Preparation of Biphenyl-Conjugated Bromotyrosine for Inhibition of PD-1/PD-L1 Immune Checkpoint Interactions

 and
and
Abstract
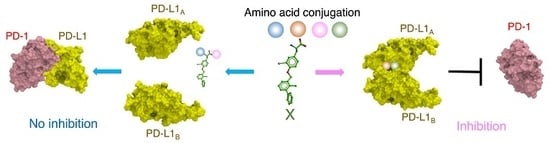
1. Introduction
2. Results
2.1. In Silico Docking Simulation and Organic Chemistry Synthesis of a Biphenyl-Conjugated Bromotyrosine
2.2. Inhibition Assays of PD-1/PD-L1 Binding by BMS-8 and X
2.3. Fragmentation of BMS-8 and Conjugation of Compounds to Prepare X
2.4. Docking Simulation and Inhibition Assay of Amino-Xs
3. Materials and Methods
3.1. Materials for Organic Chemistry Synthesis
3.2. Synthesis of a Biphenyl-Conjugated Bromotyrosine




3.3. Solid-State Peptide Synthesis
3.4. Characterization
3.5. Determination of the IC50 Value by AlphaLISA®
3.5.1. Principle of the Competitive Binding Assay
3.5.2. Preparation of Samples
3.5.3. AlphaLISA® Measurement and Analysis
3.6. Docking Simulation of Compounds
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PD-1 | Programmed cell death 1 |
| PD-L1 | Programmed cell death-ligand 1 |
| PD-L1A | PD-L1 chain A |
| PD-L1B | PD-L1 chain B |
| PD-L1AB | Homodimer of PD-L1A/PD-L1B chains |
| Alpha | Amplified Luminescence Proximity Homogeneous Assay |
| X | biphenyl-conjugated bromotyrosine |
| Amino-X | Amino acid conjugated-X |
| MALDI-TOF MS | Matrix assisted laser desorption/ionization-time of flight mass spectrometry |
| RMSD | Root mean square deviation |
| KD | Equilibrium dissociation constant |
| IC50 | 50% maximal inhibitory concentration |
| CC | Correlation coefficient |
| HTRF | Homogenous Time-Resolved Fluorescence |
References
- Rius, M.; Lyko, F. Epigenetic cancer therapy: Rationales, targets and drugs. Oncogene 2011, 31, 4257–4265. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune checkpoint blockade in cancer therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef]
- Lee, L.; Gupta, M.; Sahasranaman, S. Immune Checkpoint inhibitors: An introduction to the next-generation cancer immunotherapy. J. Clin. Pharmacol. 2016, 56, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Hoos, A. Development of immuno-oncology drugs-from CTLA4 to PD1 to the next generations. Nat. Rev. Drug Discov. 2016, 15, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Dong, H.; Zhu, G.; Tamada, K.; Chen, L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med. 1999, 5, 1365–1369. [Google Scholar] [CrossRef]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef]
- Dong, H.D.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.F.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef]
- Lee, K.M.; Chuang, E.; Griffin, M.; Khattri, R.; Hong, D.K.; Zhang, W.; Straus, D.; Samelson, L.E.; Thompson, C.B.; Bluestone, J.A. Molecular basis of T cell inactivation by CTLA-4. Science 1998, 282, 2263–2266. [Google Scholar] [CrossRef] [PubMed]
- Stamper, C.C.; Zhang, Y.; Tobin, J.F.; Erbe, D.V.; Ikemizu, S.; Davis, S.J.; Stahl, M.L.; Seehra, J.; Somers, W.S.; Mosyak, L. Crystal structure of the B7-1/CTLA-4 complex that inhibits human immune responses. Nature 2001, 410, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Drake, C.G. Basic overview of current immunotherapy approaches in urologic malignancy. Urol. Oncol. Semin. Orig. Investig. 2006, 24, 413–418. [Google Scholar] [CrossRef]
