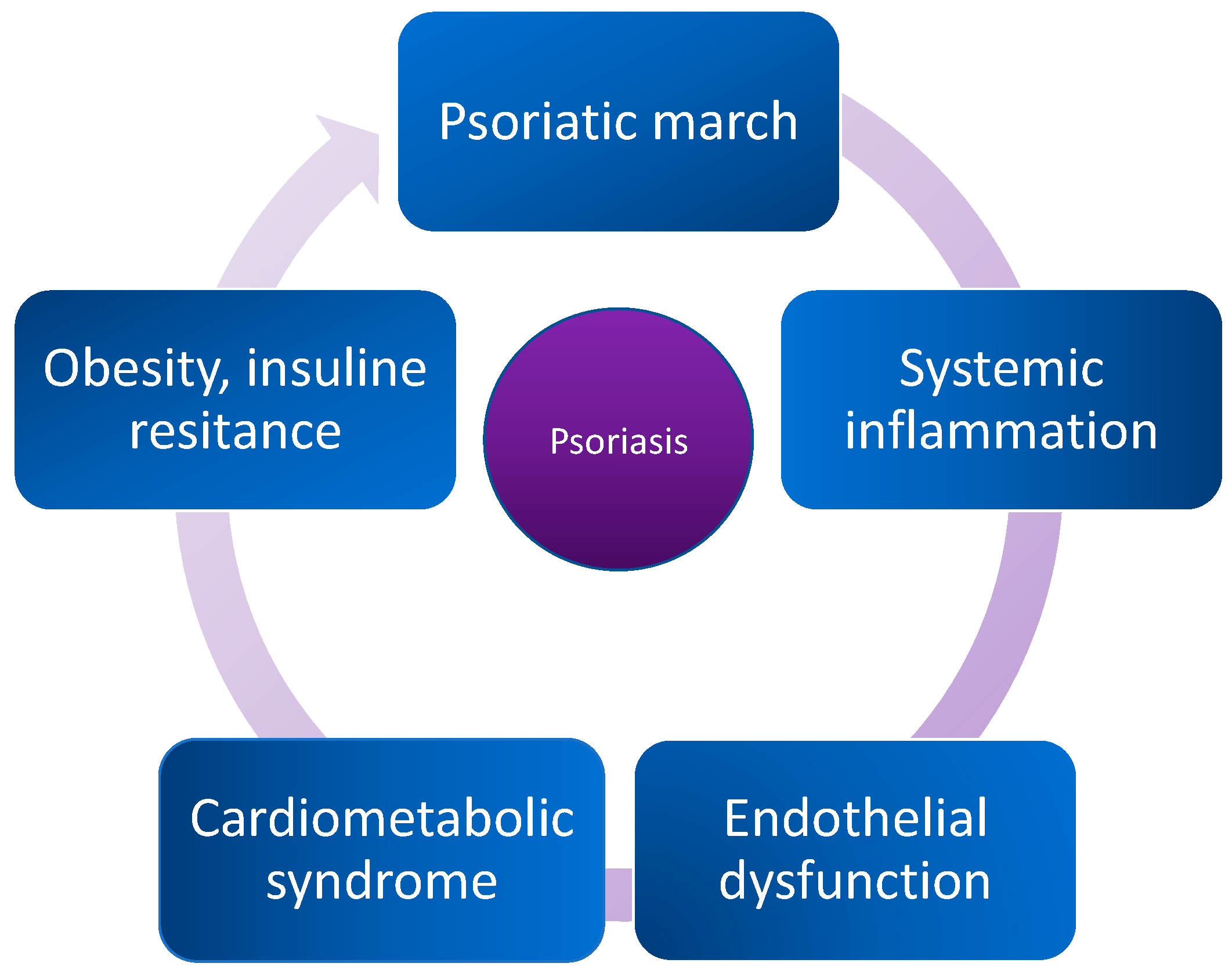
:1. Introduction
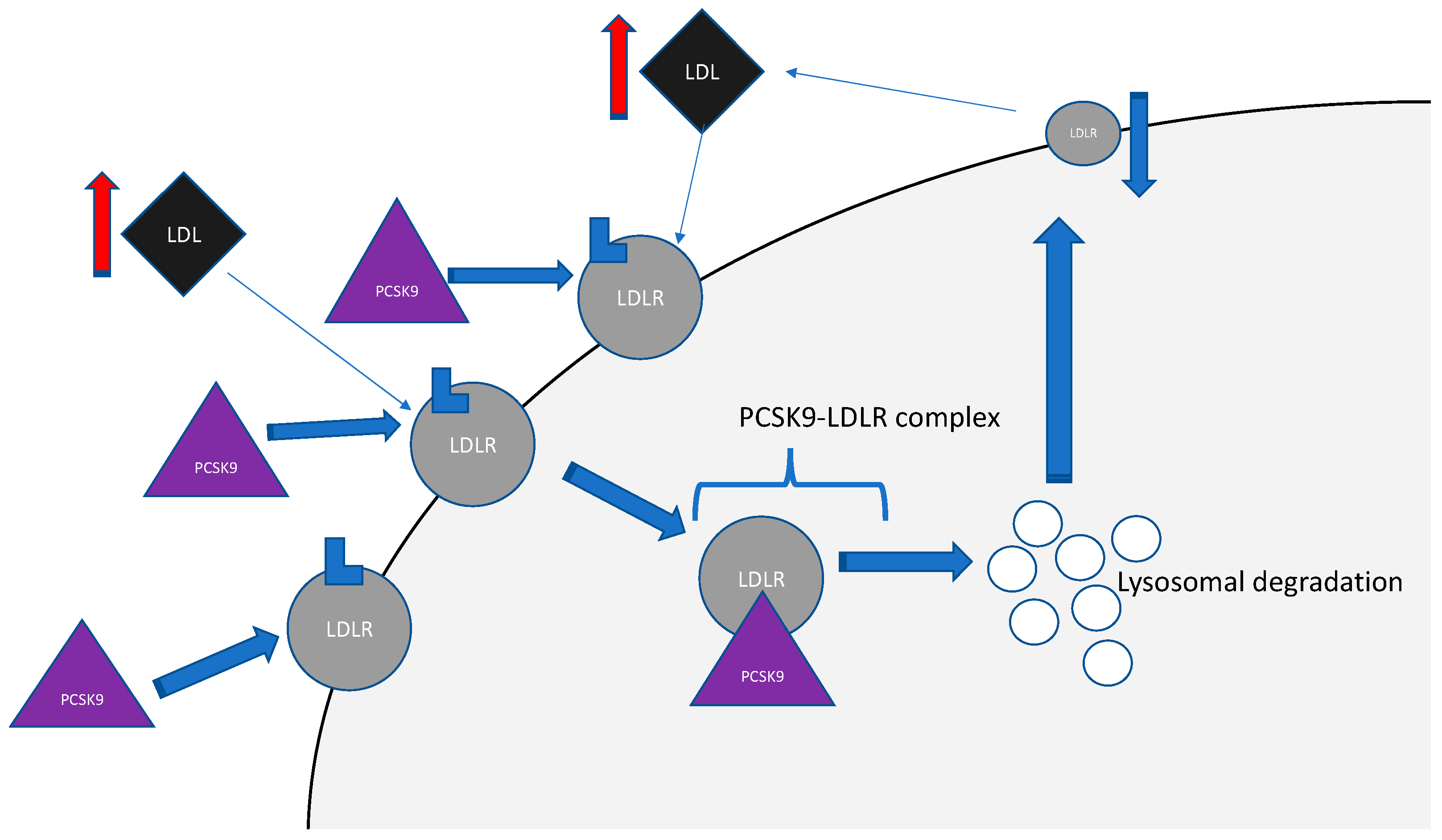
2. Proprotein Convertase Subtilisin/Kexin Type-9 (PCSK9)
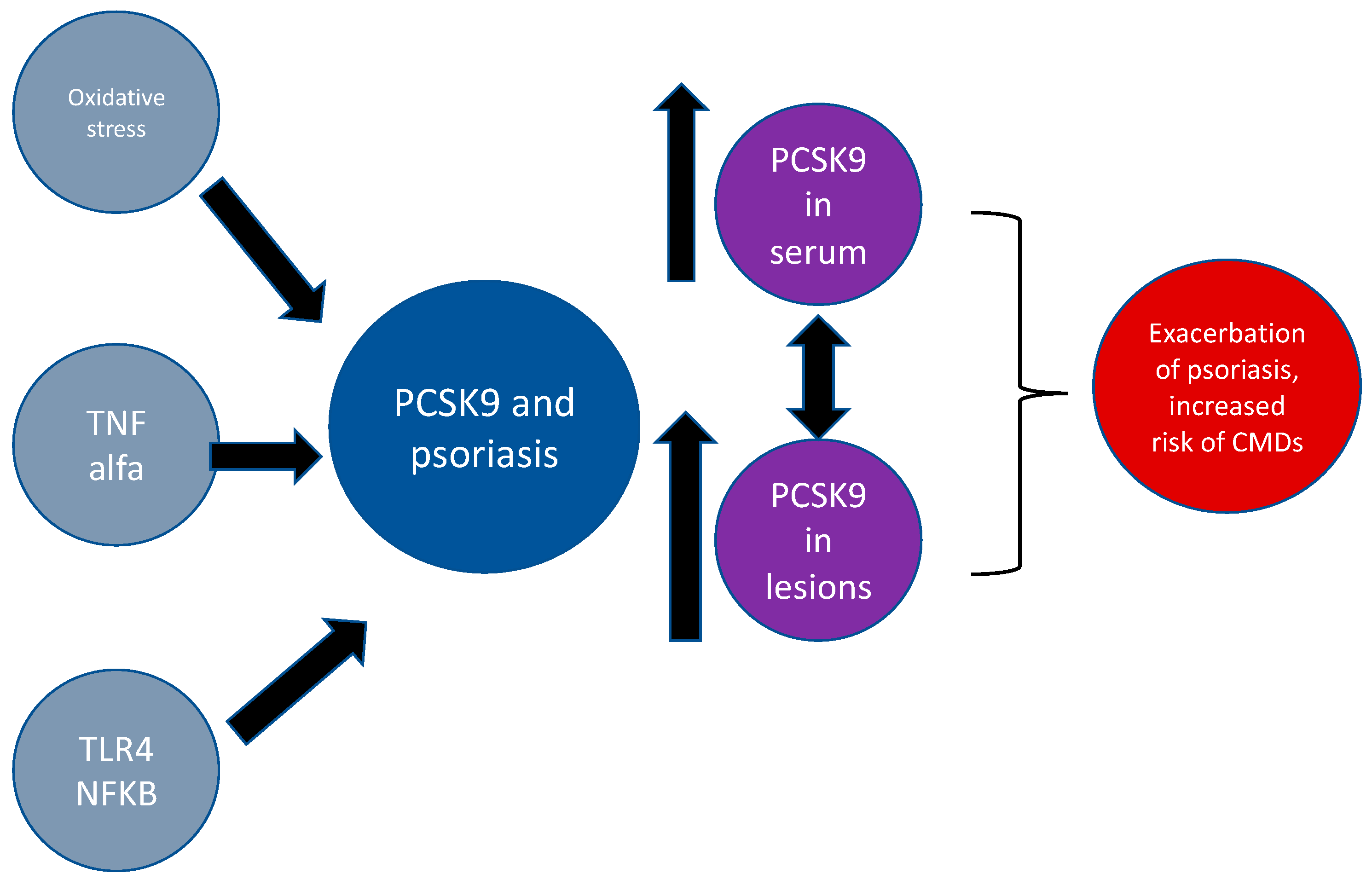
PCSK9 and Psoriasis
3. Angiopoietin-Like Protein 8 (ANGPL8)
ANGPL8 and Psoriasis
4. Sortilin
Sortilin and Psoriasis
5. Cholesteryl Ester Transfer Protein (CEPT)
Cholesteryl Ester Transfer Protein and Psoriasis
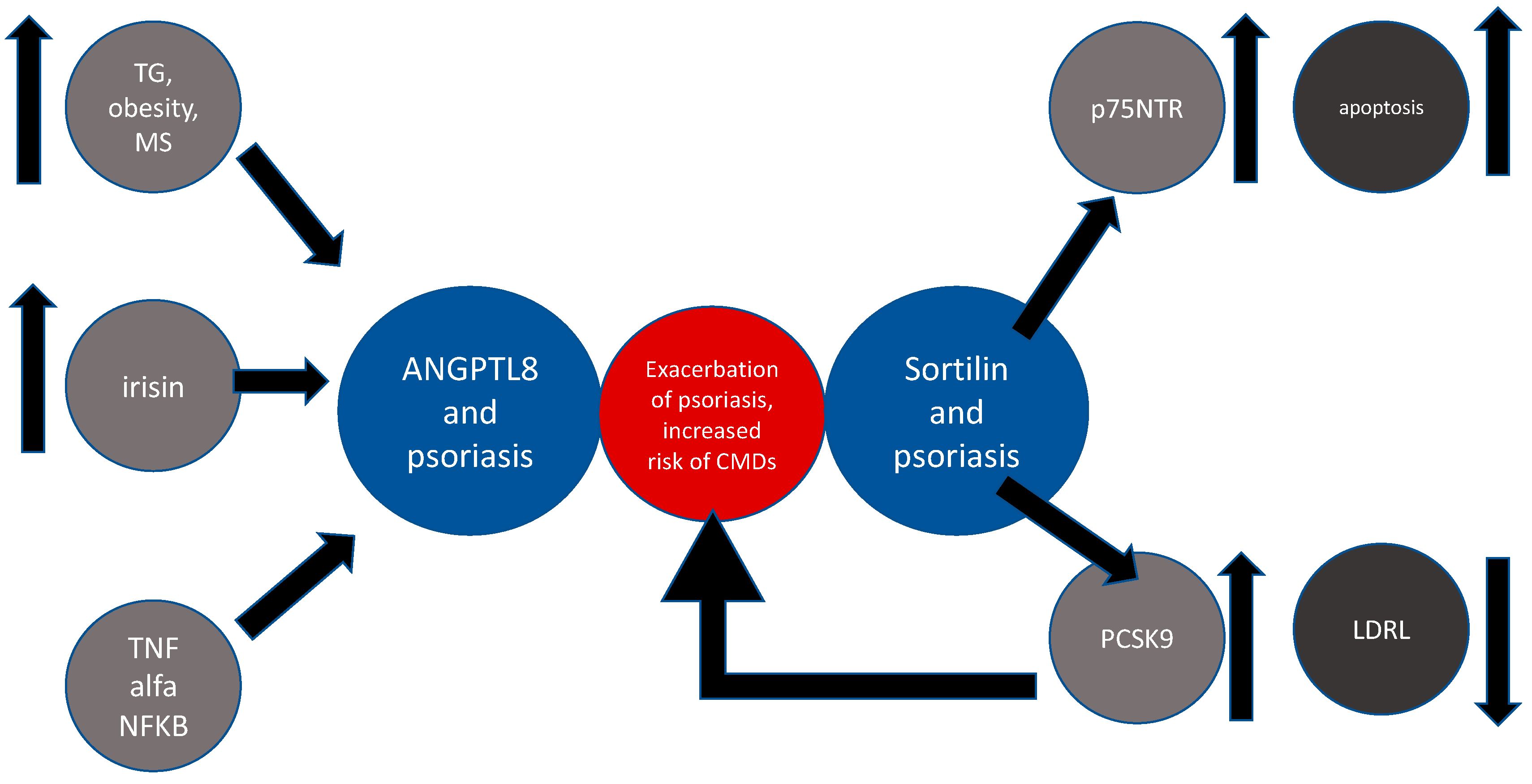
6. Conclusions
Conflicts of Interest
References
- Farley, E.; Menter, A. Psoriasis: Comorbidities and associations. Giornale Ital. Dermatol. Venereol. 2011, 146, 9–15. [Google Scholar] [PubMed]
- Fernández-Armenteros, J.; Gómez-Arbonés, X.; Buti-Soler, M.; Betriu-Bars, A.; Sanmartin-Novell, V.; Ortega-Bravo, M.; Martínez-Alonso, M.; Gari, E.; Portero-Otín, M.; Santamaria-Babi, L.; et al. Psoriasis, metabolic syndrome and cardiovascular risk factors. A population-based study. J. Eur. Acad. Dermatol. Venereol. 2018, 33, 128–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.; Dogra, S.; Shafiq, N.; Bhansali, A.; Malhotra, S. Prevalence of Metabolic Syndrome in Psoriasis and Levels of Interleukin-6 and Tumor Necrosis Factor-α in Psoriasis Patients with Metabolic Syndrome: Indian Tertiary Care Hospital Study. Int. J. Appl. Basic Med Res. 2017, 7, 169–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gisondi, P.; Tessari, G.; Conti, A.; Piaserico, S.; Schianchi, S.; Peserico, A.; Giannetti, A.; Girolomoni, G. Prevalence of metabolic syndrome in patients with psoriasis: A hospital-based case control study. Br. J. Dermatol. 2007, 157, 68–73. [Google Scholar] [CrossRef]
- Suárez-Fariñas, M.; Fuentes-Duculan, J.; Lowes, M.; Krueger, J.G. Resolved Psoriasis Lesions Retain Expression of a Subset of Disease-Related Genes. J. Investig. Dermatol. 2010, 131, 391–400. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, E.J.; Harskamp, C.T.; Armstrong, A.W. Psoriasis and Major Adverse Cardiovascular Events: A Systematic Review and Meta-Analysis of Observational Studies. J. Am. Hear. Assoc. 2013, 2, 000062. [Google Scholar] [CrossRef] [Green Version]
- Kiluk, P.; Baran, A.; Flisiak, I. Role of omentin and vaspin in metabolic diseases in association with psoriasis. Dermatol. Rev. 2017, 5, 519–528. [Google Scholar] [CrossRef]
- Wolk, R.; Bertolet, M.; Singh, P.; Brooks, M.M.; Pratley, R.E.; Frye, R.L.; Mooradian, A.D.; Rutter, M.K.; Calvin, A.D.; Chaitman, B.R.; et al. Prognostic Value of Adipokines in Predicting Cardiovascular Outcome: Explaining the Obesity Paradox. In Mayo Clinic Proceedings; Elsevier: Amsterdam, The Netherlands, 2016; Volume 91, pp. 858–866. [Google Scholar]
- Boehncke, W.-H.; Boehncke, S.; Tobin, A.M.; Kirby, B. The ‘psoriatic march’: A concept of how severe psoriasis may drive cardiovascular comorbidity. Exp. Dermatol. 2011, 20, 303–307. [Google Scholar] [CrossRef]
- Mantovani, A.; Gisondi, P.; Lonardo, A.; Targher, G. Relationship between Non-Alcoholic Fatty Liver Disease and Psoriasis: A Novel Hepato-Dermal Axis? Int. J. Mol. Sci. 2016, 17, 217. [Google Scholar] [CrossRef] [Green Version]
- Akhyani, M.; Ehsani, A.; Robati, R.M.; Robati, A. The lipid profile in psoriasis: A controlled study. J. Eur. Acad. Dermatol. Venereol. 2007, 21, 1330–1332. [Google Scholar] [CrossRef]
