FAK Family Kinases in Vascular Diseases


Abstract
1. Introduction
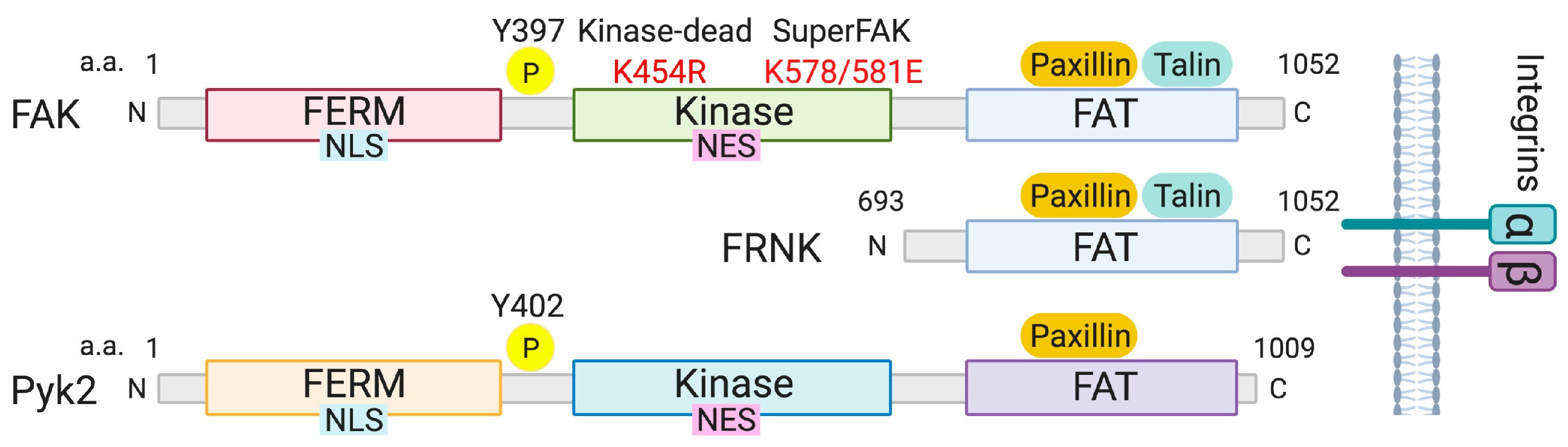
2. Structure of FAK Family Kinases
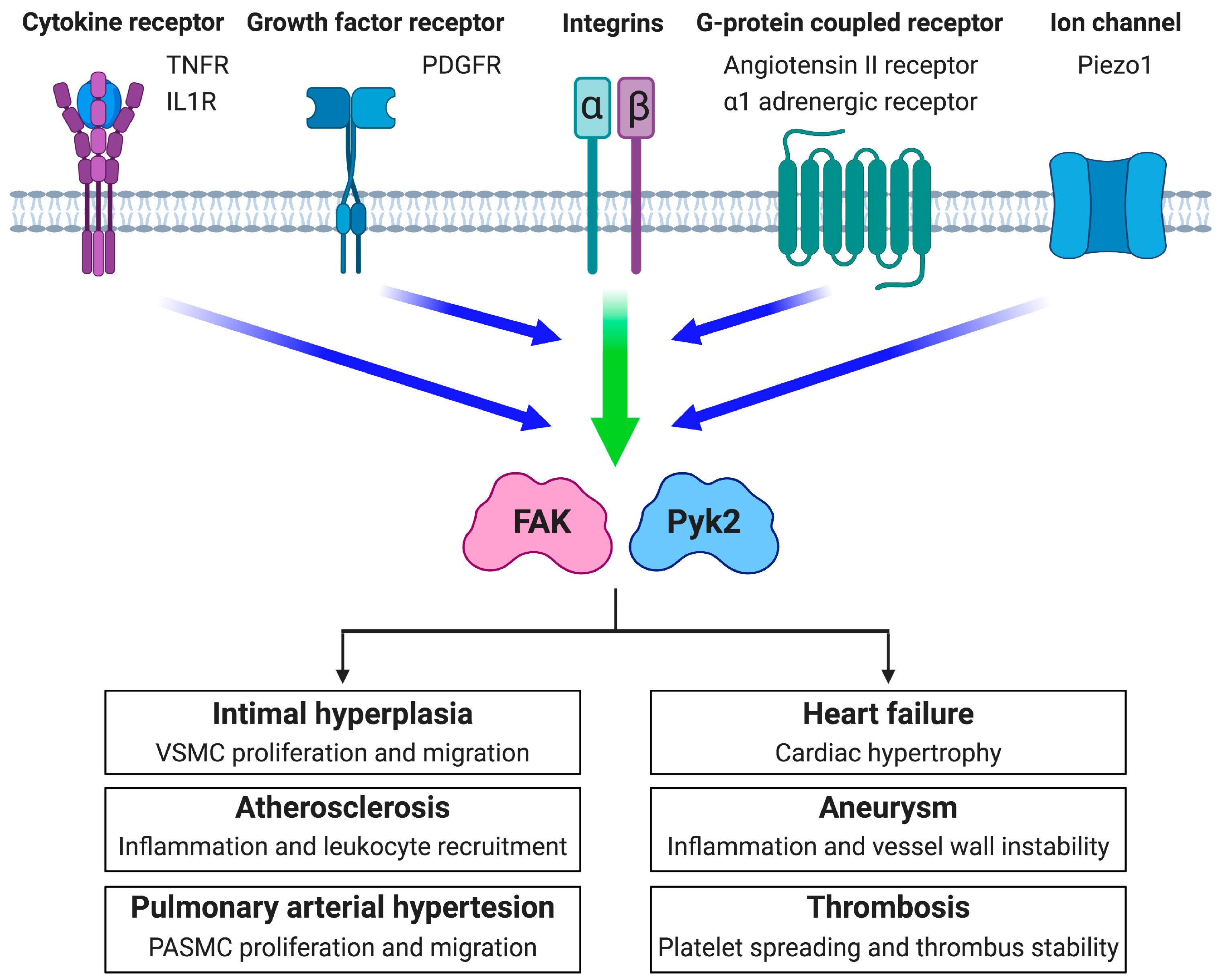
3. FAK Family Kinases in Vascular Diseases
3.1. Intimal Hyperplasia and Restenosis
3.2. Atherosclerosis
3.3. Pulmonary Arterial Hypertension
3.4. Heart Failure
3.5. Aneurysm
3.6. Thrombosis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AAA | abdominal aortic aneurysm |
| ApoE | apolipoprotein E |
| CM | cardiomyocyte |
| EC | endothelial cell |
| ECM | extracellular matrix |
| FAK | focal adhesion kinase |
| FAT | focal adhesion targeting |
| FERM | band 4.1, ezrin, radixin, moesin |
| FRNK | FAK-related nonkinase |
| HF/HC | high fat/high cholesterol |
| ICAM-1 | intercellular adhesion molecule-1 |
| IL-1β | interleukin-1β |
| KD | kinase-dead |
| KO | knockout |
| LDL | low-density lipoprotein |
| LDLR | low-density lipoprotein receptor |
| LVH | left ventricular hypertrophy |
| MMP | matrix metalloproteinase |
| oxLDL | oxidized low-density lipoprotein |
| PAH | pulmonary arterial hypertension |
| PDGF | platelet derived growth factor |
| PASMC | pulmonary artery smooth muscle cell |
| PE | phenylephrine |
| Pyk2 | proline-rich tyrosine kinase 2 |
| siRNA | small interfering RNA |
| Skp2 | S-phase kinase-associated protein 2 |
| TNF-α | tumor necrosis factor-α |
| VCAM-1 | vascular cell adhesion molecule-1 |
| VEGFR2 | vascular endothelial growth factor receptor 2 |
| VSMC | vascular smooth muscle cell |
| WT | wild-type |
References
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Chen, C.; Li, R.; Ross, R.S.; Manso, A.M. Integrins and integrin-related proteins in cardiac fibrosis. J. Mol. Cell Cardiol. 2016, 93, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Finney, A.C.; Stokes, K.Y.; Pattillo, C.B.; Orr, A.W. Integrin signaling in atherosclerosis. Cell Mol. Life Sci. 2017, 74, 2263–2282. [Google Scholar] [CrossRef] [PubMed]
- Estevez, B.; Shen, B.; Du, X. Targeting integrin and integrin signaling in treating thrombosis. Arterioscler Thromb. Vasc. Biol. 2015, 35, 24–29. [Google Scholar] [CrossRef]
- Boudreau, N.J.; Jones, P.L. Extracellular matrix and integrin signalling: The shape of things to come. Biochem. J. 1999, 339(Pt. 3), 481–488. [Google Scholar] [CrossRef]
- Harburger, D.S.; Calderwood, D.A. Integrin signalling at a glance. J. Cell Sci. 2009, 122, 159–163. [Google Scholar] [CrossRef]
- Malinin, N.L.; Pluskota, E.; Byzova, T.V. Integrin signaling in vascular function. Curr. Opin. Hematol. 2012, 19, 206–211. [Google Scholar] [CrossRef]
- Ponticos, M.; Smith, B.D. Extracellular matrix synthesis in vascular disease: Hypertension, and atherosclerosis. J. Biomed. Res. 2014, 28, 25–39. [Google Scholar] [CrossRef]
- Eliceiri, B.P. Integrin and growth factor receptor crosstalk. Circ. Res. 2001, 89, 1104–1110. [Google Scholar] [CrossRef]
- Ivaska, J.; Heino, J. Cooperation between integrins and growth factor receptors in signaling and endocytosis. Annu. Rev. Cell Dev. Biol. 2011, 27, 291–320. [Google Scholar] [CrossRef] [PubMed]
- Renshaw, M.W.; Ren, X.D.; Schwartz, M.A. Growth factor activation of MAP kinase requires cell adhesion. EMBO J. 1997, 16, 5592–5599. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.H.; Chen, Q.; Howe, A.; Juliano, R.L. Cell anchorage permits efficient signal transduction between ras and its downstream kinases. J. Biol. Chem. 1997, 272, 8849–8852. [Google Scholar] [CrossRef] [PubMed]