- Weiner, L.M.; Surana, R.; Wang, S. Monoclonal antibodies: Versatile platforms for cancer immunotherapy. Nat. Rev. Immunol. 2010, 10, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.-T.; Hou, Q.-Y.; Wang, N. Monoclonal antibodies in cancer therapy. Clin. Oncol. Cancer Res. 2011, 8, 215–219. [Google Scholar] [CrossRef]
- Pento, J.T. Monoclonal Antibodies for the Treatment of Cancer. Anticancer Res. 2017, 37, 5935–5939. [Google Scholar] [CrossRef]
- Reichert, J.M. Monoclonal Antibodies as Innovative Therapeutics. Curr. Pharm. Biotechnol. 2008, 9, 423–430. [Google Scholar] [CrossRef]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. New Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Wang, C.Y.; Thudium, K.B.; Han, M.H.; Wang, X.T.; Huang, H.C.; Feingersh, D.; Garcia, C.; Wu, Y.; Kuhne, M.; Srinivasan, M.; et al. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol. Res. 2014, 2, 846–856. [Google Scholar] [CrossRef]
- Kimiz-Gebologlu, I.; Gulce-Iz, S.; Biray-Avci, C. Monoclonal antibodies in cancer immunotherapy. Mol. Boil. Rep. 2018, 45, 2935–2940. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Ishida, T.; Yoshikawa, K.; Ueda, R. Current status of immunotherapy. Jpn. J. Clin. Oncol. 2016, 46, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, J. Current status and future directions of cancer immunotherapy. J. Cancer 2018, 9, 1773–1781. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- El-Osta, H.; Shahid, K.; Mills, G.M.; Peddi, P. Immune checkpoint inhibitors: The new frontier in non-small-cell lung cancer treatment. Onco Targets Ther. 2016, 9, 5101–5116. [Google Scholar] [CrossRef]
- Shukla, A.A.; Thommes, J. Recent advances in large-scale production of monoclonal antibodies and related proteins. Trends Biotechnol. 2010, 28, 253–261. [Google Scholar] [CrossRef]
- Chames, P.; Van Regenmortel, M.; Weiss, E.; Baty, D. Therapeutic antibodies: Successes, limitations and hopes for the future. Br. J. Pharmacol. 2009, 157, 220–233. [Google Scholar] [CrossRef]
- Bojadzic, D.; Buchwald, P. Toward small-molecule inhibition of protein-protein interactions: general aspects and recent progress in targeting costimulatory and coinhibitory (immune checkpoint) interactions. Curr. Top. Med. Chem. 2018, 18, 674–699. [Google Scholar] [CrossRef]
- Golani, L.K.; Wallace-Povirk, A.; Deis, S.M.; Wong, J.; Ke, J.; Gu, X.; Raghavan, S.; Wilson, M.R.; Li, X.; Polin, L.; et al. Tumor targeting with novel 6-substituted pyrrolo [2,3-d] pyrimidine antifolates with heteroatom bridge substitutions via cellular uptake by folate receptor alpha and the proton-coupled folate transporter and inhibition of de novo purine nucleotide biosynthesis. J. Med. Chem. 2016, 59, 7856–7876. [Google Scholar] [CrossRef]
- Chupak, L.S.; Zheng, X. Compounds Useful as Immunomodulators. Bristol-Myers Squibb Company. WO2015034820 A1, 12 March 2015. [Google Scholar]
- Zak, K.M.; Grudnik, P.; Guzik, K.; Zieba, B.J.; Musielak, B.; Dömling, A.S.S.; Dubin, G.; Holak, T.A. Holak. Structural basis for small molecule targeting of the programmed death ligand 1 (PD-L1). Oncotarget 2016, 7, 30323–30335. [Google Scholar] [CrossRef]
- Musielak, B.; Kocik, J.; Skalniak, L.; Magiera-Mularz, K.; Sala, D.; Czub, M.; Stec, M.; Siedlar, M.; Holak, T.A.; Plewka, J. CA-170-A potent small-molecule PD-L1 inhibitor or not? Molecules 2019, 24, 2804. [Google Scholar] [CrossRef] [PubMed]