- Shih, C.-M.; Huang, C.-Y.; Wang, K.-H.; Huang, C.-Y.; Wei, P.-L.; Chang, Y.-J.; Hsieh, C.-K.; Liu, K.-T.; Lee, A.-W. Oxidized Low-Density Lipoprotein-Deteriorated Psoriasis Is Associated with the Upregulation of Lox-1 Receptor and Il-23 Expression In Vivo and In Vitro. Int. J. Mol. Sci. 2018, 19, 2610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahlehoff, O.; Gislason, G.; Lindhardsen, J.; Charlot, M.G.; Jørgensen, C.H.; Olesen, J.B.; Bretler, D.-M.; Skov, L.; Torp-Pedersen, C.; Hansen, T.W. Psoriasis Carries an Increased Risk of Venous Thromboembolism: A Danish Nationwide Cohort Study. PLoS ONE 2011, 6, e18125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horreau, C.; Pouplard, C.; Brenaut, E.; Barnetche, T.; Misery, L.; Cribier, B.; Jullien, D.; Aractingi, S.; Aubin, F.; Joly, P.; et al. Cardiovascular morbidity and mortality in psoriasis and psoriatic arthritis: A systematic literature review. J. Eur. Acad. Dermatol. Venereol. 2013, 27, 12–29. [Google Scholar] [CrossRef] [PubMed]
- Seidah, N.G.; Benjannet, S.; Wickham, L.; Marcinkiewicz, J.; Jasmin, S.B.; Stifani, S.; Basak, A.; Prat, A.; Chrétien, M. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): Liver regeneration and neuronal differentiation. Proc. Natl. Acad. Sci. USA 2003, 100, 928–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seidah, N.G.; Chrétien, M. Proprotein and prohormone convertases: A family of subtilases generating diverse bioactive polypeptides. Brain Res. 1999, 848, 45–62. [Google Scholar] [CrossRef]
- Turpeinen, H.; Ortutay, Z.; Pesu, M. Genetics of the First Seven Proprotein Convertase Enzymes in Health and Disease. Curr. Genom. 2013, 14, 453–467. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.; Ahmed, S. Emerging role of proprotein convertase subtilisin/kexin type-9 (PCSK-9) in inflammation and diseases. Toxicol. Appl. Pharmacol. 2019, 370, 170–177. [Google Scholar] [CrossRef]
- Glerup, S.; Schulz, R.; Laufs, U.; Schlüter, K.-D. Physiological and therapeutic regulation of PCSK9 activity in cardiovascular disease. Basic Res. Cardiol. 2017, 112, 32. [Google Scholar] [CrossRef] [Green Version]
- Siedah, N.G. The proprotein convertases, 20 years later. Methods Mol. Biol. 2011, 768, 23–57. [Google Scholar]
- Schulz, R.; Schulter, K.D. PCSK9 targets impornatnt for lipid metabolism. Clin. Res. Cardiol. Suppl. 2017, 12, 2–11. [Google Scholar] [CrossRef] [Green Version]
- Lagace, T.A.; Curtis, D.E.; Garuti, R.; McNutt, M.C.; Park, S.W.; Prather, H.B.; Anderson, N.N.; Ho, Y.K.; Hammer, R.E.; Horton, J.D. Secreted PCSK9 decreases the numer of LDL receptors in hepatocytes and in livers of parabiotic mice. J. Clin. Investig. 2016, 116, 2995–3005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, P.N.; Defesche, J.; Fouchier, S.W.; Bruckert, E.; Luc, G.; Cariou, B.; Sjouke, B.; Leren, T.P.; Harada-Shiba, M.; Mabuchi, H.; et al. Characterization of Autosomal Dominant Hypercholesterolemia Caused by PCSK9 Gain of Function Mutations and Its Specific Treatment with Alirocumab, a PCSK9 Monoclonal Antibody. Circ. Cardiovasc. Genet. 