- Jeong, K.; Kim, J.H.; Murphy, J.M.; Park, H.; Kim, S.J.; Rodriguez, Y.A.R.; Kong, H.; Choi, C.; Guan, J.L.; Taylor, J.M.; et al. Nuclear Focal Adhesion Kinase Controls Vascular Smooth Muscle Cell Proliferation and Neointimal Hyperplasia Through GATA4-Mediated Cyclin D1 Transcription. Circ. Res. 2019, 125, 152–166. [Google Scholar] [CrossRef]
- Lim, S.T.; Chen, X.L.; Lim, Y.; Hanson, D.A.; Vo, T.T.; Howerton, K.; Larocque, N.; Fisher, S.J.; Schlaepfer, D.D.; Ilic, D. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol. Cell 2008, 29, 9–22. [Google Scholar] [CrossRef]
- Lim, S.T.; Miller, N.L.; Chen, X.L.; Tancioni, I.; Walsh, C.T.; Lawson, C.; Uryu, S.; Weis, S.M.; Cheresh, D.A.; Schlaepfer, D.D. Nuclear-localized focal adhesion kinase regulates inflammatory VCAM-1 expression. J. Cell Biol. 2012, 197, 907–919. [Google Scholar] [CrossRef]
- Ceccarelli, D.F.; Song, H.K.; Poy, F.; Schaller, M.D.; Eck, M.J. Crystal structure of the FERM domain of focal adhesion kinase. J. Biol. Chem. 2006, 281, 252–259. [Google Scholar] [CrossRef]
- Ossovskaya, V.; Lim, S.T.; Ota, N.; Schlaepfer, D.D.; Ilic, D. FAK nuclear export signal sequences. FEBS Lett. 2008, 582, 2402–2406. [Google Scholar] [CrossRef]
- Lim, S.T.; Miller, N.L.; Nam, J.O.; Chen, X.L.; Lim, Y.; Schlaepfer, D.D. Pyk2 inhibition of p53 as an adaptive and intrinsic mechanism facilitating cell proliferation and survival. J. Biol. Chem. 2010, 285, 1743–1753. [Google Scholar] [CrossRef]
- Sun, S.; Wu, H.J.; Guan, J.L. Nuclear FAK and its kinase activity regulate VEGFR2 transcription in angiogenesis of adult mice. Sci. Rep. 2018, 8, 2550. [Google Scholar] [CrossRef]
- Avraham, H.; Park, S.Y.; Schinkmann, K.; Avraham, S. RAFTK/Pyk2-mediated cellular signalling. Cell Signal. 2000, 12, 123–133. [Google Scholar] [CrossRef]
- Mitra, S.K.; Hanson, D.A.; Schlaepfer, D.D. Focal adhesion kinase: In command and control of cell motility. Nat. Rev. Mol. Cell Biol. 2005, 6, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Eliceiri, B.P.; Puente, X.S.; Hood, J.D.; Stupack, D.G.; Schlaepfer, D.D.; Huang, X.Z.; Sheppard, D.; Cheresh, D.A. Src-mediated coupling of focal adhesion kinase to integrin alpha(v)beta5 in vascular endothelial growth factor signaling. J. Cell Biol. 2002, 157, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.L.; Nam, J.O.; Jean, C.; Lawson, C.; Walsh, C.T.; Goka, E.; Lim, S.T.; Tomar, A.; Tancioni, I.; Uryu, S.; et al. VEGF-induced vascular permeability is mediated by FAK. Dev. Cell 2012, 22, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Barsukov, I.L.; Prescot, A.; Bate, N.; Patel, B.; Floyd, D.N.; Bhanji, N.; Bagshaw, C.R.; Letinic, K.; Di Paolo, G.; De Camilli, P.; et al. Phosphatidylinositol phosphate kinase type 1gamma and beta1-integrin cytoplasmic domain bind to the same region in the talin FERM domain. J. Biol. Chem. 2003, 278, 31202–31209. [Google Scholar] [CrossRef]
- Cooper, L.A.; Shen, T.L.; Guan, J.L. Regulation of focal adhesion kinase by its amino-terminal domain through an autoinhibitory interaction. Mol. Cell Biol. 2003, 23, 8030–8041. [Google Scholar] [CrossRef]
- Cai, X.; Lietha, D.; Ceccarelli, D.F.; Karginov, A.V.; Rajfur, Z.; Jacobson, K.; Hahn, K.M.; Eck, M.J.; Schaller, M.D. Spatial and temporal regulation of focal adhesion kinase activity in living cells. Mol. Cell Biol. 2008, 28, 201–214. [Google Scholar] [CrossRef]
- Goni, G.M.; Epifano, C.; Boskovic, J.; Camacho-Artacho, M.; Zhou, J.; Bronowska, A.; Martin, M.T.; Eck, M.J.; Kremer, L.; Grater, F.; et al. Phosphatidylinositol 4,5-bisphosphate triggers activation of focal adhesion kinase by inducing clustering and conformational changes. Proc. Natl. Acad. Sci. USA 2014, 111, E3177–E3186. [Google Scholar] [CrossRef]