- Abagyan, R.; Totrov, M.; Kuznetsov, D. ICM–A New method for protein modeling and design-applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 1994, 15, 488–506. [Google Scholar] [CrossRef]
- Bottegoni, G.; Kufareva, I.; Totrov, M.; Abagyan, R. Four-dimensional docking: A fast and accurate account of discrete receptor flexibility in ligand docking. J. Med. Chem. 2009, 52, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Bottegoni, G.; Kufareva, I.; Totrov, M.; Abagyan, R. A new method for ligand docking to flexible receptors by dual alanine scanning and refinement (SCARE). J. Comput. Aided. Mol. Des. 2008, 22, 311–325. [Google Scholar] [CrossRef][Green Version]
- Wang, W.; Hirano, Y.; Uzawa, T.; Liu, M.; Taiji, M.; Ito, Y. In vitro selection of a peptide aptamer that potentiates inhibition of cyclin-dependent kinase 2 by purvalanol. MedChemComm 2014, 5, 1400–1403. [Google Scholar] [CrossRef]
- Dharmatti, R.; Miyatake, H.; Nandakumar, A.; Ueda, M.; Kobayashi, K.; Kiga, D.; Yamamura, M.; Ito, Y. Enhancement of binding affinity of folate to its receptor by peptide conjugation. Int. J. Mol. Sci. 2019, 20, 2152. [Google Scholar] [CrossRef] [PubMed]
- Eglen, R.M.; Reisine, T.; Roby, P.; Rouleau, N.; Illy, C.; Bosse, R.; Bielefeld, M. The use of AlphaScreen technology in HTS: Current status. Curr. Chem. Genom. 2008, 1, 2–10. [Google Scholar] [CrossRef]
- Tan, S.; Zhang, H.; Chai, Y.; Song, H.; Tong, Z.; Wang, Q.; Qi, J.; Wong, G.; Zhu, X.; Liu, W.J.; et al. An unexpected N-terminal loop in PD-1 dominates binding by nivolumab. Nat. Commun. 2017, 8, 14369. [Google Scholar] [CrossRef]
- Guzik, K.; Zak, K.M.; Grudnik, P.; Magiera, K.; Musielak, B.; Torner, R.; Skalniak, L.; Domling, A.; Dubin, G.; Holak, T.A. Small-molecule inhibitors of the programmed cell death-1/programmed death-ligand 1 (PD-1/PD-L1) interaction via transiently induced protein states and dimerization of PD-L1. J. Med. Chem. 2017, 60, 5857–5867. [Google Scholar] [CrossRef]
- Skalniak, L.; Zak, K.M.; Guzik, K.; Magiera, K.; Musielak, B.; Pachota, M.; Szelazek, B.; Kocik, J.; Grudnik, P.; Tomala, M.; et al. Small-molecule inhibitors of PD-1/PD-L1 immune checkpoint alleviate the PD-L1-induced exhaustion of T-cells. Oncotarget 2017, 8, 72167–72181. [Google Scholar] [CrossRef]
- Magiera-Mularz, K.; Skalniak, L.; Zak, K.M.; Musielak, B.; Rudzinska-Szostak, E.; Berlicki, L.; Kocik, J.; Grudnik, P.; Sala, D.; Zarganes-Tzitzikas, T.; et al. Bioactive macrocyclic inhibitors of the PD-1/PD-L1 immune checkpoint. Angew. Chem. Int. Ed. 2017, 56, 13732–13735. [Google Scholar] [CrossRef] [PubMed]
- Lazar-Molnar, E.; Scandiuzzi, L.; Basu, I.; Quinn, T.; Sylvestre, E.; Palmieri, E.; Ramagopal, U.A.; Nathenson, S.G.; Guha, C.; Almo, S.C. Structure-guided development of a high-affinity human Programmed Cell Death-1: Implications for tumor immunotherapy. EBioMedicine 2017, 17, 30–44. [Google Scholar] [CrossRef]
- Wang, W.; Hirano, Y.; Uzawa, T.; Taiji, M.; Ito, Y. Peptide-assisted enhancement of inhibitory effects of small molecular inhibitors for kinases. Bull. Chem. Soc. Jpn. 2016, 89, 444–446. [Google Scholar] [CrossRef]