2015, 8, 823–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sucajtys-Szulc, E.; Szolkiewicz, M.; Świerczyński, J.; Rutkowski, B. Up-regulation of liver Pcsk9 gene expression as a possible cause of hypercholesterolemia in experimental chronic renal failure. Mol. Cell. Biochem. 2015, 411, 281–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, M.E.; Levenson, A.E.; Sun, X.; Liao, W.-H.; Rutkowski, J.M.; De Ferranti, S.D.; Schumacher, V.A.; Scherer, P.E.; Salant, D.J.; Biddinger, S.B.; et al. The Role of Proprotein Convertase Subtilisin/Kexin Type 9 in Nephrotic Syndrome-Associated Hypercholesterolemia. Circulation 2016, 134, 61–72. [Google Scholar] [CrossRef] [Green Version]
- Ozkan, C.; Akturk, M.; Altınova, A.E.; Cerit, E.T.; Gulbahar, O.; Yalcin, M.M.; Cakir, N.; Toruner, F.B. Proprotein convertase subtilisin/kexin type 9 (PCSK9), soluble lectin-like oxidized LDL receptor 1 (sLOX-1) and ankle brachial index in patients with differentiated thyroid cancer. Endocr. J. 2015, 62, 1091–1099. [Google Scholar] [CrossRef] [Green Version]
- Costet, P.; Cariou, B.; Lambert, G.; Lalanne, F.; Lardeux, B.; Jarnoux, A.-L.; Grefhorst, A.; Staels, B.; Krempf, M. Hepatic PCSK9 Expression Is Regulated by Nutritional Status via Insulin and Sterol Regulatory Element-binding Protein 1c. J. Boil. Chem. 2006, 281, 6211–6218. [Google Scholar] [CrossRef] [Green Version]
- Ruscica, M.; Ferri, N.; Macchi, C.; Meroni, M.; Lanti, C.; Ricci, C.; Maggioni, M.; Fracanzani, A.L.; Badiali, S.; Fargion, S.; et al. Liver fat accumulation is associated with circulating PCSK9. Ann. Med. 2016, 48, 1–8. [Google Scholar] [CrossRef]
- Jeenduang, N. Circulating PCSK9 concentrations are increased in postmenopausal women with the metabolic syndrome. Clin. Chim. Acta 2019, 494, 151–156. [Google Scholar] [CrossRef]
- Fang, C.; Luo, T.; Lin, L. Elevation of serum proprotein convertase subtilisin/kexin type 9 (PCSK9) concentrations and its possible atherogenic role in patients with systemic lupus erythematosus. Ann. Transl. Med. 2018, 6, 452. [Google Scholar] [CrossRef]
- Ferraz-Amaro, I.; López-Mejías, R.; Ubilla, B.; Genre, F.; Tejera-Segura, B.; De Vera-González, A.M.; González-Rivero, A.F.; Olmos, J.M.; Hernández, J.L.; Llorca, J.; et al. Proprotein convertase subtilisin/kexin type 9 in rheumatoid arthritis. Clin. Exp. Rheumatol. 2016, 34, 1013–1019. [Google Scholar]
- Vlachopoulos, C.; Terentes-Printzios, D.; Georgiopoulos, G.; Skoumas, I.; Koutagiar, I.; Ioakeimidis, N.; Stefanadis, C.; Tousoulis, D. Prediction of cardiovascular events with levels of proprotein convertase subtilisin/kexin type 9: A systematic review and meta-analysis. Atheroscler. 2016, 252, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.-H.; Li, T.-H.; Peng, J.; Zheng, J.; Li, T.-T.; Liu, L.-S.; Jiang, Z.; Zheng, X.-L. PCSK9: A novel inflammation modulator in atherosclerosis? J. Cell. Physiol. 2018, 234, 2345–2355. [Google Scholar] [CrossRef] [PubMed]