- Zhou, J.; Aponte-Santamaria, C.; Sturm, S.; Bullerjahn, J.T.; Bronowska, A.; Grater, F. Mechanism of Focal Adhesion Kinase Mechanosensing. PLoS Comput. Biol. 2015, 11, e1004593. [Google Scholar] [CrossRef]
- Parsons, J.T. Focal adhesion kinase: The first ten years. J. Cell Sci. 2003, 116, 1409–1416. [Google Scholar] [CrossRef]
- Zheng, C.; Xing, Z.; Bian, Z.C.; Guo, C.; Akbay, A.; Warner, L.; Guan, J.L. Differential regulation of Pyk2 and focal adhesion kinase (FAK). The C-terminal domain of FAK confers response to cell adhesion. J. Biol. Chem. 1998, 273, 2384–2389. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.M.; Mack, C.P.; Nolan, K.; Regan, C.P.; Owens, G.K.; Parsons, J.T. Selective expression of an endogenous inhibitor of FAK regulates proliferation and migration of vascular smooth muscle cells. Mol. Cell Biol. 2001, 21, 1565–1572. [Google Scholar] [CrossRef] [PubMed]
- Richardson, A.; Parsons, T. A mechanism for regulation of the adhesion-associated proteintyrosine kinase pp125FAK. Nature 1996, 380, 538–540. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.J.; Li, M.B.; Wu, X.; Wu, S.; Zhu, W.; Chen, D.; Luo, M.; Eitenmuller, I.; Kampmann, A.; Schaper, J.; et al. Activation of the integrins alpha 5beta 1 and alpha v beta 3 and focal adhesion kinase (FAK) during arteriogenesis. Mol. Cell Biochem. 2009, 322, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Zhu, Q.; Liu, H.; Zuo, C.; He, Y.; Chen, G.; Lu, A. Osteoprotegerin Disruption Attenuates HySu-Induced Pulmonary Hypertension Through Integrin alphavbeta3/FAK/AKT Pathway Suppression. Circ. Cardiovasc. Genet. 2017, 10. [Google Scholar] [CrossRef]
- Turner, C.J.; Badu-Nkansah, K.; Crowley, D.; van der Flier, A.; Hynes, R.O. alpha5 and alphav integrins cooperate to regulate vascular smooth muscle and neural crest functions in vivo. Development 2015, 142, 797–808. [Google Scholar] [CrossRef][Green Version]
- Petzold, T.; Orr, A.W.; Hahn, C.; Jhaveri, K.A.; Parsons, J.T.; Schwartz, M.A. Focal adhesion kinase modulates activation of NF-kappaB by flow in endothelial cells. Am. J. Physiol Cell Physiol 2009, 297, C814–C822. [Google Scholar] [CrossRef]
- Albarran-Juarez, J.; Iring, A.; Wang, S.; Joseph, S.; Grimm, M.; Strilic, B.; Wettschureck, N.; Althoff, T.F.; Offermanns, S. Piezo1 and Gq/G11 promote endothelial inflammation depending on flow pattern and integrin activation. J. Exp. Med. 2018, 215, 2655–2672. [Google Scholar] [CrossRef]
- Yurdagul, A., Jr.; Green, J.; Albert, P.; McInnis, M.C.; Mazar, A.P.; Orr, A.W. alpha5beta1 integrin signaling mediates oxidized low-density lipoprotein-induced inflammation and early atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1362–1373. [Google Scholar] [CrossRef]
- Hu, S.; Liu, Y.; You, T.; Heath, J.; Xu, L.; Zheng, X.; Wang, A.; Wang, Y.; Li, F.; Yang, F.; et al. Vascular Semaphorin 7A Upregulation by Disturbed Flow Promotes Atherosclerosis Through Endothelial beta1 Integrin. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 335–343. [Google Scholar] [CrossRef]
- Chen, J.; Green, J.; Yurdagul, A., Jr.; Albert, P.; McInnis, M.C.; Orr, A.W. alphavbeta3 Integrins Mediate Flow-Induced NF-kappaB Activation, Proinflammatory Gene Expression, and Early Atherogenic Inflammation. Am. J. Pathol. 2015, 185, 2575–2589. [Google Scholar] [CrossRef] [PubMed]
- Kuppuswamy, D.; Kerr, C.; Narishige, T.; Kasi, V.S.; Menick, D.R.; Cooper, G.t. Association of tyrosine-phosphorylated c-Src with the cytoskeleton of hypertrophying myocardium. J. Biol. Chem. 1997, 272, 4500–4508. [Google Scholar] [CrossRef] [PubMed]