- Neves, M.A.; Totrov, M.; Abagyan, R. Docking and scoring with ICM: The benchmarking results and strategies for improvement. J. Comput. Mol. Des. 2012, 26, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Abagyan, R.; Totrov, M. Biased probability monte-carlo conformational searches and electrostatic calculations for peptides and proteins. J. Mol. Biol. 1994, 235, 983–1002. [Google Scholar] [CrossRef] [PubMed]
- Totrov, M.; Abagyan, R. Flexible protein-ligand docking by global energy optimization in internal coordinates. Proteins 1997, 29 (Suppl. 1), 215–220. [Google Scholar] [CrossRef]
- Schapira, M.; Totrov, M.; Abagyan, R. Prediction of the binding energy for small molecules, peptides and proteins. J. Mol. Recognit. 1999, 12, 177–190. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amino Acid Length | Sequence | Score | RMSD (Å) | IC50 (μM) |
|---|---|---|---|---|
| - | BMS-8 | −49.5 | - | 7.2 |
| 1 | X | −42.96 | 0.40 | 1.5 |
| 2 | GX | −41.0 | 0.52 | 448.5 |
| XG | −46.9 | 0.28 | 2.1 | |
| XS | −42.1 | 0.60 | 2655.0 | |
| XR | −45.7 | 0.37 | 892.0 | |
| XA | −43.1 | 0.47 | 22.3 | |
| XW | −43.3 | 0.51 | 845.0 | |
| 3 | YXC | −37.1 | 0.46 | 465.0 |
| WXG | −50.6 | 0.48 | 404.8 | |
| QXQ | −37.7 | 0.73 | 1961.0 | |
| CXA | −42.0 | 0.48 | 665.0 | |
| RXN | −40.3 | 0.63 | 405.3 | |
| SXR | −36.7 | 0.58 | 796.0 | |
| NXR | −50.3 | 0.46 | 982.0 | |
| CXR | −41.5 | 0.54 | 550.0 | |
| GXG | −43.6 | 0.39 | 676.0 | |
| XNL | −43.0 | 0.58 | 855.0 | |
| XNH | −40.7 | 0.50 | 313.0 | |
| XHP | −33.5 | 0.55 | 359.0 | |
| XGG | −36.1 | 0.57 | 6505.0 | |
| 4 | XCSE | −32.6 | 0.45 | 1555.0 |
| XGGG | −53.3 | 0.51 | 6766.0 | |
| 5 | WRXNN | −38.1 | 0.38 | 157.4 |
| ERXNK | −21.3 | 0.48 | 15.6 | |
| WRXNQ | −19.4 | 0.49 | 163.2 | |
| XRRRR | −28.3 | 0.45 | 435.6 | |
| XGGGG | −41.8 | 0.75 | 647.5 | |
| 6 | XGGGGG | −45.3 | 0.48 | 846.0 |
| 7 | CERXNKM | 4.65 | 1.80 | 308.2 |
| FWRXNNI | −7.30 | 0.41 | 311.8 |
| Length | CC of Score/IC50 | CC of RMSD/IC50 |
|---|---|---|
| 1-2 | 0.40 | 0.67 |
| 1-3 | 0.35 | 0.37 |
| 1-4 | 0 | 0.28 |
| 1-7 | −0.20 | 0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, E.-H.; Kawamoto, M.; Dharmatti, R.; Kobatake, E.; Ito, Y.; Miyatake, H. Preparation of Biphenyl-Conjugated Bromotyrosine for Inhibition of PD-1/PD-L1 Immune Checkpoint Interactions. Int. J. Mol. Sci. 2020, 21, 3639. https://doi.org/10.3390/ijms21103639
Kim E-H, Kawamoto M, Dharmatti R, Kobatake E, Ito Y, Miyatake H. Preparation of Biphenyl-Conjugated Bromotyrosine for Inhibition of PD-1/PD-L1 Immune Checkpoint Interactions. International Journal of Molecular Sciences. 2020; 21(10):3639. https://doi.org/10.3390/ijms21103639
Chicago/Turabian StyleKim, Eun-Hye, Masuki Kawamoto, Roopa Dharmatti, Eiry Kobatake, Yoshihiro Ito, and Hideyuki Miyatake. 2020. "Preparation of Biphenyl-Conjugated Bromotyrosine for Inhibition of PD-1/PD-L1 Immune Checkpoint Interactions" International Journal of Molecular Sciences 21, no. 10: 3639. https://doi.org/10.3390/ijms21103639
APA StyleKim, E.-H., Kawamoto, M., Dharmatti, R., Kobatake, E., Ito, Y., & Miyatake, H. (2020). Preparation of Biphenyl-Conjugated Bromotyrosine for Inhibition of PD-1/PD-L1 Immune Checkpoint Interactions. International Journal of Molecular Sciences, 21(10), 3639. https://doi.org/10.3390/ijms21103639