- Gencer, B.; Montecucco, F.; Nanchen, D.; Carbone, F.; Klingenberg, R.; Vuilleumier, N.; Aghlmandi, S.; Heg, D.; Räber, L.; Auer, R.; et al. Prognostic value of PCSK9 levels in patients with acute coronary syndromes. Eur. Hear. J. 2015, 37, 546–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceballos-Macías, J.J.; Madriz-Prado, R.; Cárdenas, N.A.V.; Aguilar-Salinas, C.; Tusié-Luna, M.T.; Flores-Real, J.A.; Ortega-Gutiérrez, G.; Vargas-Sánchez, J.; Lara-Sánchez, C.; Hernández-Moreno, A. Use of PCSK9 Inhibitor in a Mexican Boy with Compound Heterozygous Familial Hypercholesterolemia: A Case Report. J. Endocr. Soc. 2019, 4, bvz018. [Google Scholar] [CrossRef] [Green Version]
- Dwivedi, D.J.; Grin, P.; Khan, M.; Prat, A.; Zhou, J.; Fox-Robichaud, A.E.; Seidah, N.G.; Liaw, P.C. Differential Expression of PCSK9 Modulates Infection, Inflammation, and Coagulation in a Murine Model of Sepsis. Shock 2016, 46, 672–680. [Google Scholar] [CrossRef]
- Walley, K.R.; Thain, K.R.; Russell, J.A.; Reilly, M.; Meyer, N.J.; Ferguson, J.F.; Christie, J.D.; Nakada, T.-A.; Fjell, C.; Thair, S.A.; et al. PCSK9 is a critical regulator of the innate immune response and septic shock outcome. Sci. Transl. Med. 2014, 6, 258ra143. [Google Scholar] [CrossRef] [Green Version]
- Rannikko, J.; Sanz, D.J.; Ortutay, Z.; Seiskari, T.; Aittoniemi, J.; Huttunen, R.; Syrjänen, J.; Pesu, M. Reduced plasma PCSK 9 response in patients with bacteraemia is associated with mortality. J. Intern. Med. 2019, 286, 553–561. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, R.-X.; Li, S.; Zhu, C.-G.; Guo, Y.-L.; Sun, J.; Li, J.-J. Association of plasma small dense LDL cholesterol with PCSK9 levels in patients with angiographically proven coronary artery disease. Nutr. Metab. Cardiovasc. Dis. 2015, 25, 426–433. [Google Scholar] [CrossRef]
- Koren, M.J.; Lundqvist, P.; Bolognese, M.; Neutel, J.M.; Monsalvo, M.L.; Yang, J.; Kim, J.B.; Scott, R.; Wasserman, S.M.; Bays, H.; et al. Anti-PCSK9 Monotherapy for Hypercholesterolemia. J. Am. Coll. Cardiol. 2014, 63, 2531–2540. [Google Scholar] [CrossRef] [Green Version]
- Colhoun, H.M.; Robinson, J.G.; Farnier, M.; Cariou, B.; Blom, D.J.; Kereiakes, D.J.; Lorenzato, C.; Pordy, R.; Chaudhari, U. Efficacy and safety of alirocumab, a fully human PCSK9 monoclonal antibody, in high cardiovascular risk patients with poorly controlled hypercholesterolemia on maximally tolerated doses of statins: Rationale and design of the ODYSSEY COMBO I and II trials. BMC Cardiovasc. Disord. 2014, 14, 121. [Google Scholar] [CrossRef] [Green Version]
- Cao, A.; Wu, M.; Li, H.; Liu, J. Janus kinase activation by cytokine oncostatin M decreases PCSK9 expression in liver cells. J. Lipid Res. 2010, 52, 518–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Yang, X.; Li, Q.; Zeng, P.; Liu, Y.; Liu, L.; Chen, Y.; Yu, M.; Ma, C.; Li, X.; et al. Activation of Adiponectin Receptor Regulates Proprotein Convertase Subtilisin/Kexin Type 9 Expression and Inhibits Lesions in ApoE-Deficient Mice. Arter. Thromb. Vasc. Boil. 2017, 37, 1290–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakry, O.; Seleit, I.; El Gayed, E.A.; Ghanem, M. Peroxisome proliferator-activated receptor-γ gene polymorphism in psoriasis and its relation to obesity, metabolic syndrome, and narrowband ultraviolet B response: A case–control study in Egyptian patients. Indian J. Dermatol. 2019, 64, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Schuluter, K.D. ox-LDL and Angiotensin: Cooperative effects via induction of PCSK9 in cardiomiocytes. Clin. Res. Cardiol. 2016, 105, V1278. [Google Scholar]