- Shattil, S.J.; Haimovich, B.; Cunningham, M.; Lipfert, L.; Parsons, J.T.; Ginsberg, M.H.; Brugge, J.S. Tyrosine phosphorylation of pp125FAK in platelets requires coordinated signaling through integrin and agonist receptors. J. Biol. Chem. 1994, 269, 14738–14745. [Google Scholar] [PubMed]
- Cipolla, L.; Consonni, A.; Guidetti, G.; Canobbio, I.; Okigaki, M.; Falasca, M.; Ciraolo, E.; Hirsch, E.; Balduini, C.; Torti, M. The proline-rich tyrosine kinase Pyk2 regulates platelet integrin alphaIIbbeta3 outside-in signaling. J. Thromb. Haemost. 2013, 11, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Consonni, A.; Cipolla, L.; Guidetti, G.; Canobbio, I.; Ciraolo, E.; Hirsch, E.; Falasca, M.; Okigaki, M.; Balduini, C.; Torti, M. Role and regulation of phosphatidylinositol 3-kinase beta in platelet integrin alpha2beta1 signaling. Blood 2012, 119, 847–856. [Google Scholar] [CrossRef]
- Newby, A.C. An overview of the vascular response to injury: A tribute to the late Russell Ross. Toxicol. Lett. 2000, 112–113, 519–529. [Google Scholar] [CrossRef]
- Clowes, A.W.; Reidy, M.A.; Clowes, M.M. Mechanisms of stenosis after arterial injury. Lab. Invest. 1983, 49, 208–215. [Google Scholar]
- Chaabane, C.; Otsuka, F.; Virmani, R.; Bochaton-Piallat, M.L. Biological responses in stented arteries. Cardiovasc. Res. 2013, 99, 353–363. [Google Scholar] [CrossRef]
- Drachman, D.E.; Simon, D.I. Restenosis: Intracoronary Brachytherapy. Curr. Treat. Options Cardiovasc.Med. 2002, 4, 109–118. [Google Scholar] [CrossRef]
- Morla, A.O.; Mogford, J.E. Control of smooth muscle cell proliferation and phenotype by integrin signaling through focal adhesion kinase. Biochem. Biophys Res. Commun. 2000, 272, 298–302. [Google Scholar] [CrossRef]
- Perez, J.; Torres, R.A.; Rocic, P.; Cismowski, M.J.; Weber, D.S.; Darley-Usmar, V.M.; Lucchesi, P.A. PYK2 signaling is required for PDGF-dependent vascular smooth muscle cell proliferation. Am. J. Physiol. Cell Physiol. 2011, 301, C242–C251. [Google Scholar] [CrossRef] [PubMed]
- Sayers, R.L.; Sundberg-Smith, L.J.; Rojas, M.; Hayasaka, H.; Parsons, J.T.; Mack, C.P.; Taylor, J.M. FRNK expression promotes smooth muscle cell maturation during vascular development and after vascular injury. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 2115–2122. [Google Scholar] [CrossRef] [PubMed]
- Koshman, Y.E.; Kim, T.; Chu, M.; Engman, S.J.; Iyengar, R.; Robia, S.L.; Samarel, A.M. FRNK inhibition of focal adhesion kinase-dependent signaling and migration in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2226–2233. [Google Scholar] [CrossRef] [PubMed]
- Walker, H.A.; Whitelock, J.M.; Garl, P.J.; Nemenoff, R.A.; Stenmark, K.R.; Weiser-Evans, M.C. Perlecan up-regulation of FRNK suppresses smooth muscle cell proliferation via inhibition of FAK signaling. Mol. Biol. Cell 2003, 14, 1941–1952. [Google Scholar] [CrossRef]
- Mui, K.L.; Bae, Y.H.; Gao, L.; Liu, S.L.; Xu, T.; Radice, G.L.; Chen, C.S.; Assoian, R.K. N-Cadherin Induction by ECM Stiffness and FAK Overrides the Spreading Requirement for Proliferation of Vascular Smooth Muscle Cells. Cell Rep. 2015, 10, 1477–1486. [Google Scholar] [CrossRef]
- Klein, E.A.; Yin, L.; Kothapalli, D.; Castagnino, P.; Byfield, F.J.; Xu, T.; Levental, I.; Hawthorne, E.; Janmey, P.A.; Assoian, R.K. Cell-cycle control by physiological matrix elasticity and in vivo tissue stiffening. Curr. Biol. 2009, 19, 1511–1518. [Google Scholar] [CrossRef]