- Baran, A.; Flisiak, I.; Jaroszewicz, J.; Świderska, M. Effect of psoriasis activity on serum adiponectin and leptin levels. Adv. Dermatol. Allergol. 2015, 32, 101–106. [Google Scholar] [CrossRef] [Green Version]
- Luan, C.; Chen, X.; Zhu, Y.; Osland, J.M.; Gerber, S.D.; Dodds, M.; Hu, Y.; Chen, M.; Yuan, R. Potentiation of Psoriasis-Like Inflammation by PCSK9. J. Investig. Dermatol. 2019, 139, 859–867. [Google Scholar] [CrossRef] [Green Version]
- Krahel, J.A.; Baran, A.; Kamiński, T.W.; Maciaszek, M.; Flisiak, I. Methotrexate Decreases the Level of PCSK9—A Novel Indicator of the Risk of Proatherogenic Lipid Profile in Psoriasis. The Preliminary Data. J. Clin. Med. 2020, 9, 910. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.; Peng, D. ANGPTL8: An Important Regulator in Metabolic Disorders. Front. Endocrinol. 2018, 9, 169. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.-Y.; Pang, X.-W.; Yu, S.; Su, Y.-R.; Wang, H.-C.; Yin, Y.-H.; Wang, Y.-D.; Chen, W.F. Identification of genes differentially expressed in human hepatocellular carcinoma by a modified suppression subtractive hybridization method. Int. J. Cancer 2004, 112, 239–248. [Google Scholar] [CrossRef]
- Ren, G.; Kim, J.Y.; Smas, C.M. Identification of RIFL, a novel adipocyte-enriched insulin target gene with a role in lipid metabolism. Am. J. Physiol. Metab. 2012, 303, E334–E351. [Google Scholar] [CrossRef] [Green Version]
- Abu-Farha, M.; Al-Khairi, I.; Cherian, P.; Chandy, B.; Sriraman, D.; Alhubail, A.; Al-Refaei, F.; AlTerki, A.; Abubaker, J. Increased ANGPTL3, 4 and ANGPTL8/betatrophin expression levels in obesity and T2D. Lip. Health Dis. 2016, 15, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R. Lipasin, a novel nutritionally-regulated liver-enriched factor that regulates serum triglyceride levels. Biochem. Biophys. Res. Commun. 2012, 424, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Quagliarini, F.; Wang, Y.; Kozlitina, J.; Grishin, N.V.; Hyde, R.; Boerwinkle, E.; Valenzuela, D.M.; Murphy, A.J.; Cohen, J.C.; Hobbs, H.H. Atypical angiopoietin-like protein that regulates ANGPTL3. Proc. Natl. Acad. Sci. USA 2012, 109, 19751–19756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, P.; Park, J.-S.; Melton, D.A. Retraction Notice to: Betatrophin: A Hormone that Controls Pancreatic β Cell Proliferation. Cell 2016, 168, 326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gusarova, V.; Alexa, C.A.; Na, E.; Stevis, P.E.; Xin, Y.; Bonner-Weir, S.; Cohen, J.C.; Hobbs, H.H.; Murphy, A.J.; Yancopoulos, G.D.; et al. ANGPTL8/betatrophin does not control pancreatic beta cell expansion. Cell 2014, 159, 691–696. [Google Scholar] [CrossRef] [Green Version]
- Cox, A.R.; Lam, C.J.; Bonnyman, C.W.; Chavez, J.; Rios, J.S.; Kushner, J.A. Angiopoietin-like protein 8 (ANGPTL8)/betatrophin overexpression does not increase beta cell proliferation in mice. Diabetol 2015, 58, 1523–1531. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Qin, Y.; Wang, N.; Yang, L.; Yuan, G. The Relationship between Circulating ANGPTL8/Betatrophin Concentrations and Adult Obesity: A Meta-Analysis. Dis. Markers 2019, 2019, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Abu-Farha, M.; Sriraman, D.; Cherian, P.; AlKhairi, I.; Elkum, N.; Behbehani, K.; Abubaker, J. Circulating ANGPTL8/Betatrophin Is Increased in Obesity and Reduced after Exercise Training. PLoS ONE 2016, 11, e0147367. [Google Scholar] [CrossRef]