- Bae, Y.H.; Mui, K.L.; Hsu, B.Y.; Liu, S.L.; Cretu, A.; Razinia, Z.; Xu, T.; Pure, E.; Assoian, R.K. A FAK-Cas-Rac-lamellipodin signaling module transduces extracellular matrix stiffness into mechanosensitive cell cycling. Sci. Signal. 2014, 7, ra57. [Google Scholar] [CrossRef]
- Bond, M.; Sala-Newby, G.B.; Newby, A.C. Focal adhesion kinase (FAK)-dependent regulation of S-phase kinase-associated protein-2 (Skp-2) stability. A novel mechanism regulating smooth muscle cell proliferation. J. Biol. Chem. 2004, 279, 37304–37310. [Google Scholar] [CrossRef]
- Chiu, J.J.; Chien, S. Effects of disturbed flow on vascular endothelium: Pathophysiological basis and clinical perspectives. Physiol. Rev. 2011, 91, 327–387. [Google Scholar] [CrossRef]
- Orr, A.W.; Ginsberg, M.H.; Shattil, S.J.; Deckmyn, H.; Schwartz, M.A. Matrix-specific suppression of integrin activation in shear stress signaling. Mol. Biol. Cell 2006, 17, 4686–4697. [Google Scholar] [CrossRef]
- Wang, Y.; Chang, J.; Li, Y.C.; Li, Y.S.; Shyy, J.Y.; Chien, S. Shear stress and VEGF activate IKK via the Flk-1/Cbl/Akt signaling pathway. Am. J. Physiol. Heart. Circ. Physiol. 2004, 286, H685–H692. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Flores, L.; Lu, S.; Miao, H.; Li, Y.S.; Chien, S. Shear Stress Regulates the Flk-1/Cbl/PI3K/NF-kappaB Pathway Via Actin and Tyrosine Kinases. Cell Mol. Bioeng. 2009, 2, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.E.; Tedbury, P.R.; Homer-Vanniasinkam, S.; Walker, J.H.; Ponnambalam, S. Biochemistry and cell biology of mammalian scavenger receptors. Atherosclerosis 2005, 182, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, J.P.; Major, A.S. How Oxidized Low-Density Lipoprotein Activates Inflammatory Responses. Crit Rev. Immunol. 2018, 38, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Yurdagul, A., Jr.; Sulzmaier, F.J.; Chen, X.L.; Pattillo, C.B.; Schlaepfer, D.D.; Orr, A.W. Oxidized LDL induces FAK-dependent RSK signaling to drive NF-kappaB activation and VCAM-1 expression. J. Cell Sci. 2016, 129, 1580–1591. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.M.; Jeong, K.; Rodriguez, Y.A.R.; Kim, J.H.; Ahn, E.E.; Lim, S.S. FAK and Pyk2 activity promote TNF-alpha and IL-1beta-mediated pro-inflammatory gene expression and vascular inflammation. Sci. Rep. 2019, 9, 7617. [Google Scholar] [CrossRef]
- Yamaura, T.; Kasaoka, T.; Iijima, N.; Kimura, M.; Hatakeyama, S. Evaluation of therapeutic effects of FAK inhibition in murine models of atherosclerosis. BMC Res. Notes 2019, 12, 200. [Google Scholar] [CrossRef]
- Thenappan, T.; Chan, S.Y.; Weir, E.K. Role of extracellular matrix in the pathogenesis of pulmonary arterial hypertension. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1322–H1331. [Google Scholar] [CrossRef]
- Dieffenbach, P.B.; Maracle, M.; Tschumperlin, D.J.; Fredenburgh, L.E. Mechanobiological Feedback in Pulmonary Vascular Disease. Front. Physiol. 2018, 9, 951. [Google Scholar] [CrossRef]
- Bijli, K.M.; Kang, B.Y.; Sutliff, R.L.; Hart, C.M. Proline-rich tyrosine kinase 2 downregulates peroxisome proliferator-activated receptor gamma to promote hypoxia-induced pulmonary artery smooth muscle cell proliferation. Pulm. Circ. 2016, 6, 202–210. [Google Scholar] [CrossRef]
- Fukai, K.; Nakamura, A.; Hoshino, A.; Nakanishi, N.; Okawa, Y.; Ariyoshi, M.; Kaimoto, S.; Uchihashi, M.; Ono, K.; Tateishi, S.; et al. Pyk2 aggravates hypoxia-induced pulmonary hypertension by activating HIF-1alpha. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H951–H959. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wilson, J.L.; Rupasinghe, C.; Usheva, A.; Warburton, R.; Kaplan, C.; Taylor, L.; Hill, N.; Mierke, D.F.; Polgar, P. Modulating the dysregulated migration of pulmonary arterial hypertensive smooth muscle cells with motif mimicking cell permeable peptides. Curr. Top. Pept. Protein. Res. 2015, 16, 1–17. [Google Scholar] [PubMed]
- Paulin, R.; Meloche, J.; Courboulin, A.; Lambert, C.; Haromy, A.; Courchesne, A.; Bonnet, P.; Provencher, S.; Michelakis, E.D.; Bonnet, S. Targeting cell motility in pulmonary arterial hypertension. Eur. Respir. J. 2014, 43, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Graf, K.; Do, Y.S.; Ashizawa, N.; Meehan, W.P.; Giachelli, C.M.; Marboe, C.C.; Fleck, E.; Hsueh, W.A. Myocardial osteopontin expression is associated with left ventricular hypertrophy. Circulation 1997, 96, 3063–3071. [Google Scholar] [CrossRef] [PubMed]
- Mamuya, W.; Chobanian, A.; Brecher, P. Age-related changes in fibronectin expression in spontaneously hypertensive, Wistar-Kyoto, and Wistar rat hearts. Circ. Res. 1992, 71, 1341–1350. [Google Scholar] [CrossRef]
- Terracio, L.; Rubin, K.; Gullberg, D.; Balog, E.; Carver, W.; Jyring, R.; Borg, T.K. Expression of collagen binding integrins during cardiac development and hypertrophy. Circ. Res. 1991, 68, 734–744. [Google Scholar] [CrossRef]
- Taylor, J.M.; Rovin, J.D.; Parsons, J.T. A role for focal adhesion kinase in phenylephrine-induced hypertrophy of rat ventricular cardiomyocytes. J. Biol. Chem. 2000, 275, 19250–19257. [Google Scholar] [CrossRef]
- Bayer, A.L.; Heidkamp, M.C.; Patel, N.; Porter, M.J.; Engman, S.J.; Samarel, A.M. PYK2 expression and phosphorylation increases in pressure overload-induced left ventricular hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H695–H706. [Google Scholar] [CrossRef]
- DiMichele, L.A.; Doherty, J.T.; Rojas, M.; Beggs, H.E.; Reichardt, L.F.; Mack, C.P.; Taylor, J.M. Myocyte-restricted focal adhesion kinase deletion attenuates pressure overload-induced hypertrophy. Circ. Res. 2006, 99, 636–645. [Google Scholar] [CrossRef]
- Clemente, C.F.; Tornatore, T.F.; Theizen, T.H.; Deckmann, A.C.; Pereira, T.C.; Lopes-Cendes, I.; Souza, J.R.; Franchini, K.G. Targeting focal adhesion kinase with small interfering RNA prevents and reverses load-induced cardiac hypertrophy in mice. Circ. Res. 2007, 101, 1339–1348. [Google Scholar] [CrossRef]
- Chen, J.; Kubalak, S.W.; Chien, K.R. Ventricular muscle-restricted targeting of the RXRalpha gene reveals a non-cell-autonomous requirement in cardiac chamber morphogenesis. Development 1998, 125, 1943–1949. [Google Scholar]
- Hakim, Z.S.; DiMichele, L.A.; Rojas, M.; Meredith, D.; Mack, C.P.; Taylor, J.M. FAK regulates cardiomyocyte survival following ischemia/reperfusion. J. Mol. Cell Cardiol. 2009, 46, 241–248. [Google Scholar] [CrossRef]
- Cheng, Z.; DiMichele, L.A.; Hakim, Z.S.; Rojas, M.; Mack, C.P.; Taylor, J.M. Targeted focal adhesion kinase activation in cardiomyocytes protects the heart from ischemia/reperfusion injury. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 924–933. [Google Scholar] [CrossRef]
- Bibli, S.I.; Zhou, Z.; Zukunft, S.; Fisslthaler, B.; Andreadou, I.; Szabo, C.; Brouckaert, P.; Fleming, I.; Papapetropoulos, A. Tyrosine phosphorylation of eNOS regulates myocardial survival after an ischaemic insult: Role of PYK2. Cardiovasc. Res. 2017, 113, 926–937. [Google Scholar] [CrossRef]
- Loot, A.E.; Schreiber, J.G.; Fisslthaler, B.; Fleming, I. Angiotensin II impairs endothelial function via tyrosine phosphorylation of the endothelial nitric oxide synthase. J. Exp. Med. 2009, 206, 2889–2896. [Google Scholar] [CrossRef]