- Abu-Farha, M.; Abubaker, J.; Alkhairi, I.; Cherian, P.; Noronha, F.; Kavalakatt, S.; Khadir, A.; Behbehani, K.; Al-Arouj, M.; Bennakhi, A.; et al. Circulating angiopoietin-like protein 8 (betatrophin) association with HsCRP and metabolic syndrome. Cardiovasc. Diabetol. 2016, 15, 25. [Google Scholar] [CrossRef] [Green Version]
- Boström, P.A.; Fernández-Real, J.-M. Irisin, the metabolic syndrome and follistatin in humans. Nat. Rev. Endocrinol. 2013, 10, 11–12. [Google Scholar] [CrossRef]
- Wang, S.; Hong, X.; Tu, Z.; Yuan, G. Angiopoietin-like protein 8: An attractive biomarker for the evaluation of subjects with insulin resistance and related disorders. Diabetes Res. Clin. Pr. 2017, 133, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Baran, A.; Myśliwiec, H.; Kiluk, P.; Świderska, M.; Flisiak, I. Serum irisin levels in patients with psoriasis. J. Dermatol. Treat. 2016, 28, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Guo, X.; Yan, W.; Chen, Y.; Ke, M.; Cheng, C.; Zhu, X.; Xue, W.; Zhou, Q.; Zheng, L.; et al. ANGPTL8 negatively regulates NF-κB activation by facilitating selective autophagic degradation of IKKγ. Nat. Commun. 2017, 8, 2164. [Google Scholar] [CrossRef]
- Conlon, D.M. Role of sortilin in lipid metabolism. Curr. Opin. Lipidol. 2019, 30, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Petersen, C.M.; Nielsen, M.S.; Nykjaer, A.; Jacobsen, L.; Tommerup, N.; Rasmussen, H.H.; Røigaard, H.; Gliemann, J.; Madsen, P.S.; Moestrup, S.K. Molecular Identification of a Novel Candidate Sorting Receptor Purified from Human Brain by Receptor-associated Protein Affinity Chromatography. J. Boil. Chem. 1997, 272, 3599–3605. [Google Scholar] [CrossRef] [Green Version]
- Samani, N.J.; Erdmann, J.; Hall, I.P.; Hengstenberg, C.; Mangino, M.; Mayer, B.; Dixon, R.J.; Meitinger, T.; Braund, P.; Wichmann, H.-E.; et al. Genomewide association analysis of coronary artery disease. N. Engl. J. Med. 2007, 357, 443–453. [Google Scholar] [CrossRef] [Green Version]
- Su, X.; Peng, D. New insight into sortilin in controlling lipid metabolism and the risk of atherogenesis. Boil. Rev. 2019, 95, 232–243. [Google Scholar] [CrossRef]
- Musunuru, K.; Strong, A.; Frank-Kamenetsky, M.; Lee, N.E.; Ahfeldt, T.; Sachs, K.V.; Li, X.; Li, H.; Kuperwasser, N.; Ruda, V.M.; et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature 2010, 466, 714–719. [Google Scholar] [CrossRef]
- Willnow, T.E.; Kjolby, M.; Nykjaer, A. Sortilins: New players in lipoprotein metabolism. Curr. Opin. Lipidol. 2011, 22, 79–85. [Google Scholar] [CrossRef]
- E Sparks, C.; Sparks, R.P.; Sparks, J. The enigmatic role of sortilin in lipoprotein metabolism. Curr. Opin. Lipidol. 2015, 26, 598–600. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Yang, J.; Xie, W.; Peng, T.; Lv, Y. Complicated trafficking behaviors involved in paradoxical regulation of sortilin in lipid metabolism. J. Cell. Physiol. 2019, 235, 3258–3269. [Google Scholar] [CrossRef] [PubMed]