- Quintana, R.A.; Taylor, W.R. Cellular Mechanisms of Aortic Aneurysm Formation. Circ. Res. 2019, 124, 607–618. [Google Scholar] [CrossRef]
- Yamashita, O.; Yoshimura, K.; Nagasawa, A.; Ueda, K.; Morikage, N.; Ikeda, Y.; Hamano, K. Periostin links mechanical strain to inflammation in abdominal aortic aneurysm. PLoS One 2013, 8, e79753. [Google Scholar] [CrossRef]
- Harada, T.; Yoshimura, K.; Yamashita, O.; Ueda, K.; Morikage, N.; Sawada, Y.; Hamano, K. Focal Adhesion Kinase Promotes the Progression of Aortic Aneurysm by Modulating Macrophage Behavior. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 156–165. [Google Scholar] [CrossRef]
- Shattil, S.J.; Kashiwagi, H.; Pampori, N. Integrin signaling: The platelet paradigm. Blood 1998, 91, 2645–2657. [Google Scholar] [CrossRef]
- Haimovich, B.; Kaneshiki, N.; Ji, P. Protein kinase C regulates tyrosine phosphorylation of pp125FAK in platelets adherent to fibrinogen. Blood 1996, 87, 152–161. [Google Scholar] [CrossRef]
- Hitchcock, I.S.; Fox, N.E.; Prevost, N.; Sear, K.; Shattil, S.J.; Kaushansky, K. Roles of focal adhesion kinase (FAK) in megakaryopoiesis and platelet function: Studies using a megakaryocyte lineage specific FAK knockout. Blood 2008, 111, 596–604. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Roh, M.E.; Cosgrove, M.; Gorski, K.; Hitchcock, I.S. Off-targets effects underlie the inhibitory effect of FAK inhibitors on platelet activation: Studies using Fak-deficient mice. J. Thromb. Haemost. 2013, 11, 1776–1778. [Google Scholar] [CrossRef] [PubMed]
- Weis, S.M.; Lim, S.T.; Lutu-Fuga, K.M.; Barnes, L.A.; Chen, X.L.; Gothert, J.R.; Shen, T.L.; Guan, J.L.; Schlaepfer, D.D.; Cheresh, D.A. Compensatory role for Pyk2 during angiogenesis in adult mice lacking endothelial cell FAK. J. Cell Biol. 2008, 181, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, A.C.; Pereira, A.H.M.; Ambrosio, A.L.B.; Consonni, S.R.; Rocha de Oliveira, R.; Bajgelman, M.C.; Dias, S.M.G.; Franchini, K.G. FAK Forms a Complex with MEF2 to Couple Biomechanical Signaling to Transcription in Cardiomyocytes. Structure 2016, 24, 1301–1310. [Google Scholar] [CrossRef] [PubMed]
- Tanjoni, I.; Walsh, C.; Uryu, S.; Tomar, A.; Nam, J.O.; Mielgo, A.; Lim, S.T.; Liang, C.; Koenig, M.; Sun, C.; et al. PND-1186 FAK inhibitor selectively promotes tumor cell apoptosis in three-dimensional environments. Cancer Biol. Ther. 2010, 9, 764–777. [Google Scholar] [CrossRef]
- Shapiro, I.M.; Kolev, V.N.; Vidal, C.M.; Kadariya, Y.; Ring, J.E.; Wright, Q.; Weaver, D.T.; Menges, C.; Padval, M.; McClatchey, A.I.; et al. Merlin deficiency predicts FAK inhibitor sensitivity: A synthetic lethal relationship. Sci. Transl. Med. 2014, 6, 237ra268. [Google Scholar] [CrossRef]
- Zhang, Y.; Moschetta, M.; Huynh, D.; Tai, Y.T.; Zhang, Y.; Zhang, W.; Mishima, Y.; Ring, J.E.; Tam, W.F.; Xu, Q.; et al. Pyk2 promotes tumor progression in multiple myeloma. Blood 2014, 124, 2675–2686. [Google Scholar] [CrossRef]
- Stokes, J.B.; Adair, S.J.; Slack-Davis, J.K.; Walters, D.M.; Tilghman, R.W.; Hershey, E.D.; Lowrey, B.; Thomas, K.S.; Bouton, A.H.; Hwang, R.F.; et al. Inhibition of focal adhesion kinase by PF-562,271 inhibits the growth and metastasis of pancreatic cancer concomitant with altering the tumor microenvironment. Mol. Cancer Ther. 2011, 10, 2135–2145. [Google Scholar] [CrossRef]