- Carlo, A.-S. Sortilin, a novel APOE receptor implicated in Alzheimer disease. Prion 2013, 7, 378–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, T.J.; Ahn, C.H.; Kim, B.-R.; Kim, K.M.; Moon, J.H.; Lim, S.; Park, K.S.; Lim, C.; Jang, H.C.; Choi, S.H. Circulating sortilin level as a potential biomarker for coronary atherosclerosis and diabetes mellitus. Cardiovasc. Diabetol. 2017, 16, 92. [Google Scholar] [CrossRef]
- Gustafsen, C.; Kjolby, M.; Nyegaard, M.; Mattheisen, M.; Lundhede, J.; Buttenschøn, H.; Mors, O.; Bentzon, J.F.; Madsen, P.S.; Nykjaer, A.; et al. The Hypercholesterolemia-Risk Gene SORT1 Facilitates PCSK9 Secretion. Cell Metab. 2014, 19, 310–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, D.; Yang, Y.; Peng, D.-Q. Increased sortilin and its independent effect on circulating proprotein convertase subtilisin/kexin type 9 (PCSK9) in statin-naive patients with coronary artery disease. Int. J. Cardiol. 2017, 227, 61–65. [Google Scholar] [CrossRef]
- Myśliwiec, H.; Kiluk, P.; Żelazowska-Rutkowska, B.; Baran, A.; Flisiak, I. Influence of narrowband ultraviolet B phototherapy on serum tumour necrosis factor-like weak inducer of apoptosis (TWEAK) in patients with psoriasis. Clin. Exp. Dermatol. 2017, 42, 786–790. [Google Scholar] [CrossRef]
- Truzzi, F.; Marconi, A.; Atzei, P.; Panza, M.C.; Lotti, R.; Dallaglio, K.; Tiberio, R.; Palazzo, E.; Vaschieri, C.; Pincelli, C. p75 neurotrophin receptor mediates apoptosis in transit-amplifying cells and its overexpression restores cell death in psoriatic keratinocytes. Cell Death Differ. 2010, 18, 948–958. [Google Scholar] [CrossRef] [Green Version]
- Nykjaer, A.; Lee, R.; Teng, K.; Jansen, P.; Madsen, P.S.; Nielsen, M.S.; Jacobsen, C.; Kliemannel, M.; Schwarz, E.; Willnow, T.E.; et al. Sortilin is essential for proNGF-induced neuronal cell death. Nature 2004, 427, 843–848. [Google Scholar] [CrossRef]
- Barter, P.; Faergeman, O.; Havel, R. Metabolism of cholesteryl esters of very low density lipoproteins in the guinea pig. Metabolism 1977, 26, 615–622. [Google Scholar] [CrossRef]
- Chowaniec, Z.; Skoczyńska, A. Plasma lipid transfer proteins: The role of PLTP and CETP in atherogenesis. Adv. Clin. Exp. Med. 2018, 27, 429–436. [Google Scholar] [CrossRef]
- Yamashita, S.; Matsuzawa, Y. Re-evaluation of cholesteryl ester transfer protein function in atherosclerosis based upon genetics and pharmacological manipulation. Curr. Opin. Lipidol. 2016, 27, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Ferri, N.; Corsini, A.; Sirtori, C.R.; Ruscica, M. Present therapeutic role of cholesteryl ester transfer protein inhibitors. Pharmacol. Res. 2018, 128, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Barter, P.J.; Rye, K.-A. Cholesteryl Ester Transfer Protein Inhibitors as Agents to Reduce Coronary Heart Disease Risk. Cardiol. Clin. 2018, 36, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Amer, N.N.; Shaaban, G.M. Association of Serum Cholesterol Ester Transfer Protein Levels with Taq IB Polymorphism in Acute Coronary Syndrome. Lab. Med. 2019, 51, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, A.E.M.; Maranhão, R.C.; Carvalho, P.O.; Freitas, F.R.; Silva, B.M.O.; Curiati, M.N.C.; Filho, R.K.; Pereira-Barretto, A.C. Cholesteryl ester transfer protein (CETP), HDL capacity of receiving cholesterol and status of inflammatory cytokines in patients with severe heart failure. Lipids Heal. Dis. 2018, 17, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girona, J.; Ibarretxe, D.; Plana, N.; Guaita-Esteruelas, S.; Amigó, N.; Heras, M.; Masana, L. Circulating PCSK9 levels and CETP plasma activity are independently associated in patients with metabolic diseases. Cardiovasc. Diabetol. 2016, 15, 107. [Google Scholar] [CrossRef] [Green Version]
- Barter, P.J.; Caulfield, M.J.; Eriksson, M.; Grundy, S.M.; Kastelein, J.J.; Komajda, M.; López-Sendón, J.; Mosca, L.; Tardif, J.-C.; Waters, D.D.; et al. Effects of Torcetrapib in Patients at High Risk for Coronary Events. N. Engl. J. Med. 2007, 357, 2109–2122. [Google Scholar] [CrossRef] [Green Version]
- Menon, V.; Kumar, A.; Patel, D.R.; John, J.S.; Riesmeyer, J.; Weerakkody, G.; Ruotolo, G.; E Wolski, K.; McErlean, E.; Cremer, P.C.; et al. Effect of CETP inhibition with evacetrapib in patients with diabetes mellitus enrolled in the ACCELERATE trial. BMJ Open Diabetes Res. Care 2020, 8, e000943. [Google Scholar] [CrossRef] [Green Version]
- Hovingh, G.K.; Ray, K.K.; Boekholdt, S.M. Is Cholesteryl Ester Transfer Protein Inhibition an Effective Strategy to Reduce Cardiovascular Risk? CETP as a Target to Lower CVD Risk: Suspension of Disbelief? Circulation 2015, 132, 433–440. [Google Scholar] [CrossRef]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krahel, J.A.; Baran, A.; Kamiński, T.W.; Flisiak, I. Proprotein Convertase Subtilisin/Kexin Type 9, Angiopoietin-Like Protein 8, Sortilin, and Cholesteryl Ester Transfer Protein—Friends of Foes for Psoriatic Patients at the Risk of Developing Cardiometabolic Syndrome? Int. J. Mol. Sci. 2020, 21, 3682. https://doi.org/10.3390/ijms21103682
Krahel JA, Baran A, Kamiński TW, Flisiak I. Proprotein Convertase Subtilisin/Kexin Type 9, Angiopoietin-Like Protein 8, Sortilin, and Cholesteryl Ester Transfer Protein—Friends of Foes for Psoriatic Patients at the Risk of Developing Cardiometabolic Syndrome? International Journal of Molecular Sciences. 2020; 21(10):3682. https://doi.org/10.3390/ijms21103682
Chicago/Turabian StyleKrahel, Julita Anna, Anna Baran, Tomasz W. Kamiński, and Iwona Flisiak. 2020. "Proprotein Convertase Subtilisin/Kexin Type 9, Angiopoietin-Like Protein 8, Sortilin, and Cholesteryl Ester Transfer Protein—Friends of Foes for Psoriatic Patients at the Risk of Developing Cardiometabolic Syndrome?" International Journal of Molecular Sciences 21, no. 10: 3682. https://doi.org/10.3390/ijms21103682
APA StyleKrahel, J. A., Baran, A., Kamiński, T. W., & Flisiak, I. (2020). Proprotein Convertase Subtilisin/Kexin Type 9, Angiopoietin-Like Protein 8, Sortilin, and Cholesteryl Ester Transfer Protein—Friends of Foes for Psoriatic Patients at the Risk of Developing Cardiometabolic Syndrome? International Journal of Molecular Sciences, 21(10), 3682. https://doi.org/10.3390/ijms21103682