- Walsh, C.; Tanjoni, I.; Uryu, S.; Tomar, A.; Nam, J.O.; Luo, H.; Phillips, A.; Patel, N.; Kwok, C.; McMahon, G.; et al. Oral delivery of PND-1186 FAK inhibitor decreases tumor growth and spontaneous breast to lung metastasis in pre-clinical models. Cancer Biol. Ther. 2010, 9, 778–790. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Cell Type | Integrin | Function | Reference |
|---|---|---|---|
| Vascular smooth muscle cells | α5β1 | Binds fibronectin and promotes FAK activation | [34] |
| αvβ3 | Binds fibronectin and promotes FAK activation | [34] | |
| Promotes FAK activation upon binding to osteoprotegerin under hypoxic conditions | [35] | ||
| α5 | Dual knockout using SM22α-Cre reduced FAK activity and tyrosine phosphorylation of downstream target proteins | [36] | |
| αv | |||
| Endothelial cells | α5β1 | Promotes flow-induced FAK-mediated NF-κB transcriptional activation | [37] |
| Promotes disturbed flow activation of FAK | [38] | ||
| Mediates oxidized LDL activation of FAK | [39] | ||
| α1β1 | Increases FAK activation upon binding to semaphorin 7A | [40] | |
| αvβ3 | Promotes flow-induced FAK-mediated NF-κB transcriptional activation | [37] | |
| Promotes high shear flow-induced FAK expression and inflammatory gene expression | [41] | ||
| Cardiomyocytes | β3 | Increased association with FAK in pressure-overloaded hypertrophic hearts | [42] |
| Platelets | αIIbβ3 | Activates FAK upon binding fibrinogen in conjunction with costimulatory molecules like ADP, epinephrine, and thrombin | [43] |
| Activates Pyk2 upon binding fibrinogen to promote phosphorylation of c-Cbl | [44] | ||
| α2β1 | Activates Pyk2 to promote aIIbb3 inside-out signaling | [45] |
| Target Cells (Genotype) | Specific Cre or Modification | Result | Reference |
|---|---|---|---|
| Cardiomyocytes (FAK KO) | Nkx2-5 Cre | Worsened ischemia/reperfusion infarct injury | [82] |
| Cardiomyocytes (FAK KO) | MLC2v Cre | Attenuated hypertrophy | [79] |
| Cardiomyocyte (SuperFAK transgene) | βMHC promoter drives SuperFAK expression | Protected against ischemia/reperfusion | [83] |
| Vascular smooth muscle cells (FAK KO) | Myh11 CreERT2 | Prevented N-cadherin expression and intimal hyperplasia | [55] |
| Vascular smooth muscle cells (FAK KD knock-in) | Myh11 CreERT2T2 | Prevented GATA4-induced cyclin D1 expression and intimal hyperplasia | [14] |
| Global (FRNK KO) | Deletion of FRNK promoter | SMCs were unable to re-differentiate after artery ligation | [52] |
| Endothelial cells (FAK KD knock-in) | SCL CreERT2 | Reduced macrophage recruitment and VCAM-1 expression in C57BL/6 mice fed high fat diet | [65] |
| Platelets (FAK KO) | Pf4 Cre | Impaired platelet spreading and increased tail rebleeding | [91,92] |
| Platelets (Pyk2 global KO) | Deletion of Pyk2 gene | Decreased hypoxia-induced PAH and pulmonary vessel thickening | [71] |
| Impaired platelet activation, hemostasis, and thrombosis | [44,45] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murphy, J.M.; Jeong, K.; Lim, S.-T.S. FAK Family Kinases in Vascular Diseases. Int. J. Mol. Sci. 2020, 21, 3630. https://doi.org/10.3390/ijms21103630
Murphy JM, Jeong K, Lim S-TS. FAK Family Kinases in Vascular Diseases. International Journal of Molecular Sciences. 2020; 21(10):3630. https://doi.org/10.3390/ijms21103630
Chicago/Turabian StyleMurphy, James M., Kyuho Jeong, and Ssang-Taek Steve Lim. 2020. "FAK Family Kinases in Vascular Diseases" International Journal of Molecular Sciences 21, no. 10: 3630. https://doi.org/10.3390/ijms21103630
APA StyleMurphy, J. M., Jeong, K., & Lim, S.-T. S. (2020). FAK Family Kinases in Vascular Diseases. International Journal of Molecular Sciences, 21(10), 3630. https://doi.org/10.3390/ijms21103630